
Structure and performance of the LiFePO4 cathode material: from the bulk to the surface
Jiangtao
Hu
,
Weiyuan
Huang
,
Luyi
Yang
 * and
Feng
Pan
*
* and
Feng
Pan
*
School of Advanced Materials, Peking University Shenzhen Graduate School, Shenzhen 518055, China. E-mail: yangly@pkusz.edu.cn; panfeng@pkusz.edu.cn
First published on 23rd June 2020
Abstract
Currently, LiFePO4 is one of the most successfully commercialized cathode materials in the rechargeable lithium-ion battery (LIB) system, owing to its excellent safety performance and remarkable electrochemical properties and is expected to have a broader market in the near future. Although it is widely recognized that the crystalline structure of a cathode material largely dictates its electrochemical properties (e.g. capacity, cycle life and rate capabilities), this intrinsic connection in LiFePO4 has not been systematically reviewed. Different from the previous reviews, which mainly focus on the improvement of electrochemical performance by all kinds of techniques, in this review, the relationship between its electrochemical performance and bulk/surface structure is reviewed and discussed. First, it is revealed that the intra-particle Li+ transfer is influenced by several properties of the bulk, including crystalline structures, antisite defects and electronic structures. Next, it is demonstrated that the surface/interfacial structures of LiFePO4, which can be reconstructed artificially or spontaneously, also have great impacts on the performances. Lastly, the intrinsic connection between the structure and performance is preliminarily established, showing brand-new perspectives on the strategy for further improvement and contributing to a comprehensive understanding of LiFePO4.
 Jiangtao Hu | Dr Jiangtao Hu received his Ph.D degree in 2013 from Peking University, China. His research interests mainly lie in design and development of functional materials for energy storage and conversion applications such as batteries, supercapacitors, and catalysis. |
 Weiyuan Huang | Weiyuan Hang received his B.S. degree in New Energy Materials and Devices from South China Normal University in 2017. He is pursuing his Ph.D. degree in the School of Advanced Materials, Peking University, China. His research interest includes energy storage and conversion applications including lithium ion batteries, sodium ion batteries, and supercapacitors. |
 Luyi Yang | Dr Luyi Yang received his B.S. degree from the Department of Chemistry at Xiamen University (China) in 2010, and earned a Ph.D. degree from the School of Chemistry at Southampton University (U.K.) in 2015 under the supervision of Prof. John Owen. Dr Yang is currently a researcher at the School of Advanced Materials, Peking University Shenzhen Graduate School. His research interests mainly focus on the investigation of key materials in lithium batteries including solid-state electrolytes and cathode materials. |
 Feng Pan | Prof. Feng Pan, Chair-Professor, Founding Dean of School of Advanced Materials, Peking University Shenzhen Graduate School, and Director of National Center of Electric Vehicle Power Battery and Materials for International Research, received his B.S. degree from Dept. of Chemistry, Peking University, in 1985 and Ph.D. from Dept. of P&A Chemistry, University of Strathclyde, UK, with “Patrick D. Ritchie Prize” for the best Ph.D. in 1994. Prof. Pan has been engaged in fundamental research of structure chemistry, exploring “Material Genes” for Li-ion batteries and developing novel energy conversion-storage materials and devices. He also received the 2018 ECS Battery Division Technology Award. |
1. Introduction
The cathode is regarded as a key component in lithium-ion batteries (LIBs) and the determinant of commercialization since it directly determines the electrochemical performance.1–5 As the first-generation cathode material, layered oxide LiCoO2 was commercialized with the C6Li anode by Sony in 1991. Afterwards, other kinds of materials such as layered transition metal oxides (LiNi1−y−zMnyCozO2), Li-rich layered oxides, spinel oxides (LiMn2O4), polyanion oxides (LiFePO4), etc., have emerged in recent decades.6–11 Despite the pursuit of high-energy-density battery materials, safety is still the paramount demand in battery systems for both portable electronic devices and electric vehicles (EVs). Therefore, demonstrating excellent safety features, LiFePO4 has been the focus in both scientific research and the industrial community since the first report in 1997 by Goodenough et al.12 Moreover, its moderate operating voltage (3.5 V vs. Li/Li+), moderate capacity (170 mA h g−1), flat voltage plateau, abundant material supply, low material cost, and good environmental compatibility have also made LiFePO4 a favourable cathode material for commercial LIBs. However, the poor electronic conductivity (>10−9 S cm−1), low ionic diffusivity (ca. 10−11–10−10 S cm−1), poor low temperature performance and low volumetric energy density are considered main obstacles for its wider application.13 To address these pressing issues, numerous works have been performed to investigate the synthesis, structure and defects of LiFePO4, and important breakthroughs have been achieved and adopted by the market. Generally, the low ionic and electronic conductivities can be optimized by the following three methods: conductive layer coating,14 element doping13,15 and particle nanosizing.16 As for the poor low temperature performance, optimizing the use of Li-salts, electrolyte solvents and additives can realize significant improvements;17–19 moreover, reducing the particle size and removing the impurities in the raw materials are also deterministic ways to improve the low temperature performance.20 To overcome the limitation of volumetric energy density, micro-sized LiFePO4 particle preparation is the most common method, showing a porous structure for enhancing ionic and electronic conductivities.21–23With decades of research and development of LiFePO4, major breakthroughs have been made through various material modifications, playing an important role in solving the inherent drawbacks of LiFePO4. Despite these exciting techniques and methods, it should still be noted that the electrochemical properties of LiFePO4 can be essentially traced back to its crystalline structures. Moreover, as the confined region for the charge transfer process, the surface/interface structure of LiFePO4 also dictates the reaction kinetics of LiFePO4. Although many relevant studies have been carried out, they were not systematically summarized until now. In this review, the impact of the LiFePO4 structure on its electrochemical performance will be briefly summarized from the bulk to the surface. With the aid of some graphical models, the mechanism of Li+ diffusion is clearly presented, providing not only a better understanding of fundamental questions, but also useful guide on future material design.
2. Bulk structure of LiFePO4
2.1 LiFePO4 phase structure
Most cathode materials can be seen as crystals consisting of numerous basic structure units, which can be considered as “material genes” that assemble via periodic arrangement.24 As a result, they directly determine most properties (e.g. Li+ diffusion coefficient, thermal stability and electrochemical stability) of cathode materials. For instance, LiFePO4 belongs to olivine family cathode materials with an orthorhombic lattice structure in a space group Pnma. Oxygen atoms in the structure are in hexagonal-close-packed arrangement, and phosphorus atoms and iron/lithium occupy tetrahedral and octahedral sites, which forms corner-shared FeO6 octahedra and edge-shared LiO6 octahedra parallel to the b-axis. The two octahedra are linked by the PO4 tetrahedra, forming 3D spatial network structures, named α-LiFePO4.25 During delithiation, the FePO4 phase appears in the single crystal structure, which has essentially the same crystalline structure as LiFePO4 (Fig. 1). Up to the fully charged state, the volume change content is just 6.81%, which avoids capacity attenuation caused by volume changes during long-term cycles.12 With the strong P–O covalent bond and limited volume variation, LiFePO4 shows good thermal stability, cyclability and safety. | ||
| Fig. 1 Crystal structures of LiFePO4 and FePO4. During charging, LiFePO4 changes to FePO4 by delithiation. In the discharge process, a reversible transformation from FePO4 to LiFePO4 occurs by lithiation. | ||
Two decades ago, another phase of LiFePO4 (β-LiFePO4) was synthesized under high pressure, exhibiting a space group of Cmcm.26 Owing to the lower formation energy compared with α-LiFePO4, β-LiFePO4 is usually regarded as an intermediate or metastable state, and experiences a phase change from α to β at high temperatures.27 In the Cmcm β-LiFePO4 structure, tetrahedral LiO4 shares a corner with the closed octahedral FeO6 and a corner with the closed tetrahedral PO4 (Fig. 2a). In this structure, Li+ is isolated by the nearest FeO6 and PO4, leading to high energy barriers for lithium migration, which is why β-LiFePO4 exhibits almost no electrochemical activity.28 However, proper processing, such as introducing disorder structures and creating new diffusion channels in β-LiFePO4, can also realize high capacity and stability25 (Fig. 2b). Moreover, the factors that influence the controllable synthesis of β-LiFePO4 were studied, which is helpful for us to obtain high performance olivine structure cathode materials.29
 | ||
| Fig. 2 (a) Crystal structure of β-LiFePO4. Li+ is shown in green, [FeO6] octahedron is shown in dark gray, [PO4] tetrahedron is shown in light gray, and [LiO4] tetrahedron is shown in light green. (b) Electrochemical performance of β-LiFePO4 with different ball-milling times. Reproduced from ref. 25 with permission from the American Chemical Society, copyright 2016. | ||
2.2 Antisite defects
Owing to the ordered-olivine structure, Li+ moves in 1D diffusion channels, which can be easily blocked by Li/Fe disorder or stacking faults. Generally, a proper synthesis route and doping are effective approaches to suppress cation mixing and optimize the electrochemical performance. Guo et al.30 synthesized LiFePO4/carbon hybrid microtubes using strong chelation interaction with Fe3+ and absorption interaction with Li+ of alginate, which yields very low Fe–Li antisite defects and exhibits remarkable electrochemical performance. Machida et al.31 successfully realized O-site doping in LiFePO4 by S, inducing an expanded lattice in a and b directions and suppression of antisite defects between Fe and Li owing to the larger ionic radius of S2− than O2−; hence, enhanced electrochemical properties were achieved. Furthermore, cation replacement (e.g. Nb-doping32) is another good way to improve the effective lithium mobility during the insertion/extraction reaction. Li diffusion in materials with a 1D diffusion mechanism, such as LiFePO4, has a strong dependence on particle size, illustrating a significant reduction of Li diffusivity at large particle sizes. Moreover, if the antisite defects exist in the diffusion channels, the negative effects will be amplified. The correlation between the channel length and antisite content was built and described by Ceder's group16 (Fig. 3a). | ||
| Fig. 3 (a) Expected unblocked capacity vs. channel length in LiFePO4 for various defect concentrations. (b) Variation of the Li-vacancy self-diffusion D[100] (blue), D[010] (green), and D[001] (red) with the defect concentration at T = 440 K. Reproduced from ref. 16 with permission from the American Chemical Society, copyright 2010. | ||
Ab initio molecular dynamics simulations prove that ionic migration perpendicular to the 1D channels has greater possibility than along the blocked channels by comparing the migration energy.33 In addition, the related multi-dimensional transport properties have also been demonstrated in other studies.34,35 The high dimensionality of ionic migration will be promoted by the support of channel crossover induced by high antisite content. D[100] and D[001] as a function of defect concentration were described, in which at 440 K, with higher defect concentration the Li+ diffusivity decreases in the b direction and increases in the a and c directions (Fig. 3b). Pan et al.36 synthesized α-LiMn1−yFeyPO4 with high antisite content, realizing excellent rate performance, which can be attributed to the 2D and/or 3D Li+ diffusion mechanism, resulting in the transformation of the two-phase mechanism into a solid-state mechanism.
2.3 Special properties induced by the electronic structure
LiFePO4 with an olivine structure exhibits long range antiferromagnetic order at low temperatures, below 51(1) K in LiFePO4 and below 114(1) K in FePO4.37,38 The splitting of the d-orbitals in Fe2+ (d5) and Fe3+ (d6) will occur and form doubly degenerate eg orbitals and triply degenerate t2g orbitals. Through the electron transfer between Fe2+ and Fe3+via the O 2p state exchange interaction can be realized and if both two ions are aligned ferromagnetically, then the ferromagnetical state emerges,39 which has influences on the movement of charged particles. During the electrochemical process, Fe2+ and Fe3+ coexist in the bulk structure, bringing the possibility of the magnetic field interference on electrochemical properties. Pan et al.40 provided strong proof about the relationship between magnetic domains and Li+ diffusion, in which room temperature (RT) magnetic order was detected in LixFePO4 (x < 0.12) nanocrystals, which trapped Li+ and formed sub-10 nm solid solution domains owing to the strong Lorentz force in RT magnetic domains (Fig. 4). The formation of RT magnetic order was proposed due to both Fe2+–Fe3+ super-exchange and the broken local symmetries induced by Fe2+–Li+ anti-site defects. Moreover, with the relationship between magnetic properties and element electronic structure, Werner et al. demonstrated a procedure to precisely determine the defect concentration by static magnetization measurements on LiFePO4.41 Under the hypothesis that the transition metals in LiMn1−xFexPO4 are antiferromagnetic, the impact of the super-exchange interactions on the transition metal structure sequence and magnetic structure of LiMn1−xFexPO4 was observed, suggesting the clustering arrangement of transition metals,42 which explains the previous experimental results where LiMn1−xFexPO4 generally exhibits two redox peaks, while sometimes only one can be observed.43 | ||
| Fig. 4 Magnetic structures of LiFePO4, FePO4 and RT magnetic order zone. The magnetic polaron domain marked by a dashed black box, caused by the presence of antisite defects and super-exchange between Fe2+ and Fe3+. Reproduced from ref. 40 with permission from the American Chemical Society, copyright 2019. | ||
2.4 Other olivine-type cathode materials
Attempts have also been made to improve the energy density of LiFePO4 by replacing Fe with other transition metals. LiMnPO4 has also attracted researchers due to its higher voltage plateau at approximately 4 V vs. Li/Li+. However, pristine LiMnPO4 suffers from the mismatched LiMnPO4/MnPO4 interface and even poorer electronic conductivity which is several orders of magnitude lower than that of LiFePO4.44 Moreover, the Jahn–Teller distortion of unstable Mn3+ in charged MnPO4 causes the deformation of the MnO6 octahedra and Mn dissolution, and hence the capacity fading.45,46 Alternatively, as the solid solution of LiFePO4 and LiMnPO4, LiMn1−xFexPO4 exhibits two voltage plateaus, which correspond to Fe3+/Fe2+ and Mn3+/Mn2+ redox pairs. Similar to LiFePO4 and LiMnPO4, LiMn1−xFexPO4 (LMFP) also exhibits low electronic and ionic conductivity. With high operating voltages, LiNiPO4 (5.1 V vs. Li/Li+) and LiCoPO4 (4.8 V vs. Li/Li+) are studied as potential cathode material candidates for high-energy-density batteries. However, they are both faced with not only low conductivity, but also poor chemical/electrochemical stabilities. One major issue is that their high operating voltages usually cause severe electrolyte degradation during the charging process. Different from LiFePO4, LiCoPO4 exhibits two voltage plateaus due to the formation of an intermediate Li2/3CoPO4 phase, which co-exists with both lithiated and delithiated phases.47 However, the delithiated CoPO4 is very unstable according to a first-principles calculation. Moreover, a second phase of Co2P will be formed during heat treatment, which is detrimental to its capacity.48 Compared with LiCoPO4, LiNiPO4 exhibits even worse cycle stability due to its higher charging voltage. Density functional theory (DFT) results also predict large volume changes (9.6%) between LiNiPO4 and NiPO4 during Li (de)intercalation, which induces structural instability.49Inspired by the successful commercialization of LiFePO4, iron phosphate has been tested as the host for other cations. With an acceptable theoretical capacity (154 mA h g−1) and working potential (2.9 V vs. Na/Na+), NaFePO4 has been regarded as a promising alternative for LiFePO4. Similar to LiNiPO4, olivine NaFePO4 also shows a different ion (de)intercalation mechanism to LiFePO4, which includes a Na2/3FePO4 intermediate phase due to the volumetric mismatch between NaFePO4 and FePO4.50 However, the theoretical capacity of NaFePO4 cannot be fully realized in practice due to the poor electron conductivity and limited Na diffusion path. Therefore, one improvement strategy is to synthesize amorphous NaFePO4.51 Moreover, since olivine-phase NaFePO4 exhibits lower thermodynamic stability compared to its maricite phase, which is electrochemically inert, it cannot be synthesized via traditional methods.52 Other than Na-ions, Mg-ions can also be inserted into FePO4 due to their similar ionic size to Li-ions. However, it is found that only half of the capacity can be achieved, which is due to that the Coulomb repulsions between Mg2+ in adjacent channels exert a large diffusion energy barrier for Mg-ion intercalation.53
3. Surface electrochemistry of LiFePO4
3.1 Artificial coating
Due to the poor ionic and electronic conductivity of LiFePO4, surface engineering is a very common strategy to improve the electrochemical performance of LiFePO4. A fast ion-conducting polyphosphate surface phase was obtained by Ceder's group,54 which increases lithium ion diffusion across the surface; however, the final result is suspected by J. B. Goodenough et al.55 As to the electronic conductivity, the coating substances include Cu, Ag, carbon, conducting polymers, metal oxides and polymers.56–58 Among them, carbon coating attracts more attention owing to its high conductivity, low lost, operation friendliness, and chemical stability during electrochemical processes.59–61 Sun et al.14 made a comprehensive summary on the development of carbon coating on LiFePO4 cathode materials, related to carbon content, thickness, structure, morphology and porosity. In practical applications, the total carbon coating content and thickness should be optimized for high energy density LiFePO4 LIBs. It has been previously revealed that enhancement of electronic conductivity and interparticle connectivity are the commonly held role of carbon coating;62 however, it works more than that. Pan et al.63 achieved an excess capacity of 186 and 207 mA h g−1 in LiFePO4 samples (E-LFP) with mean particle sizes of 83 nm and 42 nm, respectively, by special carbon coating. The extra lithium was stored between the carbon layer and particles, which is a very stable structure built by C–O–Fe bonds, realizing full usage of the large surface areas of nanosized particles (Fig. 5). | ||
| Fig. 5 (a, b) Crystal structures of normal LiFePO4 (LFP-N) and excess capacity LiFePO4 (LFP-E). (c, d) Transmission electron micrograph (TEM) of charged and discharged LFP-E nanoparticles, respectively. Reproduced from ref. 63 with permission from the American Chemical Society, copyright 2017. | ||
As mentioned above, Mn, Co and Ni can be used to replace Fe for higher energy densities. However, severe interfacial degradation is a pressing challenge for these materials, which can be mitigated by artificial protective layers. By preparing a single-particle (SP) electrode, Yang and co-workers identified MnF2 species on the surface of LMFP after cycling.64 As shown in Fig. 6a, it is proposed that the Mn4+ generated by Mn3+ disproportionation will react with the solvent (e.g. EC) through a ring-opening process, forming Mn(OR)2, which further combines with HF and eventually forms MnF2 passivation domains, leading to capacity fading. The result also shows that Mn dissolution is more severe in the SP electrode compared to the conventional thick electrode due to the larger contact area between LMFP and the electrolyte. In order to suppress this surface degradation, a thin layer (approximately 5 nm) of Al2O3 is homogeneously applied on the cathode material via the atomic layer deposition (ALD) technique. It can be seen from Fig. 6b and c that after coating, the cycling stability of the LMFP single-particle electrode is greatly improved by inhibiting undesirable side reactions.
 | ||
| Fig. 6 (a) Proposed surface corrosion mechanism of LMFP particles; voltage profiles of different galvanostatic cycle numbers for (b) bare LMFP and (c) Al2O3 coated LMFP. Reproduced from ref. 64 with permission from the American Chemical Society, copyright 2019. | ||
In order to avoid the direct contact between LiCoPO4 and the electrolyte, Naoi and co-workers applied a layer of FePO4 on the LiCoPO4 surface.65 Owing to the well-matched crystal structures, after coating, Fe3+ is found to partially diffuse to the bulk LiCoPO4, forming an Fe-substituted LiCoPO4 core with an Fe-rich outer layer. Such an outer layer is found to effectively suppress interfacial degradation reactions by exhibiting a capacity retention of as high as 99% after 100 cycles, which far surpasses that of the unmodified LCP materials.
Alternatively, concentration gradient hollow sphere LiMn0.5Fe0.5PO4 with an Fe-rich surface and a Mn-rich core was synthesized by Wang and co-workers through a modified precipitation process (Fig. 7a).66 In addition to higher chemical stability, the porous hollow structure could accommodate significant volume changes during repeated Li intercalation and deintercalation processes. As a result, a greatly improved cycle life was obtained (Fig. 7b). The higher discharge capacity of HCG-LMFP/C compared to C-LMFP/C is attributed to the high ionic/electric conductivity of the Fe-rich surface (Fig. 7b).
 | ||
| Fig. 7 (a) High-angle annular dark-field scanning TEM (HAADF-STEM) image and elemental mapping results from the cross-section of a hollow-structured concentration-gradient LiMn0.5Fe0.5PO4/C (HCG-LMFP/C) particle. (b) Voltage profiles of a controlled LiMn0.5Fe0.5PO4/C hollow sphere material (C-LMFP/C) and HCG-LMFP/C at 10C with 1000 cycles. Reproduced from ref. 66 with permission from the Royal Society of Chemistry, copyright 2019. | ||
3.2 Spontaneous interfacial reconstruction
The solid–solution interface is an important part in cells, which is the gate for Li+ in and out of the host materials. To understand the impact of the interface on Li+ transmission, its definition and structure should be clear. As is well-known, there is just physical contact between LiFePO4 and carbon, which results in a loose structure with amounts of internal spaces, so the solvent molecules can permeate the carbon layer.67 In the LiFePO4 bulk, FeO6 octahedra and LiO6 octahedra were linked by the PO4 tetrahedra and formed a perfect symmetry structure; however, in the LiFePO4 and FePO4 surface, it's a truncated octahedral symmetry. In this condition, how the Li+ diffuses into the host material from the broken surface becomes very important and meaningful. Pan et al.68 gave a perfect explanation both in aqueous and organic electrolytes (Fig. 8). By combining comprehensive electrochemistry tests and ab initio calculations, it was identified that the truncated symmetry of the solid LiFePO4 surface could be compensated by the chemisorbed H2O molecules or ethylene carbonate (EC) molecules, forming a half-solid (LiFePO4) and half-liquid (H2O or EC) Janus interface. This kind of interface promotes the Li desolvation process near the surface, realizing a super high rate performance, for example, in aqueous electrolyte at 600C (3600/600 = 6 s charge time, 1C = 170 mA h g−1) reaching 72 mA h g−1 energy storage (42% of the theoretical capacity).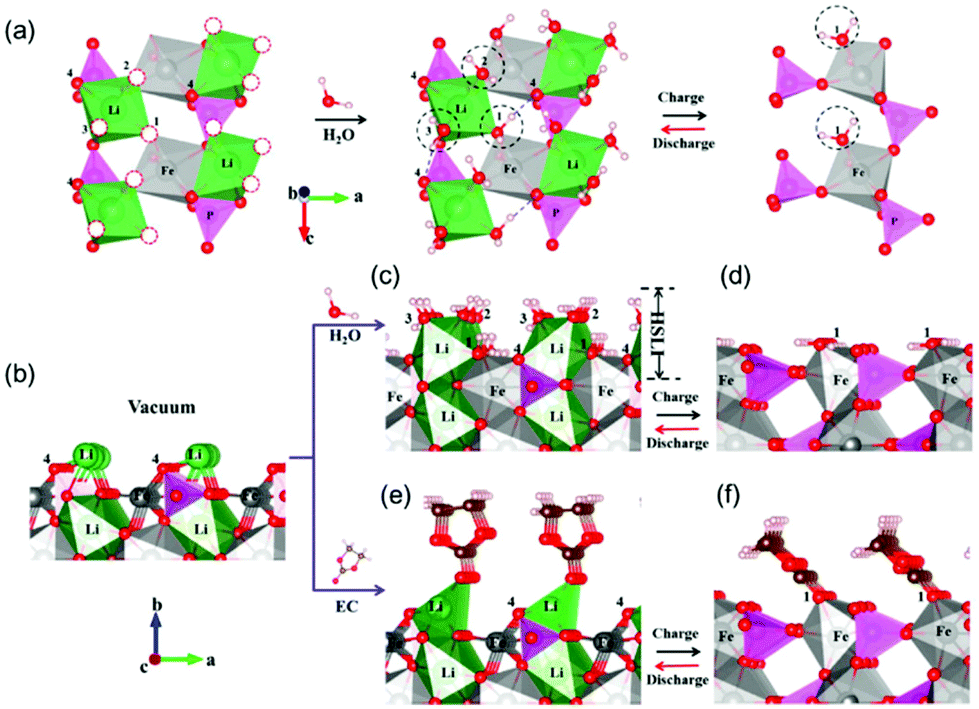 | ||
| Fig. 8 Ab initio calculated H2O/EC absorption at LiFePO4 and FePO4 surfaces. Reproduced from ref. 68 with permission from the American Chemical Society, copyright 2015. | ||
Rate performance68 and cyclic voltammetry (CV) curves69 of the LiFePO4 electrode were also compared in aqueous and organic electrolytes. It was apparent that aqueous batteries exhibit higher rate performance and lower polarization. To investigate the mechanisms, while excluding the interference ionic transport between adjacent crystallites,70 quasi-single-particle (QSP) LiFePO4 electrodes and a QSP model were developed.69 The prepared quasi monolayer electrodes consist of well separated single particles of LiFePO4, which can minimize the concentration polarization, electrochemical polarization, and other internal interference to benefit for testing the intrinsic electrochemical properties of LiFePO4. Through this model, the precise relationship between the reaction rate and voltage can be obtained by using the real potential-SOC curve of LiFePO4. After the analysis of experimental and simulation results, Pan et al. found that the intrinsic Li+ diffusion coefficients of LiFePO4 are nearly the same in water and organic electrolytes, but the interfacial rate constant in aqueous electrolyte is one order higher, accounting for the excellent rate performance in aqueous electrolyte for LiFePO4. It's also the first time of establishing the connection between the pre-exponential factor and Li+ de-solvation/solvation process, that the larger the pre-exponential factor, the higher the interfacial rate constant and the faster the diffusivity.
With the previous work, it is known that surface reconstruction is important for Li+ diffusion. To determine whether the above results have universal value owing to the complex electrolyte categories, Pan et al.71 recently explored the effects of anions on surface reconstruction and the Helmholtz plane vs. lithium-ion transport at the solid–liquid interface (Fig. 9). Through ab initio calculations and comparison, it was proved that H2O has a stronger binding energy with Fe and Li on the surface of LiFePO4 and forms a Janus interface compared with different kinds of anions in aqueous electrolytes. Moreover, they divided the solid–liquid interface into three parts, which are the surface reconstruction layer (inner Helmholtz plane (IHP), E0), anion adsorption layer (IHP, E1) and cation adsorption layer (outer Helmholtz plane (OHP), E2), and clearly illustrated the effects of the anion adsorption layer on Li+ electrochemical behaviors.
 | ||
| Fig. 9 The migration energy barrier of Li+ in the inner Helmholtz plane (IHP) and outer Helmholtz plane (OHP). Reproduced from ref. 71 with permission from Elsevier, copyright 2020. | ||
4. Conclusions
To conclude, by illustrating the detailed bulk and surface structure information of LiFePO4, its correlation with electrochemical performance is established. For the bulk structure, the effects of the phase structure, antisite defects and magnetic structure on performance have been reviewed, and all of these factors are closely related to Li+ bulk diffusivity. (1) Owing to the high energy barriers for lithium migration, the synthesis conditions should be carefully controlled to avoid the presence of β-LiFePO4. (2) Given that 2D and 3D Li+ diffusion mechanisms could co-exist under the condition of high Fe2+–Li+ antisite content, a low-antisite route might not be necessary for achieving high diffusion ability. (3) Due to both Fe2+–Fe3+ super-exchange and the broken local symmetries induced by Fe2+–Li+ antisite defects, RT magnetic order was induced, leading to the appearance of a solid–solution region and capacity loss.As for the interface structure, three factors are included and analyzed. (1) The impact of electrolyte solvents on Li+ interface kinetics. Taking an aqueous battery as an example, as shown in Fig. 10, the Li+ diffusion pathway is presented. When Li+ diffuses into the electrolyte from the anode, it will be coordinated by four water molecules in its primary solvation sheath, forming a complex cation Li+(H2O)4. With the potential difference between the cathode and anode, the complex cation groups diffuse across the separator and into the cathode side. Close to the solid–liquid interface, Li+(H2O)4 will shuffle off two H2O and approach the LiFePO4 surface, realizing particle surface reorganization by the left two H2O. After Li+ diffuses into the LiFePO4 bulk along the Li channel, the connected two H2O will desorb from the surface. By contrast, in the organic electrolyte, EC molecules will be bonded with Li+, forming Li+(EC)4. Owing to its relatively bulky size, three EC molecules will be stripped off, bringing a much higher energy barrier when Li+ crosses the interface, inducing a slower interfacial reaction constant compared with in the aqueous electrolyte. (2) The impact of the anion interfacial adsorption layer on Li+ diffusion. The ionic absorption layer has a significant influence on Li+ diffusion: the smaller the anion size, the better the diffusion ability. (3) Carbon coating affects the electronic conductivity and capacity. Carbon coating brought a breakthrough development of LiFePO4, which realized a significant improvement of electron conductivity, achieving high capacity and rate performance. Moreover, with proper coating techniques, excess capacity could be realized for high energy density LIBs.72 In actual applications, ionic conductivity is the determinant of performance owing to the lower diffusivity (ca. 10−11–10−10 S cm−1 at RT) compared with electronic diffusivity (>10−9 S cm−1), so the factors affecting ionic conductivity (e.g. size73) should be in good balance with carbon coating for realizing high energy-density and power-density LIBs.
 | ||
| Fig. 10 Li+ diffusion path from the electrolyte to the solid–liquid interface and then to LiFePO4 bulk. The factors that affect the performance were concluded. | ||
Here, by establishing the relationship between the bulk–surface structure and the electrochemical performance, we aim to provide a new perspective on olivine-type LiFePO4. However, the Li-ion diffusion behaviours at different states of charges and different places of a LiFePO4 particle still remain unclear. Therefore, in the future, a deeper understanding is needed on the atomic scale to enable fast electron and ion transport. In addition, with the mature material preparing techniques and clear structural cognition, more attention should be paid to how to realize performance improvement under extreme environments, how to improve the volumetric energy density, and if there is a cost-effective synthetic route to produce a high-quality LiFePO4 material.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2016YFB0700600), the National Natural Science Foundation of China (No. 21603007 and 51672012), and Shenzhen Science and Technology Research Grant (No. JCYJ20160531141048950 and JCYJ20150729111733470).Notes and references
- J.-T. Hu and J.-G. Zhang, Chin. J. Struct. Chem., 2020, 38, 2005–2008 Search PubMed
.
- M.-J. Zhang, Y.-S. Chen, F. Pan and Y. Ren, Chin. J. Struct. Chem., 2020, 39, 26–30 Search PubMed
- J. R. Owen, Chem. Soc. Rev., 1997, 26, 259–267 RSC
- Z. Chen, W. Zhang and Z. Yang, Nanotechnology, 2019, 31, 012001 CrossRef PubMed
- L. Yang, K. Yang, J. Zheng, K. Xu, K. Amine and F. Pan, Chem. Soc. Rev., 2020 10.1039/d0cs00137f
- M. S. Whittingham, Chem. Rev., 2014, 114, 11414–11443 CrossRef CAS PubMed
- T.-C. Liu, F. Pan and K. Amine, Chin. J. Struct. Chem., 2020, 39, 11–15 CrossRef
- Z. Zheng, M.-Y. Weng, L.-Y. Yang, Z.-X. Hu, Z.-F. Chen and F. Pan, Chin. J. Struct. Chem., 2020, 38, 2020–2026 Search PubMed
- Z.-W. Yin, J.-T. Li, L. Huang, F. Pan and S.-G. Sun, Chin. J. Struct. Chem., 2020, 39, 20–25 Search PubMed
- B. Pei, H. Yao, W. Zhang and Z. Yang, J. Power Sources, 2012, 220, 317–323 CrossRef CAS
- Q. Wang, W. Zhang, Z. Yang, S. Weng and Z. Jin, J. Power Sources, 2011, 196, 10176–10182 CrossRef CAS
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188–1194 CrossRef CAS
- S.-Y. Chung, J. T. Bloking and Y.-M. Chiang, Nat. Mater., 2002, 1, 123–128 CrossRef CAS PubMed
- J. Wang and X. Sun, Energy Environ. Sci., 2012, 5, 5163–5185 RSC
- B. Pei, Q. Wang, W. Zhang, Z. Yang and M. Chen, Electrochim. Acta, 2011, 56, 5667–5672 CrossRef CAS
- R. Malik, D. Burch, M. Bazant and G. Ceder, Nano Lett., 2010, 10, 4123–4127 CrossRef CAS PubMed
- S. S. Zhang, K. Xu and T. R. Jow, J. Power Sources, 2006, 159, 702–707 CrossRef CAS
- X. Z. Liao, Z. F. Ma, Q. Gong, Y. S. He, L. Pei and L. J. Zeng, Electrochem. Commun., 2008, 10, 691–694 CrossRef CAS
- L. Liao, X. Cheng, Y. Ma, P. Zuo, W. Fang, G. Yin and Y. Gao, Electrochim. Acta, 2013, 87, 466–472 CrossRef CAS
- N. Zhao, Y. Li, X. Zhao, X. Zhi and G. Liang, J. Alloys Compd., 2016, 683, 123–132 CrossRef CAS
- H. Wang, R. Wang, L. Liu, S. Jiang, L. Ni, X. Bie, X. Yang, J. Hu, Z. Wang, H. Chen, L. Zhu, D. Zhang, Y. Wei, Z. Zhang, S. Qiu and F. Pan, Nano Energy, 2017, 39, 346–354 CrossRef CAS
- S. W. Oh, S. T. Myung, H. J. Bang, C. S. Yoon, K. Amine and Y. K. Sun, Electrochem. Solid-State Lett., 2009, 12, A181–185 CrossRef CAS
- L. Wen, X. Hu, H. Luo, F. Li and H. Cheng, Particuology, 2015, 22, 24–29 CrossRef CAS
- J. Zheng, Y. Ye and F. Pan, Natl. Sci. Rev., 2019, 2, 242–245 Search PubMed
- H. Guo, X. Song, Z. Zhuo, J. Hu, T. Liu, Y. Duan, J. Zheng, Z. Chen, W. Yang, K. Amine and F. Pan, Nano Lett., 2016, 16, 601–608 CrossRef CAS PubMed
- O. García-Moreno, M. Alvarez-Vega, F. García-Alvarado, J. García-Jaca, J. M. Gallardo-Amores, M. L. Sanjuán and U. Amador, Chem. Mater., 2001, 13, 1570–1576 CrossRef
- G. Zeng, R. Caputo, D. Carriazo, L. Luo and M. Niederberger, Chem. Mater., 2013, 25, 3399–3407 CrossRef CAS
- T. E. Ashton, J. V. Laveda, D. A. Maclaren, P. J. Baker, A. Porch, M. O. Jones and S. A. Corr, J. Mater. Chem. A, 2014, 2, 6238–6245 RSC
- H. Guo, H. Ping, J. Hu, X. Song, J. Zheng and F. Pan, J. Mater. Chem. A, 2017, 5, 14294–14300 RSC
- Y. Zou, S. Chen, X. Yang, N. Ma, Y. Xia, D. Yang and S. Guo, Adv. Energy Mater., 2016, 6, 1601549 CrossRef
- K. Okada, I. Kimura and K. Machida, RSC Adv., 2018, 8, 5848–5853 RSC
- S. Y. Chung, S. Y. Choi, T. Yamamoto and Y. Ikuhara, Angew. Chem., Int. Ed., 2009, 48, 543–546 CrossRef CAS PubMed
- J. Yang and J. S. Tse, J. Phys. Chem. A, 2011, 115, 13045–13049 CrossRef CAS PubMed
- R. Amin, J. Maier, P. Balaya, D. P. Chen and C. T. Lin, Solid State Ionics, 2008, 179, 1683–1687 CrossRef CAS
- G. R. Gardiner and M. S. Islam, Chem. Mater., 2010, 22, 1242–1248 CrossRef CAS
- J. Hu, Y. Xiao, H. Tang, H. Wang, Z. Wang, C. Liu, H. Zeng, Q. Huang, Y. Ren, C. Wang, W. Zhang and F. Pan, Nano Lett., 2017, 17, 4934–4940 CrossRef CAS PubMed
- N. A. Chernova, G. M. Nolis, F. O. Omenya, H. Zhou, Z. Li and M. S. Whittingham, J. Mater. Chem., 2011, 21, 9865–9875 RSC
- G. Rousse, J. Rodriguez-Carvajal, S. Patoux and C. Masquelier, Chem. Mater., 2003, 15, 4082–4090 CrossRef CAS
- A. Mauger, K. Zaghib, F. Gendron and C. M. Julien, Ionics, 2008, 14, 209–214 CrossRef CAS
- J. Hu, H. Zeng, X. Chen, Z. Wang, H. Bin Wang, R. Wang, L. Wu, Q. Huang, L. Kong, J. Zheng, Y. Xiao, W. Zhang and F. Pan, J. Phys. Chem. Lett., 2019, 10, 4794–4799 CrossRef CAS PubMed
- J. Werner, C. Neef, C. Koo, S. Zvyagin, A. Ponomaryov and R. Klingeler, arXiv, 2020, 220, 1–15 Search PubMed
- Y. Liu, Y. Gu, H. Zeng, J. Zheng and F. Pan, J. Phys. Chem. C, 2019, 123, 17002–17009 CrossRef CAS
- A. Paolella, G. Bertoni, E. Dilena, S. Marras, A. Ansaldo, L. Manna and C. George, Nano Lett., 2014, 14, 1477–1483 CrossRef CAS PubMed
- C. Delacourt, L. Laffont, R. Bouchet, C. Wurm, J.-B. Leriche, M. Morcrette, J.-M. Tarascon and C. Masquelier, J. Electrochem. Soc., 2005, 152, A913 CrossRef CAS
- Z. Zhuo, J. Hu, Y. Duan, W. Yang and F. Pan, Appl. Phys. Lett., 2016, 109, 1–6 CrossRef
- Y. Deng, C. Yang, K. Zou, X. Qin, Z. Zhao and G. Chen, Adv. Energy Mater., 2017, 7, 1601958 CrossRef
- M. Zhang, N. Garcia-Araez and A. L. Hector, J. Mater. Chem. A, 2018, 6, 14483–14517 RSC
- J. Wolfenstine, J. Read and J. L. Allen, J. Power Sources, 2007, 163, 1070–1073 CrossRef CAS
- C. Li, X. Miao, W. Chu, P. Wu and D. G. Tong, J. Mater. Chem. A, 2015, 3, 8265–8271 RSC
- M. Casas-Cabanas, V. V. Roddatis, D. Saurel, P. Kubiak, J. Carretero-González, V. Palomares, P. Serras and T. Rojo, J. Mater. Chem., 2012, 22, 17421–17423 RSC
- C. Li, X. Miao, W. Chu, P. Wu and D. G. Tong, J. Mater. Chem. A, 2015, 3, 8265–8271 RSC
- J. Lu, S. C. Chung, S. I. Nishimura and A. Yamada, Chem. Mater., 2013, 25, 4557–4565 CrossRef CAS
- P. Shan, Y. Gu, L. Yang, T. Liu, J. Zheng and F. Pan, Inorg. Chem., 2017, 56, 13411–13416 CrossRef CAS PubMed
- B. Kang and G. Ceder, Nature, 2009, 458, 190 CrossRef CAS PubMed
- K. Zaghib, J. B. Goodenough, A. Mauger and C. Julien, J. Power Sources, 2009, 194, 1021–1023 CrossRef CAS
- F. Croce, A. D'Epifanio, J. Hassoun, A. Deptula, T. Olczac and B. Scrosati, Electrochem. Solid-State Lett., 2002, 5, 47–50 CrossRef
- Y. Jin, C. P. Yang, X. H. Rui, T. Cheng and C. H. Chen, J. Power Sources, 2011, 196, 5623–5630 CrossRef CAS
- Y. H. Huang and J. B. Goodenough, Chem. Mater., 2008, 20, 7237–7241 CrossRef CAS
- L.-L. Jia, Y.-C. Ji, K. Yang, Z.-J. Wang, H.-B. Chen and F. Pan, Chin. J. Struct. Chem., 2020, 39, 200–205 CAS
- R. Dominko, M. Bele, M. Gaberscek, M. Remskar, D. Hanzel, S. Pejovnik and J. Jamnik, J. Electrochem. Soc., 2005, 152, A607 CrossRef CAS
- S. W. Oh, S. T. Myung, S. M. Oh, K. H. Oh, K. Amine, B. Scrosati and Y. K. Sun, Adv. Mater., 2010, 22, 4842–4845 CrossRef CAS PubMed
- N. Ravet, Y. Chouinard, J. F. Magnan, S. Besner, M. Gauthier and M. Armand, J. Power Sources, 2001, 97–98, 503–507 CrossRef CAS
- Y. Duan, B. Zhang, J. Zheng, J. Hu, J. Wen, D. J. Miller, P. Yan, T. Liu, H. Guo, W. Li, X. Song, Z. Zhuo, C. Liu, H. Tang, R. Tan, Z. Chen, Y. Ren, Y. Lin, W. Yang, C. M. Wang, L. W. Wang, J. Lu, K. Amine and F. Pan, Nano Lett., 2017, 17, 6018–6026 CrossRef CAS PubMed
- W. Huang, J. Hu, L. Yang, W. Zhao, Z. Wang, H. Wang, Z. Guo, Y. Li, J. Liu, K. Yang and F. Pan, ACS Appl. Mater. Interfaces, 2019, 11, 957–962 CrossRef CAS PubMed
- N. Okita, E. Iwama, Y. Takami, S. Abo, W. Naoi, M. Thomas, H. Reid and K. Naoi, Electrochemistry, 2019, 87, 156–161 CrossRef CAS
- T. Ruan, B. Wang, F. Wang, R. Song, F. Jin, Y. Zhou, D. Wang and S. Dou, Nanoscale, 2019, 11, 4073–4082 RSC
- K. Zaghib, M. Dontigny, P. Charest, J. F. Labrecque, A. Guerfi, M. Kopec, A. Mauger, F. Gendron and C. M. Julien, J. Power Sources, 2008, 185, 698–710 CrossRef CAS
- J. Zheng, Y. Hou, Y. Duan, X. Song, Y. Wei, T. Liu, J. Hu, H. Guo, Z. Zhuo, L. Liu, Z. Chang, X. Wang, D. Zherebetskyy, Y. Fang, Y. Lin, K. Xu, L. W. Wang, Y. Wu and F. Pan, Nano Lett., 2015, 15, 6102–6109 CrossRef CAS PubMed
- J. Hu, W. Li, Y. Duan, S. Cui, X. Song, Y. Liu, J. Zheng, Y. Lin and F. Pan, Adv. Energy Mater., 2017, 7, 1601894 CrossRef
- K. T. Lee, W. H. Kan and L. F. Nazar, J. Am. Chem. Soc., 2009, 131, 6044–6045 CrossRef CAS PubMed
- J. Hu, W. Ren, X. Chen, Y. Li, W. Huang, K. Yang, L. Yang, Y. Lin, J. Zheng and F. Pan, Nano Energy, 2020, 74, 104864 CrossRef CAS
- C. Lu, X. Jiang, W. Sun, Z. Wang, K. Sun, D. W. Rooney and J. Wang, J. Mater. Chem. A, 2017, 5, 24636–24644 RSC
- M. Gaberscek, R. Dominko and J. Jamnik, Electrochem. Commun., 2007, 9, 2778–2783 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2020 |