
Electrochemical synthesis of selenyl-dihydrofurans via anodic selenofunctionalization of allyl-naphthol/phenol derivatives and their anti-Alzheimer activity†
Marcos R.
Scheide
 a,
Alex R.
Schneider
a,
Guilherme A. M.
Jardim
a,
Guilherme M.
Martins
a,
Daniele C.
Durigon
a,
Sumbal
Saba
b,
Jamal
Rafique
c and
Antonio L.
Braga
*a
a,
Alex R.
Schneider
a,
Guilherme A. M.
Jardim
a,
Guilherme M.
Martins
a,
Daniele C.
Durigon
a,
Sumbal
Saba
b,
Jamal
Rafique
c and
Antonio L.
Braga
*a
aDepartamento de Química, Universidade Federal de Santa Catarina – UFSC, Florianopolis, 88040-900, SC, Brazil. E-mail: braga.antonio@ufsc.br; Fax: +55 48 3721 6427; Tel: +55 48 3721 6427 Web: http://labselen.ufsc.br
bCentro de Ciências Naturais e Humanas-CCNH, Universidade Federal do ABC – UFABC, Santo André, 09210-580, SP, Brazil
cInstituto de Química, Universidade Federal do Mato Grosso do Sul – UFMS, Campo Grande, 79074-460, MS, Brazil
First published on 21st April 2020
Abstract
Herein, we report an eco-friendly, electrosynthetic approach for the intramolecular oxyselenylation of allyl-naphthol/phenol derivatives. This reaction proceeds with 0.2 equiv. of nBu4NClO4 as an electrolyte and Pt working electrodes in an undivided cell, resulting in the selenyl-dihydrofurans in good to excellent yields. Furthermore, several of the synthesized products presented a high percentage of acetylcholinesterase (AChE) inhibition, highlighting their potential anti-Alzheimer activity.
The dihydrobenzofuran (DHB) core, found in many natural bioactive products and pharmaceuticals, represents an important “privileged scaffold”.1 Indeed, many bioactive compounds from natural sources carry a 2,3-dihydrobenzofuran core (Fig. 1), such as (+)-decursivine (I),2a (+)-lithospermic acid (II),2b (−)-linderol A (III),2c pterocarpin (IV),2d (+)-conocarpan (V),2e heliannuol G (VI).2f DHBs are well-known for their potential applications as pharmacological agents, being associated with antioxidant, antitumor, anti-platelet, anti-malarial, anti-inflammatory, anti-depressant and anti-convulsant properties.1,2 Several of the putative entactogen drugs, e.g., 5-APDB, 5-MAPDB, 6-APDB, MBPV (Fig. 1),3 have the DHB moiety in their core structure.
 | ||
| Fig. 1 Biologically-relevant DHBs. | ||
Similarly, organoselenium compounds are an important class of molecules that attract great interest, due to their biological properties.4 In recent decades, reports of their antioxidant, anti-inflammatory, antitumor and antiviral activities have led to increased interest in these compounds.4,5 Iwaoka has recently demonstrated the importance of organoselenium moiety in the preparation of selenoinsulin.5f Also, in the past few years, several authors have highlighted the importance of the organoselenyl moiety in the inhibition of AChE for anti-Alzheimer activity.6 Furthermore, organoselenium compounds are ubiquitous in organic chemistry, acting as catalysts,7 photosensitizers,8 building blocks9 and reaction intermediates.10
Considering the biological relevance of DHB and the wide spectrum of therapeutic properties of organoselenides, relatively few synthetic methods for the construction of organoselenyl-DHB have been reported in the literature (Scheme 1A). The addition of organoselenium cations to unsaturated compounds is the most common approach and reactive selenated species in the presence of oxidants is also frequently used.11 These methods offer interesting features but some protocols have drawbacks, which include the use of non-green solvents, pre-functionalized coupling partners, low atom economy, unstable substrates and elaborate multi-step processes.
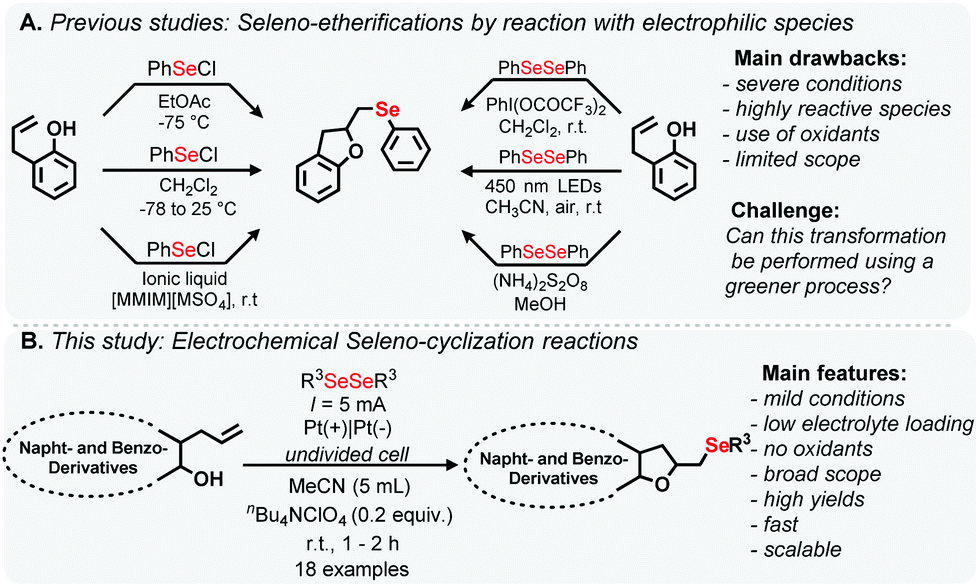 | ||
| Scheme 1 Overview. | ||
Furthermore, in recent years, intensive research has been carried out in the area of organic reactions using electrosynthesis.12 Reactions promoted in an electrochemical cell can lead to a variety of in situ generated intermediates, which are not usually accessible by conventional methods.13 These findings are related to some principles of green chemistry, such as preventing waste and diminishing the risk of accidents.14 In this context, the formation of C–Se bonds through the use of an electrical current has been gaining attention recently.15
Thus, in connection with our continuing interest in the development of new eco-friendly processes and C(sp2)–H bond selenylation of biologically compounds,16,17 herein, we report a straightforward protocol for the seleno-cyclization of allylic derivatives to access organoselenyl-DHB based heterocycles using electrosynthetic procedures (Scheme 1B). This more sustainable approach involves mild reaction conditions and a short reaction time, and provides the selenylated products in good yields, under open-to-air conditions. A range of biologically-relevant products were prepared using this atom-economic and scalable route.
Electrochemical reactions based on the anodic oxidation of diorganoyl dichalcogenides are well described in the literature.18 Based on our previous experience of electrochemical reactions,16a 1-allylnaphthalen-2-ol 1a and diphenyl diselenide 2a were used as standard substrates for electrochemical oxidative cyclization, in the presence of a stoichiometric amount of the electrolyte to be tested, for the optimization in an undivided cell reactor using platinum electrodes as the anode and cathode. To our delight, the first reaction attempt using 0.5 equiv. of nBu4NPF6 as the electrolyte and a constant current of 5 mA provided organoselenyl DHB 3a in 93% yield (Table 1, entry 1). Applying a higher current (10 mA) led to a decrease in yield and the use of a small amount of solvent (5 mL) did not adversely affect the reaction efficiency (entry 3). A series of supporting electrolytes were then tested, including nBu4NBF4, KI and nBu4NClO4. Surprisingly, the reaction with nBu4NClO4 (0.5 equiv.) led to 99% yield (entry 5). In some cases the size of electrolyte could influence in the formation of intimate ionic pair.18d In our case, we found that nBu4NBF6, nBu4NBF4, and nBu4NClO4 presented better results. The result is in accordance with the previous report,18e where similar effect was observed, considering that the relative ion's sizes are approximately equivalent. We tested in the next step, fine tuning of the electrolyte loading revealed (entries 5 vs. 6, 7) that 0.2 equiv. of nBu4NClO4 was sufficient to promote the oxidative cyclization, and 3a was obtained in 99% yield (entry 7). Furthermore, the use of a lesser amount of solvent (3 mL) resulted in decreased yield (entries 7 vs. 8). Such decrease could be observed in previous studies,18e with demonstrates that with the increase in concentration of the substrates, the signal shift on CV to cathodic field and this could lead in the formation of other intermediates of the reaction and ultimately result in lower yield. Attempts with other solvents did not afforded satisfactory yields (entries 9–12). This behavior can be noticed in previous reports where MeCN presented good results in electrosynthesis. MeCN has a good thermal and electrical stability with great dielectric constant, making it an excellent solvent for this type of transformation.12f–h,15 In order to investigate alternative conductors for the process, a graphite electrode was employed as the cathode and anode in three possible combinations (Table ESI,† entries 16–18), resulting in a small decrease in the yields. Finally, reducing the amount of 2a to 0.5 equiv. led to lower yield (Table 1, entry 13).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent (mL) | Electrolyte (equiv.) | Current |
|
Yielda (%) |
| General reaction conditions: Pt plate electrode (10 mm × 10 mm × 0.05 mm); graphite rod (Φ 4 mm); (1a) (0.2 mmol), (2a) (0.2 mmol).a Isolated yields; N.R. = for all cases, starting material was recovered. | |||||
| 1 | MeCN (10) | n Bu4NPF6 (0.5) | 5 mA | 1.0 equiv. | 93 |
| 2 | MeCN (10) | n Bu4NPF6 (0.5) | 10 mA | 1.0 equiv. | 88 |
| 3 | MeCN (5) | n Bu4NBF4 (0.5) | 5 mA | 1.0 equiv. | 74 |
| 4 | MeCN (5) | KI (0.5) | 5 mA | 1.0 equiv. | N.R. |
| 5 | MeCN (5) | n Bu4NClO4 (0.5) | 5 mA | 1.0 equiv. | 99 |
| 6 | MeCN (5) | n Bu4NClO4 (0.1) | 5 mA | 1.0 equiv. | 86 |
| 7 | MeCN (5) | n Bu 4 NClO 4 (0.2) | 5 mA | 1.0 equiv. | 99 |
| 8 | MeCN (3) | n Bu4NClO4 (0.2) | 5 mA | 1.0 equiv. | 80 |
| 9 | DMC (5) | n Bu4NClO4 (0.2) | 5 mA | 1.0 equiv. | N.R. |
| 10 | DMSO (5) | n Bu4NClO4 (0.2) | 5 mA | 1.0 equiv. | Trace |
| 11 | DCM (5) | n Bu4NClO4 (0.2) | 5 mA | 1.0 equiv. | 17 |
| 12 | EtOH (5) | n Bu4NClO4 (0.2) | 5 mA | 1.0 equiv. | 28 |
| 13 | MeCN (5) | n Bu4NClO4 (0.2) | 5 mA | 0.5 equiv. | 73 |


In order to investigate the methodology, a set of diselenides with different substituents were synthesized in order to evaluate the scope and limitations of the optimized reaction. Scheme 2 outlines the preparation of selenium-containing 1,2-dihydronaphthofurans with different electronic and structural features. First, the use of diselenides bearing electron-donating and electron-withdrawing substituents in ortho, meta and para positions afforded selenated products in average to high yields.
 | ||
| Scheme 2 Scope of electrochemical oxidative cyclization of 1a with several diselenides. | ||
The reaction proceeded smoothly for diselenides containing hydrogen, methoxy and methyl groups in the para position, and derivatives 3a, 3b and 3c were obtained in excellent yields. The method showed great compatibility with the electron-withdrawing group –F in the para position (derivative 3d, 79% yield), except for the chlorinated product 3e, which was obtained in 37% yield. When ortho-methylated and methoxylated diselenides were used, products 3f and 3g were obtained in 88% and 80% yield, respectively. With the electron-withdrawing group CF3 in the meta position, only traces of the desired product 3h were obtained. In the next step, diselenides bearing benzylic and aliphatic substituents were successfully employed in the reaction, affording derivatives 3i and 3j in 99% and 70% yields. In case of di-tert-butyl diselenide, traces of the desired product 3k were formed. Moderate yields were observed for naphthyl and thiophene derivatives 3l and 3m, and no reaction was observed when pyridine-substituted diselenide 2n was used. The reactivity of 1-allylnaphthalen-2-amine was also evaluated; however, product 3o was not observed (Scheme 2).
The method was also applied to substrates bearing modifications in the aromatic portion. For example, reaction with allyl phenol 4a provided derivative 5a in 55% yield. Similar results were observed for ketone substrate 4b (derivative 5b, 57% yield). The product 5c, based on the substrate 4c was obtained in a 70% yield. The derivative 4d, based on the vanillin skeleton, provided the selenylated compound 5d in moderate yield. The reaction showed tolerance for allyl and methoxy groups, and derivative 5e was obtained in good yield. The reaction performed with substrate 4f showed a different behavior and the yield was decreased, almost by half, compared with the model substrate 1a (derivative 5f, 55% yield). When substrate 4g, bearing two reaction sites, was employed, derivative 5g was obtained in 45% yield. Surprisingly, no reaction was observed when the nitrogen-containing substrate 4h was used (Scheme 3).
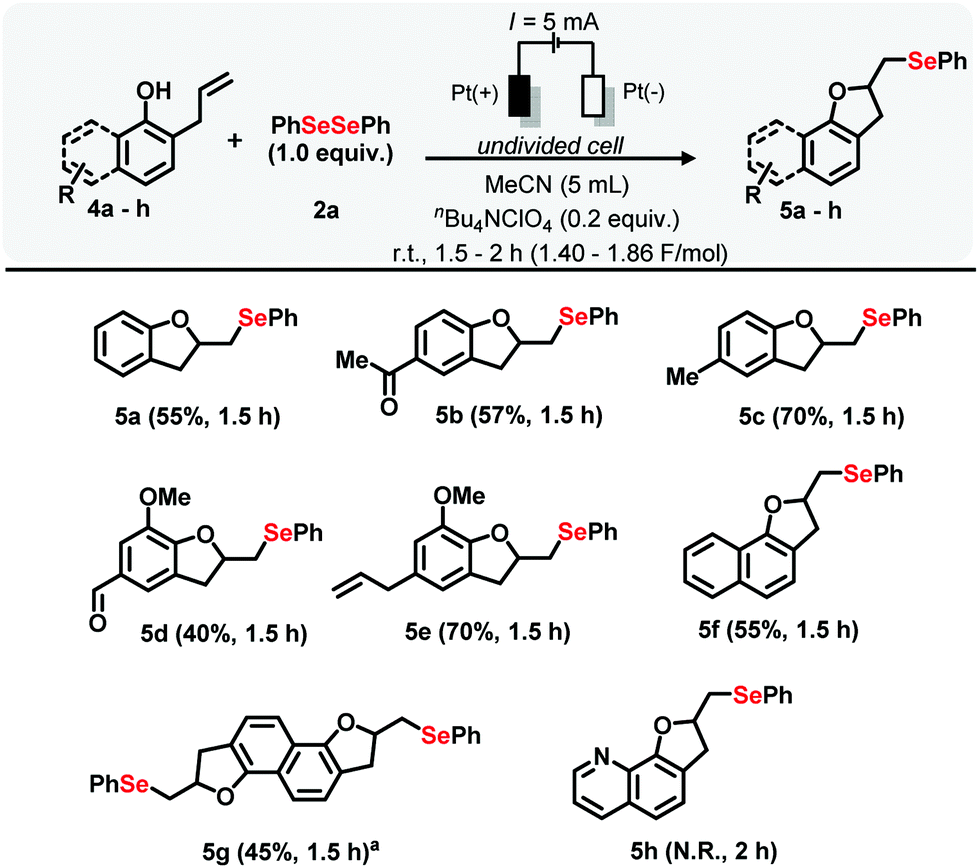 | ||
| Scheme 3 Scope of electrochemical oxidative cyclization of 4a–h with 2a. a2.0 Equivalents of 2a. | ||
To evaluate the applicability of the present method, the electrochemical oxidative oxyselenation of alkenes with 1-allylnaphthalen-2-ol 1a and diphenyl diselenide 2a was carried out in gram-scale synthesis (6 mmol), affording the product 3a in 64% yield after 15 h (Scheme 4). The reaction proved effective on large scale, highlighting its potential for industrial application. However, the cyclic voltammetry of 3a (Fig. ESI†) shows two oxidations peaks at 1.42 V and 1.75 V (vs. NHE), which may be associated with a process of degradation of the selenylated dihydrofuran, resulting in a lower yield for the gram-scale procedure.
 | ||
| Scheme 4 Gram-scale synthesis. Conditions: Platinum electrodes, constant current (5 mA), 1a (6 mmol), 2a (6 mmol), nBu4NClO4 (0.2 equiv.), MeCN (150 mL), r.t., air. Yield isolated by column chromatography. | ||
Although the fine details of the reaction mechanism remain unknown, several aspects observed during the experiments (Scheme 5 and Fig. 1) and in previous reports,19 guided us to propose a plausible mechanism (Scheme 6). In fact, two reaction routes can be proposed. Firstly, it is known that in the case of diphenyl diselenide (2a), oxidation and reduction processes may be involved in the catalytic cycle, suggesting that the reaction begins with the formation of a cationic radical intermediate Bvia anodic oxidation. Subsequently, we suggest that the addition of the radical intermediate B to the alkene moiety provides the radical intermediate D, anodic oxidation leads to the intermediate E and an intramolecular nucleophilic attack, followed by deprotonation, delivers product 3a (Pathway I). According to the control reactions, the medium was completely inhibited by the addition of 3.0 equiv. of the radical scavenger TEMPO (entry A – Scheme 5), which verifies that this process proceeds via a radical pathway. However, we cannot rule out the pathway via the phenyl selenium cation C (Pathway II). Through the formation of a reversible seleniranium intermediate F, followed by intramolecular nucleophilic attack, this route would also provide the final product 3a. This pathway was elucidated through the control reactions (entries B and C – Scheme 5).
 | ||
| Scheme 5 Control experiments. Standard conditions: Platinum electrodes, constant current (5 mA), 1a (0.20 mmol), 2a (0.20 mmol), nBu4NClO4 (0.2 equiv.), MeCN (5 mL), r.t., air. Yield isolated by column chromatography. | ||
 | ||
| Scheme 6 Proposed mechanism for electrochemical oxidative oxyselenylation of alkenes with 1-allylnaphthalen-2-ol 1a and diphenyl diselenide 2a. | ||
Cyclic voltammetry was performed and the anodic peak potentials (Epa vs. NHE) of diphenyl diselenide 2a and 1-allylnaphthalen-2-ol were obtained (Fig. ESI†). Oxidation peaks were observed for 2a (1.63 V) and 1-allylnaphthalen-2-ol 1a (1.77 V). Thus, we can assume that the oxidation of diphenyl diselenide occurs, which is in accordance with the plausible mechanism proposed in Scheme 6.
In relation to our interest in discovering potential therapeutics for Alzheimer disease,6b,d a number of structurally diverse selenylated products 3 and 5 from Schemes 2 and 3 were screened as inhibitors of the enzyme acetylcholinesterase (AChE). The Ellmann method20 was employed for the screening, galantamine was used as a standard and the IC50 results are shown in Table 2. It should be noted that all of the selenylated products tested showed a good percentage inhibition of AChE. Compounds containing a 2-methoxy substituent (3g), benzyl moiety (3i) and 2-thiophene (3m) were the most active selenides and exhibited significant inhibitory activity against AChE.
| Entry | Compounds | Acetylcholinesterase inhibitiona |
|---|---|---|
| IC50 (μM) ± SEM | ||
| a The compounds were tested in the range of 5–50 μM. Results are expressed as 50% inhibitory concentration (IC50) for AChE. Mean ± SEM of n = 3 independent experiments performed in duplicate. Data were analyzed by one-way analysis of variance (ANOVA), followed by Tukey's multiple comparisons test. | ||
| 1 | 3a | 18.16 ± 0.38 |
| 2 | 3b | 20.77 ± 3.34 |
| 3 | 3d | 21.46 ± 1.79 |
| 4 | 3g | 10.6 ± 2.45 |
| 5 | 3i | 11.6 ± 0.45 |
| 6 | 3m | 9.97 ± 0.71 |
| 7 | 5a | 13.44 ± 3.55 |
| 8 | 5b | 12.05 ± 0.34 |
| 9 | 5d | 21.28 ± 2.65 |
| 10 | 5l | 16.17 ± 2.28 |
| 11 | Galantamine | 11.55 ± 2.76 |
Conclusions
In summary, we have developed a new method for the intramolecular electrochemical oxidative oxyseleno-cyclization of allylnaphthol and allylphenol derivatives with diselenides under greener conditions. This regioselective strategy showed good compatibility of the functional group, being effective for both allylic derivatives, providing a series of organoselenium-containing dihydrofurans in good to excellent yields. In addition, the gram-scale experiment was successfully conducted, indicating the potential value of this protocol for industrial application. Furthermore, several of the selenylated products synthesized exhibited inhibition against the enzyme AChE comparable to that of galanthamine (commercially-available drug), demonstrating their potential as promising therapeutic agents for the treatment of Alzheimer's disease.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully acknowledge CAPES (001), CNPq, CERSusChem GSK/FAPESP (grant 2014/50249-8), INCT-Catálise and FAPESC-Pronex for financial support. M. R. S. would like to acknowledge CNPq for a doctoral fellowship and J. R. acknowledges CNPq (433896/2018-3) for funding. The authors are also grateful to CEBIME-UFSC for the HRMS analysis.Notes and references
- Z. Chen, M. Pitchakuntla and Y. Jia, Nat. Prod. Rep., 2019, 36, 666 RSC
.
-
(a) H. Zhang, S. Qiu, P. Tamez, G. T. Tan, Z. Aydogmus, N. V. Hung, N. M. Cuong, C. Angerhofer, D. D. Soejarto, J. M. Pezzuto and H. H. S. Fong, Pharm. Biol., 2002, 40, 221 CrossRef CAS
- A. Rickli, S. Kopf, M. C. Hoener and M. E. Liechti, Br. J. Pharmacol., 2015, 172, 3412 CrossRef CAS PubMed
-
(a) C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 104, 6255 CrossRef CAS PubMed
-
(a) D. Manna, G. Roy and G. Mugesh, Acc. Chem. Res., 2013, 46, 2706 CrossRef CAS PubMed
-
(a) J. M. Roldán-Peña, D. Alejandre-Ramos, Ó. López, I. Maya, I. Lagunes, J. M. Padrón, L. E. Peña-Altamira, M. Bartolini, B. Monti, M. L. Bolognesi and J. G. Fernández-Bolaños, Eur. J. Med. Chem., 2017, 138, 761 CrossRef PubMed
- F. V. Singh and T. Wirth, Catal. Sci. Technol., 2019, 9, 1073 RSC
-
(a) S. Mondal and G. Mugesh, Chem. – Eur. J., 2019, 25, 1773 CrossRef CAS PubMed
-
(a)
E. John, Nature's Building Blocks: An A-Z Guide to the Elements, Oxford University Press, New York, NY, New edn, 2004 Search PubMed
-
S. Kumar and H. B. Singh, in Organoselenium Compounds in Biology and Medicine, Royal Society of Chemistry, Cambridge, 2017, pp. 150–177 Search PubMed
-
(a) D. L. J. Clive, G. Chittattu, N. J. Curtis, W. A. Kiel and C. K. Kwong, J. Chem. Soc., Chem. Commun., 1977, 20, 725 RSC
-
(a) M. Yan, Y. Kawamata and P. S. Baren, Chem. Rev., 2017, 117(21), 13230 CrossRef CAS PubMed
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594 CrossRef CAS PubMed
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686 CrossRef CAS PubMed
- G. M. Martins, A. G. Meirinho, N. Ahmed, A. L. Braga and S. R. Mendes, ChemElectroChem, 2019, 6, 5928 CrossRef CAS
-
(a) A. G. Meirinho, V. F. Pereira, G. M. Martins, S. Saba, J. Rafique, A. L. Braga and S. R. Mendes, Eur. J. Org. Chem., 2019, 6465 CrossRef CAS
-
(a) S. Saba, C. R. dos Santos, B. R. Zavarise, A. A. S. Naujorks, M. S. Franco, A. R. Schneider, M. R. Scheide, R. F. Affeldt, J. Rafique and A. L. Braga, Chem. – Eur. J., 2020, 26, 4461 CrossRef CAS PubMed
-
(a) P. Röse, S. Emge, J. I. Yoshida and G. Hilt, Beilstein J. Org. Chem., 2015, 11, 174 CrossRef PubMed
-
(a) S. Torii, K. Uneyama, M. Ono and T. Bannou, J. Am. Chem. Soc., 1981, 103, 4606 CrossRef CAS
- G. L. Ellman, K. D. Courtney, V. Andres Jr. and R. M. Featherstone, Biochem. Pharmacol., 1961, 7, 88 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob00629g |
| This journal is © The Royal Society of Chemistry 2020 |