The role of a suburban forest in controlling vertical trace gas and OH reactivity distributions – a case study for the Seoul metropolitan area†
Saewung
Kim
 *a,
Roger
Seco‡
a,
Dasa
Gu§
a,
Dianne
Sanchez
a,
Daun
Jeong
a,
Alex B.
Guenther
a,
Youngro
Lee
b,
John E.
Mak
c,
Luping
Su
c,
Dan Bi
Kim
d,
Youngjae
Lee
d,
Joon-Young
Ahn
d,
Tom
Mcgee
e,
John
Sullivan
e,
Russell
Long
f,
William H.
Brune
g,
Alexander
Thames
g,
Armin
Wisthaler
hi,
Markus
Müller
i,
Thomas
Mikoviny
h,
Andy
Weinheimer
j,
Melissa
Yang
k,
Jung-Hun
Woo
l,
Soyoung
Kim
d and
Hyunjoo
Park
d
*a,
Roger
Seco‡
a,
Dasa
Gu§
a,
Dianne
Sanchez
a,
Daun
Jeong
a,
Alex B.
Guenther
a,
Youngro
Lee
b,
John E.
Mak
c,
Luping
Su
c,
Dan Bi
Kim
d,
Youngjae
Lee
d,
Joon-Young
Ahn
d,
Tom
Mcgee
e,
John
Sullivan
e,
Russell
Long
f,
William H.
Brune
g,
Alexander
Thames
g,
Armin
Wisthaler
hi,
Markus
Müller
i,
Thomas
Mikoviny
h,
Andy
Weinheimer
j,
Melissa
Yang
k,
Jung-Hun
Woo
l,
Soyoung
Kim
d and
Hyunjoo
Park
d
aDepartment of Earth System Science, School of Physical Sciences, University of California, Irvine, CA 92697, USA. E-mail: saewung.kim@uci.edu; Fax: +1-949-824-8794; Tel: +1-949-824-4531
bSchool of Earth and Atmospheric Sciences, College of Natural Sciences, Georgia Institute of Technology, Atlanta, GA 30332, USA
cSchool of Marine and Atmospheric Sciences, Stony Brook University, Stony Brook, NY 11794, USA
dClimate and Air Quality Research Department, National Institute of Environmental Research, Incheon 22689, South Korea
eGoddard Space Flight Center, National Aeronautics and Space Administration, Greenbelt, MD 20771, USA
fNational Exposure Research Laboratory, Office of Research and Development, Environmental Protection Agency, Durham, NC 27709, USA
gDepartment of Meteorology and Atmospheric Science, Pennsylvania State University, University Park, PA 16802, USA
hDepartment of Chemistry, University of Oslo, Oslo, Norway
iInstitute of Ion Physics and Applied Physics, University of Innsbruck, Innsbruck, Austria
jNational Center for Atmospheric Research, Boulder, Colorado 80301, USA
kNational Suborbital Education and Research Center, National Aeronautics and Space Administration, Langley, VA 23681, USA
lDepartment of Technology Fusion Engineering, Konkuk University, Seoul 05029, Korea
First published on 28th September 2020
Abstract
We present trace gas vertical profiles observed by instruments on the NASA DC-8 and at a ground site during the Korea-US air quality study (KORUS) field campaign in May to June 2016. We focus on the region near the Seoul metropolitan area and its surroundings where both anthropogenic and natural emission sources play an important role in local photochemistry. Integrating ground and airborne observations is the major research goal of many atmospheric chemistry field campaigns. Although airborne platforms typically aim to sample from near surface to the free troposphere, it is difficult to fly very close to the surface especially in environments with complex terrain or a populated area. A detailed analysis integrating ground and airborne observations associated with specific concentration footprints indicates that reactive trace gases are quickly oxidized below an altitude of 700 m. The total OH reactivity profile has a rapid decay in the lower part of troposphere from surface to the lowest altitude (700 m) sampled by the NASA DC-8. The decay rate is close to that of very reactive biogenic volatile organic compounds such as monoterpenes. Therefore, we argue that photochemical processes in the bottom of the boundary layer, below the typical altitude of aircraft sampling, should be thoroughly investigated to properly assess ozone and secondary aerosol formation.
1. Introduction
A thorough investigation on the vertical distributions of trace gases is an essential task to evaluate the impact of surface emissions on regional air quality.1 The negative impacts of these emissions are of great concern as deteriorating regional air quality is a public health issue,2 as well as economic issue from effects such as reduction in crop yields.3 Recently, major air pollutants such as ozone and fine particles have also been a concern due to their designation as short-lived climate forcers (SLCFs).4,5 Although the assessed radiative forcing from SLCFs is smaller than those from long-lived climate forcers such as carbon dioxide, the uncertainty is still relatively high.5Therefore, the capability to simulate the horizontal and vertical distribution of photochemical reaction products and their precursors is a necessary first step to properly diagnose their impacts on regional and global air quality, which requires two different directions of research. First, the capability to simulate complex land use in a model framework to determine accurate reactive trace gas emissions from various anthropogenic activities and natural processes. Second, the ability to accurately represent transport and photochemical oxidation processes. In this context, Shindell and colleagues4 claimed emission-based assessments of climate forcers requires an accurate process level understanding of tropospheric photochemistry. A recent assessment on the status of climate change by the Intergovernmental Panel on Climate Change (IPCC) clearly accepted this notion. As a result, an extensive discussion of gas–aerosol–cloud interactions is presented in the most recent IPCC Assessment Report (AR5).5
A megacity embedded in a forest environment is a unique testbed to examine the complicated photochemical processes from various emission sectors to photochemical degradation during vertical and horizontal transport processes.6 Moreover, considering the ever growing number of people residing in urban regions,7 it is important to understand urban photochemical processes and subsequent secondary product formation such as ozone and aerosols. This motivated us to examine vertical heterogeneity of trace gas distributions and total OH reactivities over the Seoul metropolitan area (SMA), South Korea. We present ground and airborne observations conducted as part of the Korea-US air quality study (KORUS-AQ) campaign in the late spring and early summer of 2016. The focus is placed on examining vertical heterogeneity over a forested region downwind of the megacity. Prior studies have clearly demonstrated the complicated nature of oxidation capacity and trace gas reactivity of the suburban forest region near the SMA. Kim and colleagues8 demonstrated the potential of high oxidation capacity from higher than expected HONO levels in the suburban forest. Kim and colleagues6 also reported that the observed reactive trace gases can only account for about 30% of the measured total OH reactivity demonstrating that the majority of reactive compounds cannot be observed with standard measurement techniques. Both studies have attributed natural processes, particularly biogenic volatile organic compounds (BVOCs), as the main driver determining oxidation capacity and reactivity in this region with strong anthropogenic influences of a large megacity with a population of 25 million.
During the KORUS-AQ campaign,9 the flight path of the NASA DC-8 airborne laboratory was well coordinated with the ground sites in order to obtain a statistically relevant airborne dataset to compare with the ground observational datasets. In this study, we compare observations from a suburban forest ground research site, Taehwa Research Forest (TRF), and aircraft boundary layer measurements above the suburban forest to examine interactions between the biosphere and atmosphere in an area with high background anthropogenic pollution.
2. Methods
2.1. Taehwa Research Forest and NASA DC-8 during the KORUS-AQ campaign
The TRF, shown in Fig. 1, was selected as a super site during the KORUS-AQ campaign for several reasons. The first was the geographic proximity to the SMA. TRF is located 40 km from the city center and 20 km from the nearest edge of the SMA in the southeastern direction. Previous studies have illustrated that this proximity provides a background of photochemically processed urban air at the TRF site. For example, Kim and colleagues10 reported substantially lower levels of CO and NOx at the TRF site in comparison with the levels observed at the SMA urban sites. On the other hand, secondary photochemical products, such as ozone, are observed at similar levels or even higher at the TRF in comparison to urban sites. The second reason was to investigate the roles of vegetation, particularly BVOC oxidation and photochemistry. With the high NOx conditions downwind of the megacity, the highly reactive nature of BVOCs can enhance the photochemical production of aerosols and ozone. The site is also located in the middle of a monoterpene-emitting pine tree plantation (200 m by 200 m) surrounded by a natural mixed forest including isoprene-emitting oaks. The gradient observations consistently indicate homogeneous distributions of isoprene inside (18 m or below) and right above the canopy (30 m)11 from a year-long observation, which indicates that the observations at the site can represent the concentration footprint of the airshed.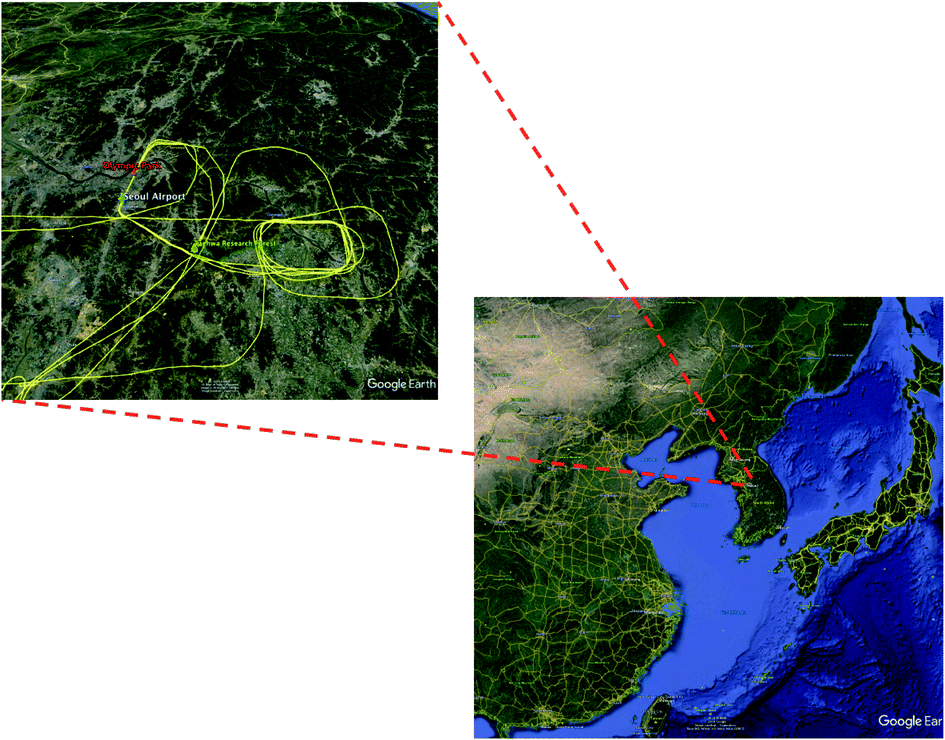 | ||
| Fig. 1 Location of the two ground super sites and stereo manoeuvring for NASA DC-8 for the KORUS-AQ field campaign in 2016. The top left map shows the flight track over the SMA. The circular flight pattern on the left of the TRF illustrates spiral profiling. | ||
2.2. Instrumental configurations
In Table 1, instrumental configurations for the dataset presented in this paper are summarized. The key parameters presented in this study, VOCs and total OH reactivity, were quantified both on the ground and on the NASA DC-8 airborne laboratory. VOCs were quantified by a proton transfer reaction-time of flight-mass spectrometer (PTR-ToF-MS).12 The ground observation was conducted by a commercial instrument (manufactured by IONICON Analytik GmbH). On the other hand, airborne measurements of VOCs were carried out using a custom-made PTR-ToF-MS instrument. The instrument described by Müller and colleagues13 was upgraded with an ion funnel and a hexapole ion guide to improve ion transmission into the mass spectrometer, thus providing higher sensitivity.14 Both the aircraft and ground instruments were periodically calibrated during the measurement period using a dynamically diluted certified VOC standard (Apel-Riemer Environmental Inc., Miami, FL, USA).| Instrument | Parameters | |
|---|---|---|
| Ground | Comparative reactivity method-chemical ionization spectroscopy (CRM-CIMS) | OH reactivity |
| Thermo Scientific 42i | NO | |
| Cavity ring down spectroscopy (Los Gatos) | NO2 | |
| Thermo Scientific 49i | O3 | |
| Lufft 501 C | Temperature | |
| Thermo Scientific 48i TLE | CO | |
| Thermo Scientific 43i TLE | SO2 | |
| Proton transfer reaction-time of flight-mass spectrometer (PTR-TOF-MS) | VOCs and OVOCs | |
| Flow tube-laser induced florescence technique15 | OH reactivity | |
| NASA DC-8 | Chemiluminescence technique36 | NOx |
| UV absorption spectroscopy36 | O3 | |
| Differential absorption CO measurement37 | CO | |
| PTR-ToF-MS | VOCs and OVOCs |
Total OH reactivity was measured on the aircraft by a flow tube-laser induced florescence (FT-LIF) instrument characterizing OH decay inside of a flow tube with ambient air.15 On the ground, total OH reactivity was measured using the comparative reactivity method with a chemical ionization mass spectrometer using the hydronium reagent ion system (CRM-CIMS).16 The identical analytical system has been thoroughly described and successfully utilized in previous field campaigns.6,17,18 This includes a thorough description of the analytical characteristics of the CRM-CIMS system18 and a comparison of the CRM-CIMS with an FT-LIF based OH reactivity system (the southern oxidant and aerosol study, SOAS, in Brent, Alabama, USA, in 2013)19 that is similar to the instrument integrated on the NASA DC-8.15 The results indicate that the data from both instruments agree within their analytical uncertainty although they reported a systematic bias likely caused by differences in inlet configurations. Based on the findings from that study, we shortened the inlet length and residence time of the ground-based instrument to prevent sample loss (4 m of ¼” PFA tubing with less than 0.5 s of the sample residence time). Over the field campaign period, we conducted periodic calibrations on both the CRM-CIMS OH reactivity system and the PTR-ToF-MS system. The PTR-ToF-MS system was routinely calibrated using a standard gas manufactured by Apel-Riemer Environmental Inc. containing isoprene, acetone, acetaldehyde, α-pinene, benzene, and toluene. After the field work, the standard gas was recalibrated by a gas chromatography-mass spectrometer system operated by the Blake Lab in the Department of Chemistry at the University of California, Irvine, USA. The laboratory also collected whole air samples for VOC analysis during the KORUS-AQ campaign on the NASA DC-8 aircraft. This analytical system and procedure are described in ref. 20. A detailed data analysis on the ground VOCs and OH reactivity observations at TRF can be found in Sanchez and colleagues.21
3. Results and discussion
3.1 The boundary layer evolution and vertical distributions of trace gases
In Fig. 2, we present the averaged vertical distribution of potential temperature (in the top far left panel) observed by the NASA DC-8 during the KORUS-AQ campaign over the SMA. The airborne dataset was filtered to represent data collected in the area shown in Fig. 1 to illustrate the observed vertical profiles over the SMA. The overall flight paths for the KORUS-AQ science flights are shown in Fig. S1 in the ESI.† The ground observation has been maintained to maximize the overlap between the ground and the airborne datasets. The seamless coordination between the different platforms can be found in previous publications.22,23 | ||
| Fig. 2 The averaged vertical profiles quantified on the NASA DC-8 during the KORUS-AQ campaign over the SMA. The blue, the red, and the dark yellow profiles illustrate average profiles for morning (8 am to 11 am), mid-day (11 am to 2 pm), and late afternoon (2 pm to 5 pm), respectively. | ||
As presented in Fig. 2, in the morning (8 am to 11 am) the troposphere is in a stable condition. As solar radiation causes thermal turbulence at the surface, the boundary layer evolves into the late afternoon. Therefore, we can expect active vertical transport and mixing of trace gases emitted from the surface through the entire boundary layer up to ∼2 km above the ground, which is well corroborated by the average daily curtain plot of ozone shown in Fig. S2,† from an ozone LIDAR instrument located at the TRF during the KORUS-AQ campaign. The trace gas distributions, shown in Fig. 2 illustrate the mixing processes.
The CO profiles clearly demonstrate the temporal evolution of the vertical mixing processes. The morning profile (8 to 11 am) shows the stagnated nature of the lower troposphere, but turbulent mixing inside of the boundary layer becomes more prominent in the afternoon. The mid-day (11 am to 2 pm) profile shows moderate vertical mixing. Furthermore, the afternoon (2 pm to 5 pm) presents substantial vertical mixing causing a well-mixed boundary layer below the altitude of 2 km. The ozone production associated with the vertical transport of ozone precursors, particularly NOx, can be interpreted by comparing their vertical profiles. The morning stagnation is conspicuous from the observations of ozone titration at the surface by the high NO emission from the ground. Consequently, pronounced NO2 was observed in the lowest part of the boundary layer and the concentration sharply decreases at higher altitude. Towards mid-day and the afternoon, however, elevated ozone is observed throughout the boundary layer, which can be explained by the photochemical production of ozone in the boundary layer from convected NO2 and VOCs. Some of the reactive anthropogenic and biogenic VOC profiles are also presented in Fig. 2.
The morning profiles reflect a stratified lower troposphere as previously discussed. On the other hand, when vertical mixing gets active in the afternoon, the average VOC profiles consistently illustrate that the photochemical lifetime and vertical mixing time scale are both critical determinants for the vertical distribution of trace gases. Benzene (kOH = 1.28 × 10−12 cm3 molecule−1 s−1 at 298 K), a relatively less reactive VOC, is evenly spread throughout the boundary layer, but a more reactive VOC such as toluene (kOH = 6.16 × 10−12 cm3 molecule−1 s−1 at 298 K) has a mixing ratio that gradually decreases as a function of altitude in the boundary layer. The most reactive gas among those presented in Fig. 2 is isoprene (kOH = 1.0 × 10−10 cm3 molecule−1 s−1 at 298 K) showing the most rapid degradation in the boundary layer as a function of altitude. The photochemical degradation of isoprene is expected to be dominated by OH producing methyl vinyl ketone (MVK; kOH = 1.85 × 10−11 cm3 molecule−1 s−1 at 298 K) and methacrolein (MACR; kOH = 3.07 × 10−11 cm3 molecule−1 s−1 at 298 K). These compounds have longer lifetimes than isoprene due to their slower reaction rates, which causes a clear accumulation of MVK and MACR in the boundary layer. As a result, the ratio of MVK + MACR to isoprene can be used to estimate the boundary layer transport times from a photochemical perspective.24 A similar vertical distribution can be found in the case of acetone originating from both direct emissions and as a by-product of VOC oxidation. Singh and colleagues25 reported that the relative source strength between primary emission and VOC oxidation is around 1 to 2 on a global scale. The biogenic primary sources are estimated to be around nine times higher than those from anthropogenic sources on a global scale.26 The vertical distribution of acetone presented in Fig. 2 is quite similar to that of MVK + MACR except that the concentration of the lowest part of the profile does not substantially change over the day for acetone in comparison to MVK + MACR, which may reflect the relative importance of surface emission of acetone in the morning from traffic. As the day progresses, the relatively slow reaction rate with OH (kOH = 2.31 × 10−13 molecule−1 s−1 cm3), causes acetone from both primary and secondary sources to appear well-mixed through the boundary layer. Furthermore, the substantial level (1–2 ppb) in the free troposphere can be attributed to its long chemical lifetime. There are two distinctive features in the OH reactivity vertical distribution. First, there is the substantial presence of OH reactivity in the free troposphere, which can be explained by contributions from relatively long-lived trace gases such as CO and acetone, as most reactive trace gases were observed to have insignificant levels compared with the values in the boundary layer. Second, enhanced OH reactivity in the boundary layer is observed in the afternoon similar to the OVOC vertical profiles such as MVK + MACR and acetone, which illustrates a homogeneous distribution throughout the boundary layer in the afternoon. In contrast, VOCs directly emitted from anthropogenic and biogenic activities such as benzene, toluene, isoprene and monoterpenes illustrate a pronounced mixing ratio near the surface. Therefore, one can speculate that the relative contribution from OVOCs to OH reactivity become more important in the upper part of the boundary layer and the free troposphere.
The normalized trace gas and OH reactivity profiles shown in Fig. 3 illustrate the observed boundary layer behavior in further detail. Fig. 3 (left) shows the vertical distributions of CO, benzene, toluene, isoprene and monoterpenes below 4 km in the late afternoon normalized by the lowest altitude observed values. A more pronounced vertical gradient in the boundary layer can be noticed for the reactive compounds such as isoprene and monoterpenes, in comparison to the less reactive compounds such as CO, benzene, and toluene. The OVOC distributions in the late afternoon such as acetone and MVK + MACR, shown in Fig. 3 (right), illustrate the broadly elevated levels in the boundary layer, and divergence in the free troposphere. OH reactivity distributions in the afternoon follow those of acetone, a longer lived OVOC.
 | ||
| Fig. 3 The normalized profiles of trace gases observed on the NASA DC-8 during the KORUS-AQ in the SMA. | ||
3.2 Combining ground site and airborne observations
The trace gas observations on the aircraft provide detailed information on interactions between boundary layer mixing processes and photochemical degradation of trace gases of various reactivity. One important fact that we should remind ourselves is that the lower levels of the vertical distribution of trace gases and other parameters are not typically observed on the aircraft. Other than during a missed approach, or a flight pattern to approach to a runway with landing gear on very close to the surface without actually landing then taking off again, at Seoul Airport, the surface air layer where most compounds are emitted, is never sampled by the instrumentation on most research aircraft. We attempt systematic comparisons between datasets collected at the lowest NASA DC-8 altitude and the TRF ground site. As shown in Fig. 1 and S1,† the overall flight tracks over the SMA consistently followed a prescribed flight path that consists of a missed approach at Seoul Airport then a turn to the southeastern direction to pass over the TRF. Typically, the DC-8 overpass was conducted at ∼700 m above the ground.We filtered the data points for the Taehwa overpasses of DC-8. The applied spatial filter for this analysis is shown in Fig. S3.† The average VOC concentrations in the afternoon (14:00 to 17:00 local time) for the overpasses, along with standard deviation error-bars, are shown in Fig. 2. The levels of most VOCs with various atmospheric reactivity are in the range of the averaged concentrations in the vertical profiles in the SMA. This consistency implies that the TRF site is representative of the average regional characteristics of trace gas distributions. It may come as a surprise that a forest site near a megacity can reflect the regional characteristics. However, as shown in Fig. 1, a substantial fraction of the flight track is over the forest. On top of that, the prevailing wind direction during the campaign was mostly easterly (Fig. 4), where forest is the dominant land cover. Lee and colleagues27 presented the diurnal circulation pattern of Seoul’s city center and its surroundings. In the analysis, Lee and colleagues27 illustrated that the airmass from the surrounding forest is circulated into the urban area. Therefore, the quantitative understanding of photochemistry in the suburban forest is also critical to properly simulate urban air quality.
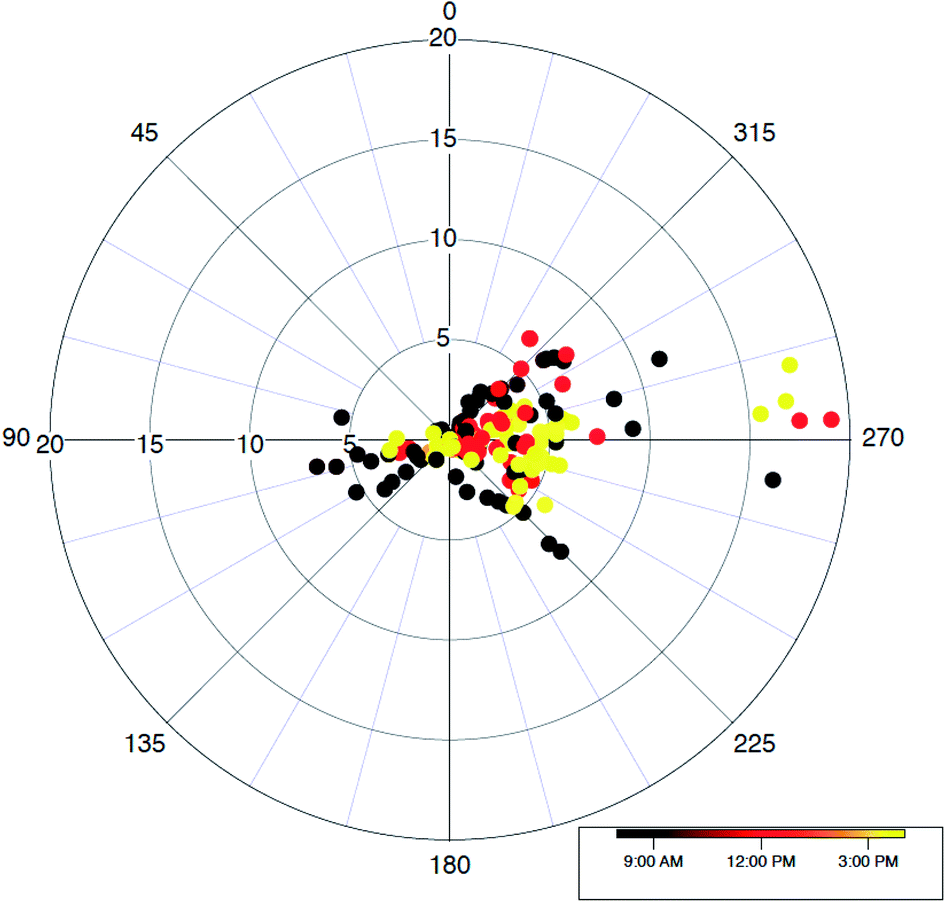 | ||
| Fig. 4 A wind rose showing observed wind distributions near the TRF. The wind data is filtered using the geographical filter shown in Fig. S3.† | ||
To examine the differences in various observed trace gas levels at the TRF and on the NASA DC-8, we present diurnal variations of VOCs from primary emissions of toluene, isoprene, and monoterpenes and OH reactivity in Fig. 5. The average diurnal variations are based on the ground observational dataset at the TRF for the dates when the NASA DC-8 conducted its science flights. The data points in the plots are averages of the airborne observational datasets when the NASA DC-8 flew near the TRF. Fig. S4† illustrates exemplary temporal variations of isoprene and OH reactivity observed at the TRF and the DC-8 over the TRF filtered by the spatial filter illustrated in Fig. S3.† The presented dataset for this analysis is based upon 14 days of science flights over a month.
 | ||
| Fig. 5 Diurnal average of (a) isoprene, (b) OH reactivity, (c) monoterpenes, and (d) toluene observed at the TRF (red trace) for the dates when NASA-DC 8 flights were conducted, and on the NASA DC-8 during the TRF overpasses (green triangles). | ||
Most of the observed trace gases, regardless of biogenic or anthropogenic origins, illustrate a clear difference between the ground and the airborne observations due to the stagnation processes discussed above (Fig. 5). As the boundary layer develops towards the afternoon, moderately long-lived VOCs such as toluene were observed at comparable levels at the TRF on the ground and at the DC-8 700 m altitude. However, the ground observations of the reactive trace gases such as isoprene and monoterpenes are substantially higher during the afternoon than those from airborne observations.
For further examination, we reconstruct the vertical distribution of reactive VOCs such as isoprene and monoterpenes and OH reactivity, by assuming first-order decay. For this analysis, we assume that the airmass that the NASA DC-8 sampled is from the forest. Again, it can be justified by the fact that the horizontal wind direction governing advection was predominantly from the forest area. As the TRF site was established in the middle of a pine plantation, which has relatively high monoterpene emissions, it is possible that monoterpenes are over-represented relative to the other local landscapes. However, site survey results indicated that monoterpene concentrations observed outside of the plantation in the surrounding research forest were at similar levels to those inside of the plantation.11 In addition, a substantial fraction of the surrounding natural forest is composed of conifers (∼50%) in South Korea.11 Therefore, the comparison of the reactive trace gas datasets between TRF and the overpass airborne measurements provides insight on the vertical divergence of reactive trace gases in the lower planetary boundary layer in this region.
The first order decay constants of isoprene, monoterpenes, and OH reactivity are calculated with the following equation.
| [concentration]z = [concentration]0 × e−kz | (1) |
The first order decay constant (k) can be deduced from the concentrations observed on the DC-8 overpass and at the TRF in the late afternoon (2 pm to 4 pm) and are presented in Fig. 6. The specific input values and deduced decay constants are summarized in Table 2.
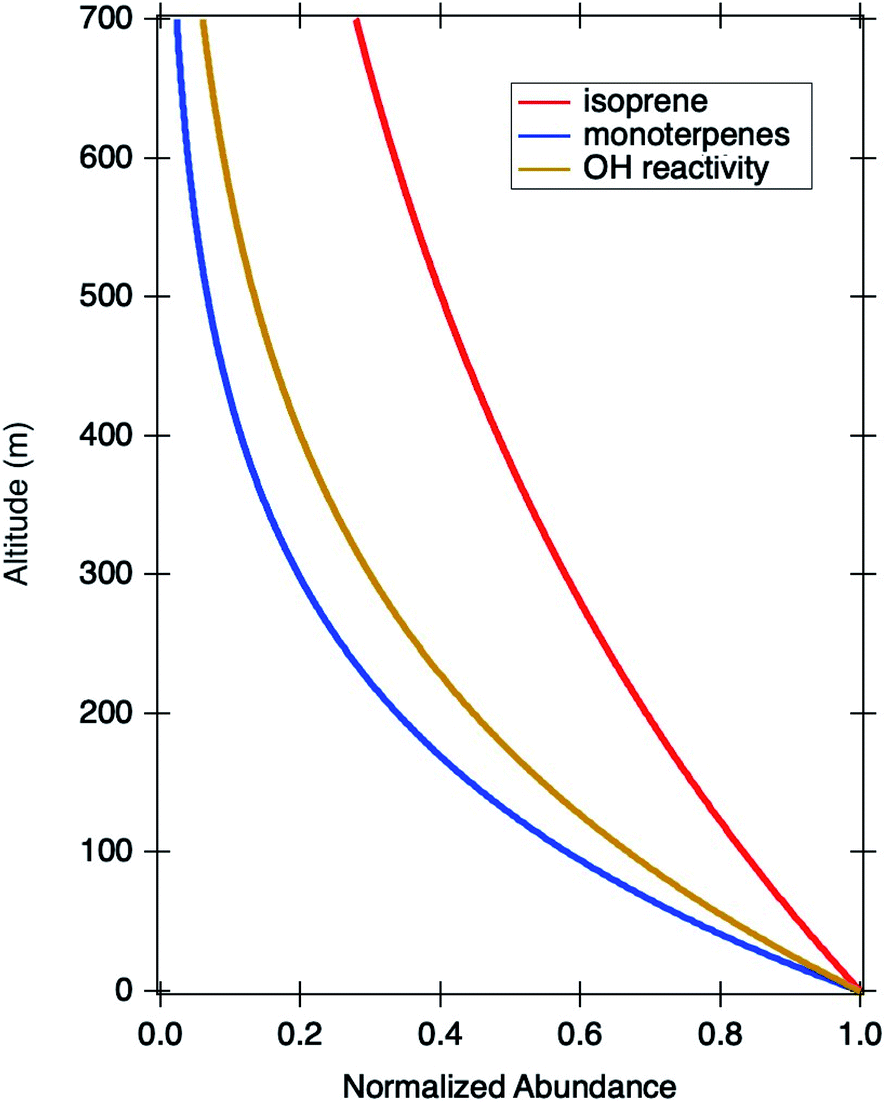 | ||
| Fig. 6 An illustration of the vertical decays of isoprene, monoterpenes, and OH reactivity using afternoon averaged datasets at TRF and NASA DC-8 during the TRF overpasses in the late afternoon (2 pm to 5 pm). | ||
| Altitude | Monoterpenes (ppb) | OH reactivity (s−1) | Isoprene (ppb) | Toluene (ppb) |
|---|---|---|---|---|
| 0 m (TRF) | 0.2 | 27.8 | 0.36 | 1.28 |
| 700 m (NASA DC-8) | 0.03 | 6.8 | 0.19 | 1.04 |
The detailed analysis by Karl and colleagues28 quantitatively describes the impact of physical parameters such as vertical heat flux, and chemical aspects such as chemical lifetime, that are mostly governed by hydroxyl radical concentrations. The OH reactivity vertical divergence clearly illustrates that the reactive compound divergence is mostly governed by chemical reactions since it more sharply decreases in its magnitude than that of isoprene. Since we know the reactivity of isoprene (kisoprene–OH = 1.0 × 10−10 cm3 molecules−1 s−1 at 25 °C), we can relate this to the average reactivity of monoterpenes, consisting of many different isomers, Table 3. It is clear that monoterpenes have a chemical divergence that is as much as three times faster than isoprene. It is notable that the dominant monoterpenes identified by the gas-chromatography technique at this site, β-pinene, α-pinene, and 3-carene,8,29,30 are not very reactive compared with isoprene. Therefore, the rapid decay of monoterpenes illustrated in Fig. 3 leads us to conclude that the total monoterpene measurement of the PTR-ToF-MS technique may mostly be detecting very reactive monoterpene species, with lifetimes of a few minutes such as ocimene (cis-β and trans-β31), that are difficult to quantify by offline GC techniques as a number of previous studies have reported.32 The decay rate of OH reactivity is faster than that of isoprene (∼twice) but slower than that of monoterpene. Therefore, this analysis quantitatively illustrates that a substantial amount of reactive trace gases are composed of highly reactive species, which directs us to investigate potential sources for compounds causing missing OH reactivity.33 Indeed, a detailed analysis by Sanchez and colleagues20 reported 61% of total OH reactivity was missing at the TRF during the KORUS campaign. It is also notable that ozone may play a significant role in the fast vertical decay of monoterpenes. In general, higher reactivity towards OH for specific monoterpene isomers warrants higher reactivity towards ozone (e.g. Kim and colleagues34), and as shown in Fig. 2, substantial ozone was present in the boundary layer over SMA.
| Monoterpenes | OH reactivity | Isoprene | |
|---|---|---|---|
| Rate constant (m−1) | 2.7 × 10−3 | 2.0 × 10−3 | 9.0 × 10−4 |
| Ratio | 3.0 | 2.2 | 1 |
Therefore, the observed high level of missing OH reactivity in the canopy during the KORUS-AQ campaign is likely to be mainly driven by highly reactive VOCs, such as reactive monoterpenes. These compounds would be quickly oxidized very close to the surface to produce less reactive oxidation products. If they are monoterpenes or similar compounds, then their oxidation products will likely be partitioned into the particle phase and will not have any further impact on oxidation capacity. Therefore, it is plausible that these highly reactive compounds have only limited impact on oxidation capacity near the surface, possibly inside of the forest canopy, which would serve as an important constraint to evaluate regional photochemistry such as NOx lifetime (e.g. Loughner and Cohen35).
4. Conclusion
The examination of the vertical evolution of collective trace gas reactivity illustrates that the ground reactivity is dominated by very reactive gases consistent with reactive BVOCs. This observation suggests that most reduced trace gases emitted from the surface of the suburban forest are mostly oxidized near the surface below the region probed by a large research aircraft such as the NASA DC-8, due to flight restrictions. Therefore, we urge the deployment of additional platforms to investigate photo-oxidation processes in the lower troposphere, such as tall towers, tethered balloons and unmanned aerial vehicles to directly measure and investigate the rapid photochemical processes occurring in this region. These results also provide a critical constraint to evaluate model performance to assess the role of megacities in the global climate system for producing short-lived climate forcers such as ozone and aerosols.Data availability
The dataset utilized in this study is open to the public and can be downloaded at https://www-air.larc.nasa.gov/missions/korus-aq/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by NASA (NNX15AT90G) and NIER. PTR-MS measurements aboard the NASA DC-8 during KORUS-AQ were supported by the Austrian Federal Ministry for Transport, Innovation and Technology (bmvit) through the Austrian Space Applications Programme (ASAP) of the Austrian Research Promotion Agency (FFG, # 847967). P. Eichler and L. Kaser are acknowledged for field support. S. Kim would like to acknowledge funding support from the Brain Pool Program of the National Research Foundation Korea (NRF) Funded by the Ministry of Science ICT (# 2020H1D3A2A01060699) and the National Strategic Project-Fine Particle of the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT), the Ministry of Environment (ME), and the Ministry of Health and Welfare (MOHW) (2019M3D8A1067406).References
- S. R. Davis, R. Talbot, H. T. Mao and J. A. Neuman, Atmosphere, 2014, 5, 973–1001 CrossRef
.
- L. B. Lave and E. P. Seskin, Science, 1970, 169, 723–733 CrossRef CAS PubMed
- W. L. Chameides, X. S. Li, X. Y. Tang, X. J. Zhou, C. Luo, C. S. Kiang, J. St John, R. D. Saylor, S. C. Liu, K. S. Lam, T. Wang and F. Giorgi, Geophys. Res. Lett., 1999, 26, 867–870 CrossRef CAS
- D. T. Shindell, G. Faluvegi, D. M. Koch, G. A. Schmidt, N. Unger and S. E. Bauer, Science, 2009, 326, 716–718 CrossRef CAS PubMed
-
IPCC, Climate Change 2013: The Physical Science Basis. Contribution of Working Group 1 to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge, U.K., New York, NY, USA, 2013 Search PubMed
- S. Kim, D. Sanchez, M. Wang, R. Seco, D. Jeong, S. Hughes, B. Barletta, D. R. Blake, J. Jung, D. Kim, G. Lee, M. Lee, J. Ahn, S.-D. Lee, G. Cho, M.-Y. Sung, Y.-H. Lee, D. B. Kim, Y. Kim, J.-H. Woo, D. Jo, R. Park, J.-H. Park, Y.-D. Hong and J.-H. Hong, Faraday Discuss., 2016, 189, 231–251 RSC
- S. X. Zhao, N. S. Guo, C. L. K. Li and C. Smith, World Dev., 2017, 98, 257–289 CrossRef
- S. Kim, S.-Y. Kim, M. Lee, H. Shim, G. M. Wolfe, A. B. Guenther, A. He, Y. Hong and J. Han, Atmos. Chem. Phys., 2015, 15, 4357–4371 CrossRef CAS
-
J. A. Al-Saadi, G. Carmichael, J. Crawford, L. Emmons, S. Kim, C.-K. Song, L.-S. Chang, G. Lee, J. Kim and R. Park, NASA Contributions to KORUS-AQ: An International Cooperative Air Quality Field Study in Korea, NASA, 2015 Search PubMed
- S. Y. Kim, X. Y. Jiang, M. Lee, A. Turnipseed, A. Guenther, J. C. Kim, S. J. Lee and S. Kim, Atmos. Environ., 2013, 70, 447–453 CrossRef CAS
-
S.-Y. Kim, H. J. Park, Y. D. Hong and J. S. Han, The study on the flux of carbon and biogenic volatile organic compounds in a forest (II), National Institute of Environmental Research, Incheon, South Korea, 2013 Search PubMed
- M. Graus, M. Muller and A. Hansel, J. Am. Soc. Mass Spectrom., 2010, 21, 1037–1044 CrossRef CAS PubMed
- M. Müller, T. Mikoviny, S. Feil, S. Haidacher, G. Hanel, E. Hartungen, A. Jordan, L. Märk, P. Mutschlechner, R. Schottkowsky, P. Sulzer, J. H. Crawford and A. Wisthaler, Atmos. Meas. Tech., 2014, 7, 3763–3772 CrossRef
- B. Yuan, A. R. Koss, C. Warneke, M. Coggon, K. Sekimoto and J. A. de Gouw, Chem. Rev., 2017, 117, 13187–13229 CrossRef CAS PubMed
- J. Mao, X. Ren, W. H. Brune, J. R. Olson, J. H. Crawford, A. Fried, L. G. Huey, R. C. Cohen, B. Heikes, H. B. Singh, D. R. Blake, G. W. Sachse, G. S. Diskin, S. R. Hall and R. E. Shetter, Atmos. Chem. Phys., 2009, 9, 163–173 CrossRef CAS
- V. Sinha, T. G. Custer, T. Kluepfel and J. Williams, Int. J. Mass Spectrom., 2009, 282, 108–111 CrossRef CAS
- S. Kim, A. Guenther, T. Karl and J. Greenberg, Atmos. Chem. Phys., 2011, 11, 8613–8623 CrossRef CAS
- D. Sanchez, D. Jeong, R. Seco, I. Wrangham, J. H. Park, W. Brune, A. Koss, J. B. Gilman, J. de Gouw, P. Misztal, A. H. Goldstein, K. Baumann, P. O. Wennberg, F. Keutsch, A. Guenther and S. Kim, Atmos. Environ., 2018, 174, 227–236 CrossRef CAS
- T. A. Kovacs and W. H. Brune, J. Atmos. Chem., 2001, 39, 105–122 CrossRef CAS
-
D. Sanchez, PhD thesis, University of California, Irvine, 2019
- D. Sanchez, R. Seco, D. Gu, A. Guenther, J. Mak, Y. Lee, D. Kim, J. Ahn, D. Blake, S. Herndon, D. Jeong, J. T. Sullivan, T. Mcgee and S. Kim, Atmos. Chem. Phys. Discuss., 2020 DOI:10.5194/acp-2020-174
- D. A. Peterson, E. J. Hyer, S. O. Han, J. H. Crawford, R. J. Park, R. Holz, R. E. Kuehn, E. Eloranta, C. Knote, C. E. Jordan and B. L. Lefer, Elem. Sci. Anth., 2019, 7, 57 CrossRef
- J. R. Schroeder, J. H. Crawford, J. Y. Ahn, L. Chang, A. Fried, J. Walega, A. Weinheimer, D. D. Montzka, S. R. Hall, K. Ullmann, A. Wisthaler, T. Mikoviny, G. Chen, D. R. Blake, N. J. Blake, S. C. Hughes, S. Meinardi, G. Diskin, J. P. Digangi, Y. Choi, S. E. Pusede, G. L. Huey, D. J. Tanner, M. Kim and P. Wennberg, Elem. Sci. Anth., 2020, 8, 3 CrossRef
- U. Kuhn, M. O. Andreae, C. Ammann, A. C. Araujo, E. Brancaleoni, P. Ciccioli, T. Dindorf, M. Frattoni, L. V. Gatti, L. Ganzeveld, B. Kruijt, J. Lelieveld, J. Lloyd, F. X. Meixner, A. D. Nobre, U. Poschl, C. Spirig, P. Stefani, A. Thielmann, R. Valentini and J. Kesselmeier, Atmos. Chem. Phys., 2007, 7, 2855–2879 CrossRef CAS
- H. B. Singh, D. Ohara, D. Herlth, W. Sachse, D. R. Blake, J. D. Bradshaw, M. Kanakidou and P. J. Crutzen, J. Geophys. Res.: Atmos., 1994, 99, 1805–1819 CrossRef CAS
- A. Guenther, C. N. Hewitt, D. Erickson, R. Fall, C. Geron, T. Graedel, P. Harley, L. Klinger, M. Lerdau, W. A. McKay, T. Pierce, B. Scholes, R. Steinbrecher, R. Tallamraju, J. Taylor and P. Zimmerman, J. Geophys. Res., 1995, 100, 8873–8892 CrossRef CAS
- K. Y. Lee, K. H. Kwak, Y. H. Ryu, S. H. Lee and J. J. Baik, Atmos. Environ., 2014, 96, 209–219 CrossRef CAS
- T. Karl, P. K. Misztal, H. H. Jonsson, S. Shertz, A. H. Goldstein and A. B. Guenther, J. Atmos. Sci., 2013, 70, 3277–3287 CrossRef
- S. Kim, M. Lee, S. Kim, S. Choi, S. Seok and S. Kim, Asia-Pac. J. Atmospheric Sci., 2013, 49, 325–331 CrossRef
- S.-Y. Kim, X. Jiang, M. Lee, A. Turnipseed, A. Guenther, J.-C. Kim, S.-J. Lee and S. Kim, Atmos. Environ., 2013, 70, 447–453 CrossRef CAS
- A. B. Jardine, K. J. Jardine, J. D. Fuentes, S. T. Martin, G. Martins, F. Durgante, V. Carneiro, N. Higuchi, A. O. Manzi and J. Q. Chambers, Geophys. Res. Lett., 2015, 42, 1576–1583 CrossRef CAS
- A. Lee, G. W. Schade, R. Holzinger and A. H. Goldstein, Atmos. Chem. Phys., 2005, 5, 505–513 CrossRef CAS
- Y. D. Yang, M. Shao, X. M. Wang, A. C. Nolscher, S. Kessel, A. Guenther and J. Williams, Atmos. Environ., 2016, 134, 147–161 CrossRef CAS
- D. Kim, P. S. Stevens and R. A. Hites, J. Phys. Chem. A, 2011, 115, 500–506 CrossRef PubMed
- J. L. Laughner and R. C. Cohen, Science, 2019, 366, 723–727 CrossRef CAS PubMed
-
UCAR/NCAR-Earth Observing Laboratory, Nitric Oxide Chemiluminescence Ozone Instrument, 2015, DOI:10.5065/D6SN070H
- G. W. Sachse, G. F. Hill, L. O. Wade and M. G. Perry, J. Geophys. Res.: Atmos., 1987, 92, 2071–2081 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0fd00081g |
| ‡ Current affiliation: Terrestrial Ecology Section, Department of Biology, Center for Permafrost (CENPERM), Department of Geosciences and Natural Resource Management, University of Copenhagen, Copenhagen, Denmark. |
| § Current affiliation: Division of Environment and Sustainability, Hong Kong University of Science and Technology, Clear Water Bay, Hong Kong SAR, China. |
| This journal is © The Royal Society of Chemistry 2021 |