
Fluid actuation and buoyancy driven oscillation by enzyme-immobilized microfluidic microcapsules†
Rohit
Varshney
,
Arshdeep Kaur
Gill
,
Mujeeb
Alam
,
Chinmayee
Agashe
and
Debabrata
Patra
 *
*
Institute of Nano Science and Technology, Sector-81, Knowledge City, Sahibzada Ajit Singh Nagar, Punjab 140306, India. E-mail: patra@inst.ac.in
First published on 2nd October 2021
Abstract
Mimicking microorganism's locomotion and actuation under fluid is difficult to realize. To better comprehend the motility in non-living matter, self-propelled synthetic systems are being developed as a fast-growing area of research. Inspired by the self-powered enzyme micropumps where the enzyme catalysis was harnessed to create motion, herein, enzyme-immobilized microfluidic microcapsules (MCs) were used as a microscale engine to maneuver the fluid flow. The fluid actuation was tuned by various parameters such as substrate concentration, reaction rate, diameter of MCs and the population of the MCs inside the flow chamber. The same MCs, when suspended in a solution, showed buoyancy driven motility by creating oxygen bubbles via an enzymatic reaction and the velocity of the MCs was directly dependent on the number of nucleated oxygen bubbles generated on the MC surface.
Introduction
Actuation and locomotion under fluid conditions are the prevalent features inhabited by many micro-organisms.1,2 Actuation can be created in a stationary phase and it helps the micro-organisms to send signals to the environment.3 However, locomotion is the displacement of the motile organism and it is the key to fetching nutrients in a fluidic environment.4 With the initial aim to understand motility in non-living systems, nano/micro motors, often called active colloids, were developed using Au–Pt bimetallic nanorods that were self-propelled by harnessing energy from localized chemical reactions.5 Several other designs have emerged to create autonomous motion either by the catalytic decomposition of fuels6 or by reaction-generated gradient7,8 for applications in cargo delivery,9,10 pollutant degradation,11 and sensing.12 The directional control of these self-propelled nano-machines was achieved by coupling ultrasound13 and magnetic fields14 with the existing propulsion pathway.15 Later, the discovery of enzyme nanomotors where free swimming enzymes exhibited enhanced diffusion has fuelled this research area.16 It encouraged researchers to couple enzymes with larger synthetic structures, and the self-propulsion of these bio-hybrid structures was achieved via an enzymatic reaction.17Similar to the self-propelled synthetic machines, the actuation of fluid powered by a chemical reaction was first demonstrated by tethering the catalytic systems on the surface, thus transferring the mechanical force to the surrounding fluid.18 It was demonstrated by immersing an Au–Ag bimetallic patch in a hydrogen peroxide solution and flow was created due to self-diffusiophoresis.19 A concentration gradient of neutral species can also cause the diffusiophoretic movement and therefore induce pumping as observed during the depolymerization reaction.20 Recent observations concluded that the fluid flow can also be generated as a result of the inhomogeneous distribution of local fluid density due to thermal/solutal buoyancy effects.21 One such example is the self-powered enzyme micropumps where surface-immobilized enzymes function as self-powered micropumps and turn on the fluid flow in the presence of a substrate.22 It mimics the analyte responsive microscopic flow generation23,24 or fluid actuation by microorganism.25 In spite of tremendous progress in developing bioinspired locomotion and actuation using synthetic materials, there still remains a critical need in integrating these two to simulate life-like motion.
Herein, emulsion-templated enzyme-immobilized MCs were fabricated by a one-step microfluidic approach to demonstrate the fluid actuation powered by enzyme catalysis. The MCs triggered self-powered convective fluid pumping, which was maneuvered by the number of MCs, diameter of the MCs and substrate concentration. These enzyme-based micropumps were able to transport fluid both spatially and temporally. In another instance, we have shown that the enzyme-immobilized MCs produced buoyancy driven motility by generating oxygen bubbles through an enzymatic reaction and the speed of the MCs was dictated by the number of nucleated oxygen bubbles at the MC surface.
The microfluidic synthesis of NP–enzyme conjugate-stabilized microcapsules (MCs) is depicted in Fig. 1.26 In a double Y-shaped microchannel, the continuous phase containing an aqueous solution of cationic Au NPs and catalase flowed via the horizontal Y-shaped channel to make low-valent complexes through electrostatic interactions. Similarly, the Grubb's catalyst and dicyclopentadiene (DCPD) monomer were flowed simultaneously in two arms of the vertical-Y-shaped channel to constitute the dispersed phase. As the oil phase sheared off the aqueous phase, oil droplets were formed and concomitantly the low valent complexes27 migrated to the oil–water interface providing stabilization to the MCs. In situ ring opening polymerization of the oil phase produced solid-core enzyme-immobilized MCs. This microfluidic approach produced monodisperse biocatalytic emulsions in a simple way with higher activity and excellent reusability. The size tunability of the MCs was achieved by changing the flow rate of the aqueous phase. The aqueous phase flow rate (Qw) was varied over a range of 200–1000 μL min−1 while the flow rate of the oil phase (Qo) was kept constant at 100 μL min−1. The images in Fig. 2(a)–(c) correspond to the optical microscope images of the droplets collected at Qw = 200 μL min−1, 600 μL min−1, and 1000 μL min−1 respectively. The inset images of the red fluorescent MCs represent the encapsulation of oil soluble Nile red dye and it also confirmed the formation of oil-in-water type emulsions. From the image analysis, it was evident that the size of the MCs decreased with an increase in the aqueous phase flow rate, i.e., the MC diameter decreased from 715 μm (@ Qw = 200 μL min−1) to 122 μm (@ Qw = 1000 μL min−1), as depicted in Fig. 2d. The polymerized MCs were later examined under a scanning electron microscope (SEM), and the corresponding image demonstrated the uniform size particles with wrinkled and rough surfaces (see ESI,† Fig. S1).
 | ||
| Fig. 1 Schematic representation showing the formation of oil in water droplets in a double Y-shaped microfluidic chip. The water phase has enzyme–NP conjugates, which are formed when passed through the horizontal Y-shaped channel and mixture of the Grubb's catalyst and DCPD are dissolved in the oil phase. The oil phase is sheared by the steady pumping of water phase, resulting in the breakage of oil phase into spherical and robust droplets. | ||
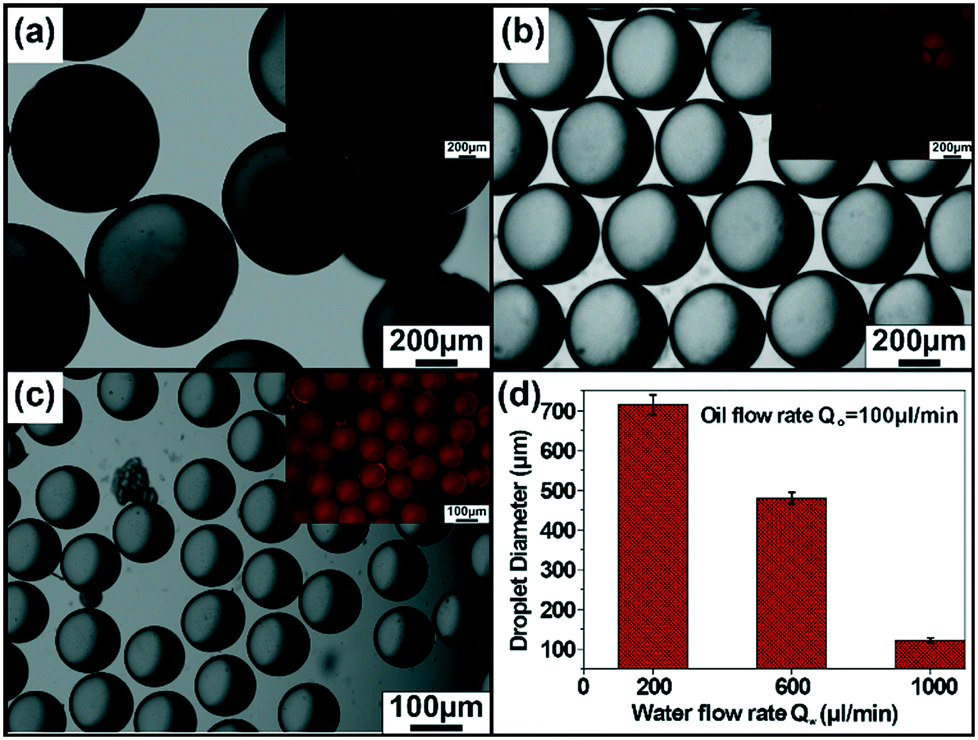 | ||
| Fig. 2 Optical microscope images of the polymerizable oil droplets generated for varied flow rates of the water phase. The optical microscope images of polymerized MCs formed at different flow rates. (a) Qw = 200 μl min−1, Qo = 100 μl min−1, (b) Qw = 600 μl min−1, Qo = 100 μl min−1, (c) Qw = 1000 μl min−1, Qo = 100 μl min−1. Inset images show fluorescent microscope images of corresponding MCs. (d) Average droplet diameter as a function of the flow rate of the aqueous phase. | ||
The ability of enzyme-immobilized MCs to generate self-powered motion was investigated by placing the MCs inside a custom-made chamber (area = 1 cm2; h = 1.8 mm) fabricated on a glass slide. The capsule was manually stuck to the side wall of the chamber to prevent from floating during the flow experiment. Later, a transparent PET sheet was placed on top of the chamber to make it leak-proof. Once the imaging chamber was ready, the substrate solution containing polystyrene tracer particles (5 μm in diameter) was injected into the chamber and the fluid flow was monitored using an optical microscope. The catalase immobilized MCs acted as an ATP-independent microengine, which was able to drive large scale fluid flow in the presence of hydrogen peroxide. The fluid flow study was carried out separately with three different sizes of MCs (122 μm, 480 μm and 715 μm) and the experiment was performed placing a single MC inside the chamber. It was observed that the smallest size MC was unable to generate any directional fluid flow in the presence of 5 mM H2O2 substrate concentration. In the case of 480 μm sized MC, an inward motion of tracer particles with an average velocity of 2.99 ± 0.24 μm s−1 towards the MC was observed when viewed at the bottom of the glass surface. As the fluid flow was observed in a closed system, so by fluid continuity, the fluid flow showed an outwards motion when viewed from the top confirming the convective flow, as shown in Fig. 3a. The thermal gradient derived from enzyme–substrate reactions and density gradient originated from the density difference between the substrate and product are the major driving forces for the chemically powered motion.21 Similarly, the directional fluid flow was also observed for large-sized MCs (715 μm) with an average velocity of 5.25 ± 0.67 μm s−1 that was higher than the smaller-sized MCd (Fig. 3b, see SV1†). As the size of the MC increased, the number of enzymes covering the surface of MCs also increased. It resulted in an enhanced the reaction rate (4.21 μM s−1 to 9.20 μM s−1), which contributed to the increase in the tracer velocity (see ESI† S2). Next, the effect of the number of MCs on the pumping velocity was explored, and the velocity was observed to increase linearly as the number of the MCs increased. This was due to the increment in the enzymatic reaction rate as the total number of enzymes was higher for the multi-MC system (see SV2 and SV3†). The experiment was performed using 480 μm diameter MCs and the tracer velocity was 5.22 ± 0.84 μm s−1 and 7.13 ± 0.72 μm s−1 for two and three MC systems, respectively (see Fig. 3c).
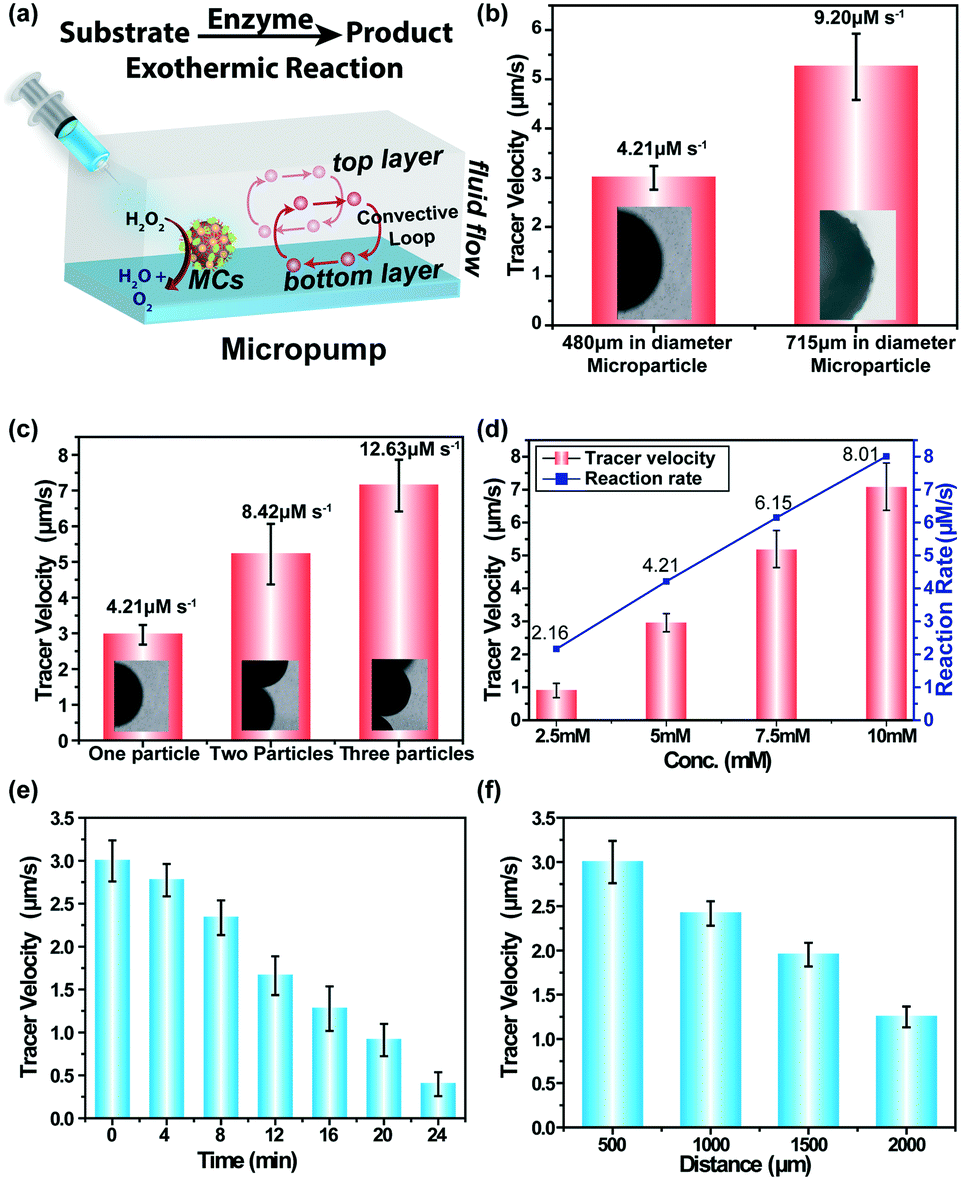 | ||
| Fig. 3 (a) Schematic of enzyme-triggered micropumps; catalase-immobilized MCs inside the chamber turn on convective fluid flow in the presence of the substrate. (b) Tracer velocity vs. size of the MCs at 5 mM substrate concentration; corresponding reaction rates are mentioned over the bar plot. (c) Tracer velocity vs. MCs number in the presence of 5 mM substrate; corresponding reaction rates are mentioned over the bar plot. (d) Substrate concentration as well as reaction rate dependence of tracer velocity. (e) Variation of the temporal velocity in the presence of 5 mM H2O2 concentration. (f) Spatial distribution of tracer velocity in the presence of 5 mM substrate concentration. | ||
Further, the effect of the substrate concentration on the rate of the reaction was examined as well as the pumping velocity using 480 μm diameter MCs. With an increase in the substrate concentration, the rate of the reaction increased linearly, as shown in Fig. 3d. For example, the rate of reaction increased up to four-fold (2.16 μM s−1 to 8.01 μM s−1) when the substrate concentration was four times higher than the previous concentration (2.5 mM to 10 mM) (see ESI† S2). The pumping velocity also increased linearly in accordance with the reaction rate. In the presence of 2.5 mM H2O2, the tracer velocity was 0.90 ± 0.21 μm s−1 and it reached up to 7.08 ± 0.72 μm s−1 when the peroxide concentration was 10 mM (see SV4–SV7†). The temporal velocity profile of the MC-powered fluid flow was investigated at a fixed substrate concentration of 5 mM. The velocity was tracked for 24 min at an interval of 4 min. As the substrate was continuously consumed by the enzymes over time, the reaction rate gradually decreased. It accounted for the gradual decrease of the tracer velocity, as shown in Fig. 3e. It is important to mention that the fluid flow came to a halt after 30 min due to the complete consumption of the substrate but reactivated later with the addition of a fresh substrate solution to the pump chamber. It was repeated multiple times to establish the reusable nature of these microscale pumps (see ESI,† Fig. S3). To understand the spatial distribution of fluid flow from the reaction epicentre, the fluid-pumping velocity was examined at set distances moving away from the enzyme-immobilized MCs. The pumping velocity did not show any major variation in short distances (50 μm to 400 μm), but decreased significantly at long distances, i.e., 2000 μm away from MCs (Fig. 3f). It can be hypothesized that the density gradient gradually faded away at long distances.
Usually, catalase decomposes hydrogen peroxide (H2O2) and produces oxygen bubbles in a solution. The freshly prepared catalase-immobilized MCs of all sizes showed a similar behaviour when placed inside an imaging well filled with the H2O2 solution. For further investigation, we choose 480 μm diameter MCs and the nucleation of bubbles was observed by systematically varying the substrate concentration. The microscopic bubbles started appearing when the substrate concentration was 10 mM and above. The nucleation of one, two, three and higher order of discrete gas bubbles (Fig. 4a) was observed at 20 mM and 30 mM of the substrate concentration. Fig. 4b represents the nucleation and growth of a single microbubble on the MC surface at the 10 mM substrate concentration. The microbubble appeared as a discrete spherical microstructure after 1 minute of addition of H2O2. It was optically transparent and grew in size with time due to pressure generation inside the bubbles. The growth continued for 8 min until the MCs were lifted from the resting position to the air–water interface. It eventually led to the rupture of the microbubble and MCs settled at the bottom of the imaging well. The nucleation and growth of the microbubble was repeated till the fuel was exhausted. Fig. 4c presents the growth kinetics of a single microbubble and it was observed that the final diameter of the microbubble expanded upto 8 times before it ruptured at the interface. It is important to mention that the MC diameter was unaltered during the entire event as the core was polymerized.
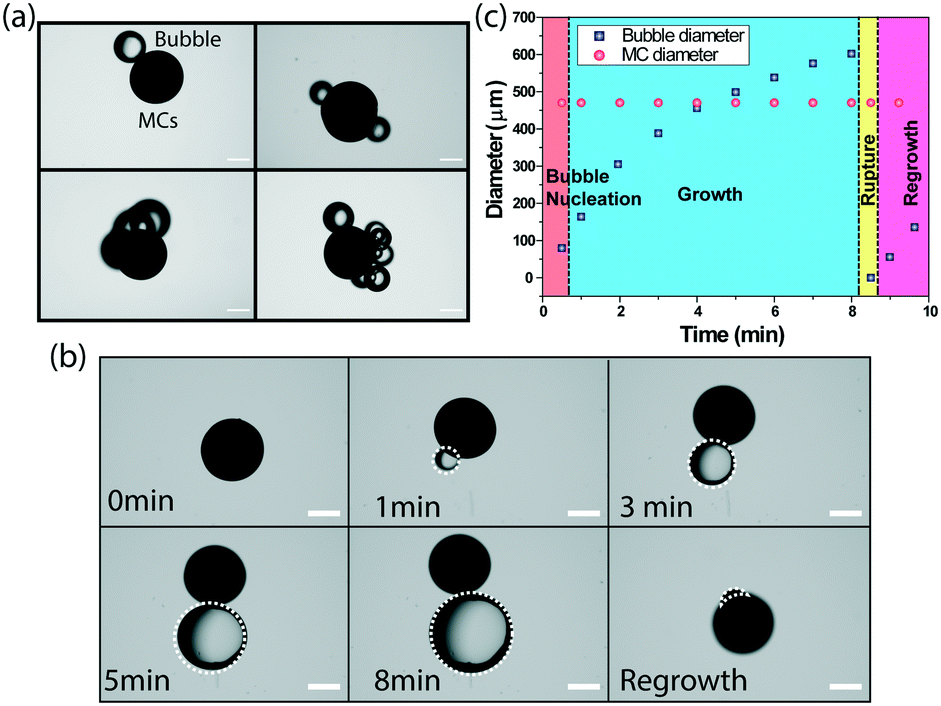 | ||
| Fig. 4 (a) The optical microscopy images of one, two, three and higher order discrete gas bubbles present on MCs. Scale bars, 200 μm. (b) The evolution of a single bubble on the MCs. Scale bar 200 um. (c) The growth kinetics of a single microbubble on MCs. | ||
The cycle of nucleation, growth and rupture of the bubbles are the key for generating oscillating motion of the MCs (Fig. 5a). To demonstrate the autonomous buoyancy driven motion, MCs (480 μm diameter) were suspended in a 20 mM hydrogen peroxide solution taken in a plastic tube filled up to 65 mm of length, as shown in ESI† Fig. S4. The generation of bubbles on the MC surface was observed immediately. As a result, MCs were able to harness buoyant force to move against gravity and started moving towards the air–water interface. As soon as the MCs reached the interface, the rupture of the bubbles occured and MCs started to sink under the influence of gravity. During the downward motion, more oxygen bubbles started to appear at the MC surface and the buoyant force again lifted the MCs against gravity. The repetition of these chain of events caused the oscillation of MCs (see SV8†). The buoyancy driven oscillation of the MCs inside the tube is presented in Fig. 5b, which considered the formation of one microbubble during the entire journey. The bubble appeared immediately on the MC surface after the addition of the peroxide solution; however, the MCs took off after 20–25 s and reached the air–water interface in 10 s. No residence time at the interface was observed as the bubble collapsed after reaching the line of tension and the MCs returned to the base in 3 s (see SV9†). This process occurred repeatedly till the fuel was exhausted. Fig. 5c compares the ascendant velocity of the MCs containing different number of bubbles and it is quite evident that MCs with greater number of bubbles experienced a higher buoyant force. For example, MCs containing one microbubble reached the interface at an average velocity of 6.8 mm s−1, whereas MCs comprising three bubbles attained a maximum velocity of 24 mm s−1. In all cases, the gravity influenced downward motion of the MCs was 19 mm s−1 as mass of the MCs was same during the oscillation motion.
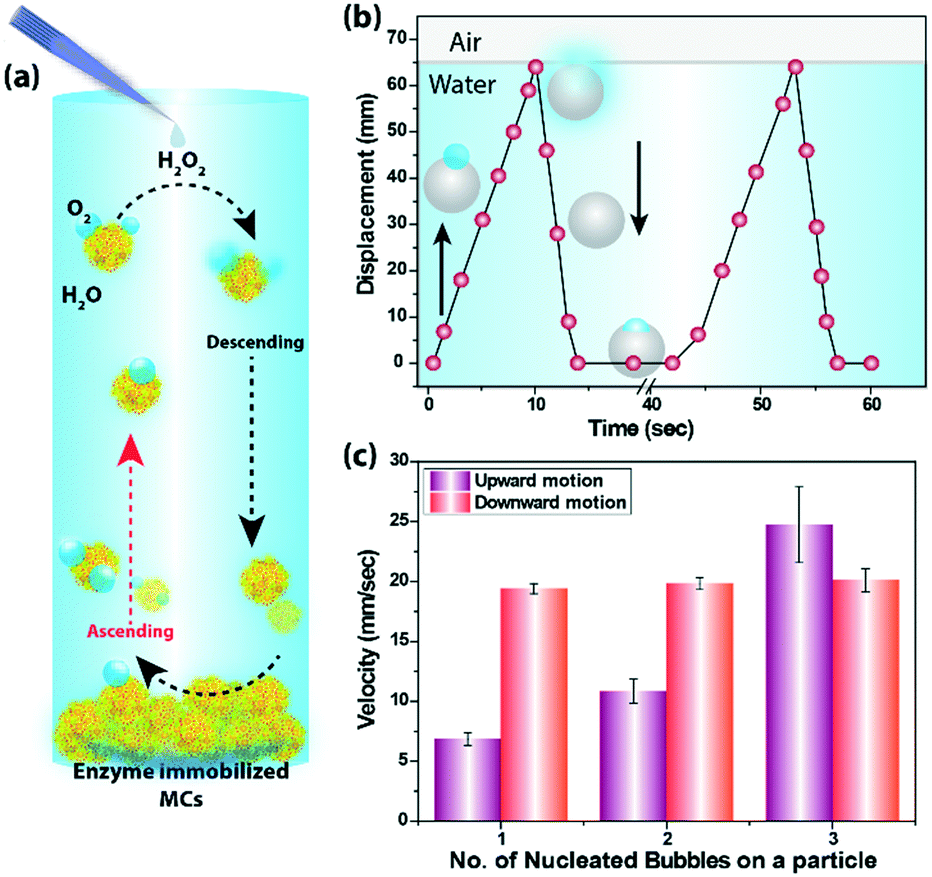 | ||
| Fig. 5 (a) The schematic of buoyancy driven oscillation of catalase immobilized MCs in the presence of substrate solution. (b) The plot shows time-dependent changes in the vertical displacement for a single MC undergoing enzyme mediated oscillatory movement. The grey sphere indicates MCs and the arrows show the direction of oscillation of MCs. (c) The plot shows velocity as a function of number of nucleated bubble on MCs. | ||
Conclusions
A one-step microfluidic route was adopted to synthesize catalase-Au NP conjugate-stabilized polymerized MCs for the demonstration of ATP-independent microengines. These MCs were able to actuate fluid in the surrounding environment by harnessing enzymatic reactions. The density-driven motion was tuned by substrate concentration, MC diameter and the population of MCs. These self-powered micropumps can be used to attain both spatial and temporal control over the fluid transport. Besides fluid actuation, the MCs showed buoyancy driven oscillation against gravity and the sustained motion was powered by oxygen gas bubbles produced during the enzymatic reaction.This system represents a step towards designing a complex microorganism structure working under non-equilibrium conditions and the autonomous motions can be generated by the catalytic harnessing of chemical energy. We envision that the fluid actuation by microcompartment structures can be utilized to release active encapsulants, which coupled with vertical motion would enable to establish complex and emergent behaviours between microcompartments. Such miniaturized systems might also find an application in sensing toxins in the environment by fluid actuation and could potentially speed up the remediation using buoyancy driven motility under a low flow environment.
Author contributions
R. V. and A. K. G. carried out materials fabrication and characterization. They contributed equally to the manuscript. M. A. and C. A. helped in micropump studies. D. P. supervised the project and helped to structure the manuscript. DP acknowledges the financial support from SERB-DST (ECR/2017/000442).Conflicts of interest
The authors declare no competing financial interest.Notes and references
- D. B. Dusenbery, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 10949–10954 CrossRef CAS PubMed
.
- S. Palagi and P. Fischer, Nat. Rev. Mater., 2018, 3, 113–124 CrossRef CAS
- L. Ricotti, B. Trimmer, A. W. Feinberg, R. Raman, K. K. Parker, R. Bashir, M. Sitti, S. Martel, P. Dario and A. Menciassi, Sci. Robot., 2017, 2, eaaq0495 CrossRef PubMed
- H. W. Huang, F. E. Uslu, P. Katsamba, E. Lauga, M. S. Sakar and B. J. Nelson, Sci. Adv., 2019, 5, eaau1532 CrossRef PubMed
- W. F. Paxton, K. C. Kistler, C. C. Olmeda, A. Sen, S. K. St. Angelo, Y. Cao, T. E. Mallouk, P. E. Lammert and V. H. Crespi, J. Am. Chem. Soc., 2004, 126, 13424–13431 CrossRef CAS PubMed
- S. Maiti, O. E. Shklyaev, A. C. Balazs and A. Sen, Langmuir, 2019, 35, 3724–3732 CrossRef CAS PubMed
- J. J. McDermott, A. Kar, M. Daher, S. Klara, G. Wang, A. Sen and D. Velegol, Langmuir, 2012, 28, 15491–15497 CrossRef CAS PubMed
- M. Alam, R. Varshney, C. Agashe, A. K. Gill and D. Patra, Chem. Commun., 2021, 57, 4584–4587 RSC
- H. Xu, M. Medina-Sánchez, M. F. Maitz, C. Werner and O. G. Schmidt, ACS Nano, 2020, 14, 2982–2993 CrossRef CAS PubMed
- R. Lin, W. Yu, X. Chen and H. Gao, Adv. Healthcare Mater., 2021, 10, 2001212 CrossRef CAS PubMed
- L. Soler, V. Magdanz, V. M. Fomin, S. Sanchez and O. G. Schmidt, ACS Nano, 2013, 7, 9611–9620 CrossRef CAS PubMed
- L. Liu, Y. Dong, Y. Sun, M. Liu, Y. Su, H. Zhang and B. Dong, Nano Res., 2016, 9, 1310–1318 CrossRef CAS
- T. Xu, L.-P. Xu and X. Zhang, Appl. Mater. Today, 2017, 9, 493–503 CrossRef
- H. Yu, W. Tang, G. Mu, H. Wang, X. Chang, H. Dong, L. Qi, G. Zhang and T. Li, Micromachines, 2018, 9, 540 CrossRef PubMed
- H. Ceylan, J. Giltinan, K. Kozielski and M. Sitti, Lab Chip, 2017, 17, 1705–1724 RSC
- J. Sun, M. Mathesh, W. Li and D. A. Wilson, ACS Nano, 2019, 13, 10191–10200 CrossRef CAS PubMed
- B. P. Kumar, A. J. Patil and S. Mann, Nat. Chem., 2018, 10, 1154–1163 CrossRef CAS PubMed
- C. Zhou, H. Zhang, Z. Li and W. Wang, Lab Chip, 2016, 16, 1797–1811 RSC
- T. R. Kline, W. F. Paxton, Y. Wang, D. Velegol, T. E. Mallouk and A. Sen, J. Am. Chem. Soc., 2005, 127, 17150–17151 CrossRef CAS PubMed
- H. Zhang, K. Yeung, J. S. Robbins, R. A. Pavlick, M. Wu, R. Liu, A. Sen and S. T. Phillips, Angew. Chem., Int. Ed., 2012, 51, 2400–2404 CrossRef CAS PubMed
- L. Valdez, H. Shum, I. Ortiz-Rivera, A. C. Balazs and A. Sen, Soft Matter, 2017, 13, 2800–2807 RSC
- S. Sengupta, D. Patra, I. Ortiz-Rivera, A. Agrawal, S. Shklyaev, K. K. Dey, U. Córdova-Figueroa, T. E. Mallouk and A. Sen, Nat. Chem., 2014, 6, 415–422 CrossRef CAS PubMed
- R. Varshney, M. Alam, C. Agashe, R. Joseph and D. Patra, Chem. Commun., 2020, 56, 9284–9287 RSC
- A. K. Gill, R. Varshney, M. Alam, C. Agashe and D. Patra, ACS Appl. Bio Mater., 2021, 4(8), 6203–6208 CrossRef CAS
- E. Lushi, H. Wioland and R. E. Goldstein, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9733–9738 CrossRef CAS PubMed
- R. Varshney, S. Sharma, B. Prakash, J. K. Laha and D. Patra, ACS Omega, 2019, 4, 13790–13794 CrossRef CAS PubMed
- D. Patra, A. Sanyal and V. M. Rotello, Chem. – Asian J., 2010, 5, 2442–2453 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1lc00699a |
| This journal is © The Royal Society of Chemistry 2021 |