
Rapidly reconstructing the active surface of cobalt-based perovskites for alkaline seawater splitting†
Ruigan
Hu‡
a,
Mengyuan
Zhao‡
b,
He
Miao
 *a,
Fuyue
Liu
a,
Jiaqun
Zou
a,
Chunfei
Zhang
a,
Qin
Wang
c,
Ziqi
Tian
b,
Qiuju
Zhang
*b and
Jinliang
Yuan
a
*a,
Fuyue
Liu
a,
Jiaqun
Zou
a,
Chunfei
Zhang
a,
Qin
Wang
c,
Ziqi
Tian
b,
Qiuju
Zhang
*b and
Jinliang
Yuan
a
aFaculty of Maritime and Transportation, Ningbo University, Ningbo 315211, PR China. E-mail: miaohe@nbu.edu.cn
bNingbo Institute of Materials Technology and Engineering, Chinese Academy of Sciences, Ningbo, 315211, PR China. E-mail: zhangqj@nimte.ac.cn
cDepartment of Microelectronic Science and Engineering, Faculty of Science, Ningbo University, Ningbo 315211, PR China
First published on 23rd June 2022
Abstract
As a potential oxygen evolution reaction (OER) catalyst, Co-based perovskites have received intensive attention. However, Sr readily accumulates on their surface, and makes them inert toward the OER. Herein, we propose a simple but versatile electrochemical reduction method to reconstruct the active surface of Co-based perovskites within a few seconds. By this method, Sr rapidly precipitates from Co-based perovskites, accompanied by the introduction of Sr and oxygen vacancies. After reconstruction, the electrochemical active surface areas of Co-based perovskites greatly increase, and the OER overpotential of the optimized SrNb0.1Co0.7Fe0.2O3−δ (ER-SNCF-20s) reaches 278 mV at 10 mA cm−2. This can be explained by the decrease of overpotentials at the rate-determining step. Using ER-SNCF-20s, the splitting voltage of alkaline natural seawater can reach 1.56 V at 10 mA cm−2, and remains steady for 300 h. This effort offers a feasible method for reconstructing the active surface of Co-based perovskites.
1. Introduction
Water splitting can convert the intermittent but regenerative electrical energy from solar or wind energy to hydrogen energy, and plays a positive role in the emission reduction of CO2 and effective energy utilization.1 Theoretically, water splitting can be achieved with a standard voltage of 1.23 V, whereas the actual voltage of water splitting is much higher than this theoretical value as a result of the presence of a large overpotential at both electrodes.2,3 The water splitting reaction is a combination of the hydrogen evolution reaction (HER) at the cathode and the oxygen evolution reaction (OER) at the anode. Comparing with the HER, the OER is a complex 4-electron reaction with a much more sluggish kinetics.4–6 Therefore, the overpotential at the anode is much higher than that at the cathode during water electrolysis, which seriously limits the advancement of water splitting technology.7–9The large-scale hydrogen production by water electrolysis will inevitably consume a large amount of vital fresh water resources which are becoming more and more scarce in many regions of our planet.10 Seawater may be one of the most plentiful natural resources on the planet, accounting for about 96.5% of the world's entire water, and it is nearly inexhaustible if we can electrolyze seawater to produce hydrogen.11 Furthermore, the utilization of hydrogen derived from seawater combined with fuel cells not only provides energy but also generates fresh water, and realizes seawater desalination. Compared with fresh water electrolysis, there are two main bottlenecks in seawater splitting.12,13 One is that the oxidation of chloride ions may compete with the OER during water electrolysis as a result of the presence of considerable amounts of chloride ions in seawater (∼0.5 mol L−1).14–16 Theoretically, the electrochemical oxidation of chloride ions to hypochlorite under alkaline conditions requires a voltage of 1.72 V which is just 490 mV above the theoretical OER voltage.16–18 Secondly, chlorides, as well as some small particles, bacteria and microorganisms, can corrode and poison the electrode, and destroy its long-term stable operation. Therefore, developing a highly active and stable anode is indispensable for seawater splitting.
To improve the activity and stability of the anode for water splitting, various OER catalysts, including transition metal oxides,19,20 hydroxides,21–23 nitrides,24–26 phosphides,27,28 sulphides,29,30 and selenides,31,32 have been developed over the past decades. In particular, cobalt-based (Co-based) perovskites have attracted intensive attention as a result of their excellent activities and stabilities as well as low cost.33–35 Co-based perovskites as ABO3-type metal oxides with a cubic structure generally consist of rare earth or alkaline earth metals in the A-site and transition metals such as Co in the B-site.33 Currently, various strategies for promoting the OER catalytic activities and stabilities of these Co-based perovskites have been developed, such as doping,36 morphological design,37–39 vacancy engineering,33,40 and surface modification or reconstruction.41 For example, Shao et al. synthesized SrNb0.1Co0.7Fe0.2O3−δ nanofibers (SNCF-NR) by means of electrostatic spinning with significantly improved OER activity.33 At 10 mA cm−2, the overpotential of SNCF-NR is merely 0.39 V, which is far below those of SNCF (0.50 V) and IrO2 (0.45 V). Zhu et al. introduced a substantial number of oxygen vacancies by doping P and S elements, which can greatly improve the OER activity of SrCoO3−δ, and a low overpotential of merely 480 mV was obtained at 10 mA cm−2.34 Even so, the OER catalytic activities of Co-based perovskites are still inferior to those of some well-known OER catalysts such as NiFe-LDH.42–44
It is well known that the OER occurs on the surface of an oxygen catalyst; therefore the active surface plays an important role in their OER catalytic activity. It is well believed that Sr readily segregates and a Sr rich layer forms on the surface of some Sr-containing cobalt-based perovskites, such as La0.6Sr0.4Co0.8Fe0.2O3 (LSCF), Ba0.5Sr0.5Co0.8Fe0.2O3 (BSCF) and SrNb0.1Co0.7Fe0.2O3 (SNCF), during their preparation process.45 This Sr rich layer seriously deteriorates the surface activity of Co-based perovskites and is detrimental to their OER activity. Eliminating the Sr rich layer and reconstructing the active surface of cobalt-based perovskites by acid etching or reducing atmosphere annealing can greatly enhance their OER activity.46,47 Gong et al.46 reconstructed the active surface of LSCF by dilute nitric acid etching, which caused A-site defects, destroyed the Sr rich layer, introduced a substantial number of oxygen vacancies and greatly enhanced the specific surface area of LSCF. Accordingly, the overpotential of LSCF decreased from 460 to 385 mV at 10 mA cm−2. Luo et al.47 constructed the active surface of PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) by reducing atmosphere annealing at 300 °C, which created many oxygen vacancies and introduced the second phase of CoP with high OER activity. Compared to the pristine PBSCF, the overpotential of the surface-restructured sample (CoP-PBSCF) decreased by 120 mV at 100 mA cm−2. Unfortunately, these above surface reconstruction methods are somewhat tedious, time-consuming, and environmentally unfriendly and require high energy consumption.
Herein, we proposed a facile but universal method of electrochemical reduction to rapidly reconstruct the active surface of Co-based perovskites in a few seconds. By this method, Sr was quickly precipitated from the surface of Co-based perovskites, accompanied by the generation of A-site defects and a substantial number of oxygen vacancies. Owing to the active surface reconstruction, the OER catalytic activity of Co-based perovskites could be greatly enhanced. Taking SrNb0.1Co0.7Fe0.2O3−δ as an example, the OER overpotential of SNCF after electrochemical reduction (ER-SNCF-20s) was 278 mV which was even lower than that of IrO2 (344 mV) in 1 M KOH at 10 mA cm−2. Using ER-SNCF-20s and Pt/C as the anode and cathode, respectively, the voltage for splitting the alkaline natural seawater could reach 1.56 V at 10 mA cm−2 at room temperature. More importantly, our ER-SNCF-20s was very robust and showed no degradation after splitting seawater for 300 hours.
2.Results and discussion
Fig. 1a shows the XRD patterns of SNCF and ER-SNCF-20s. The characteristic peaks of SNCF correspond to the (110), (111), (200), (211), (220) and (310) crystal planes of the SrFeO3−δ phase (JCPDS card no. 34-0641), indicating that SNCF is SrFeO3−δ doped with Co and Nb ions.48 For the samples treated with the different electrochemical reduction times (10–60 s, Fig. S1†) and voltages (0.8–1.4 V, Fig. S2†), their phase structures remain almost the same as that of the pristine SNCF. The initial SNCF is agglomerated particles with a diameter of 2–3 μm (Fig. S3†). As shown in Fig. 1b and c, after electrochemical reduction, a number of nano-needles with the length of 50–200 nm appear on the surface of ER-SNCF-20. The lattice spacing of the nano-needles in the high-resolution transmission electron microscopy (HRTEM) image is 0.297 nm (Fig. 1d), corresponding to the (111) crystal plane of SrO. Furthermore, the diffraction rings of the nano-needles in the SAED pattern (Fig. 1e) correspond to the (111), (200), (220) and (222) crystal planes of SrO. The STEM and EDS elemental maps indicate that the main elements of the nano-needles are Sr and O (Fig. 1g and Fig. S4†), further confirming that the nano-needles are SrO. Compared with that of SNCF, the HRTEM image of ER-SNCF-20s also prove the co-presence of SrO and SrFeO3−δ phases (Fig. 1f). Due to the content of SrO is much lower compared with that of the perovskite phase, it does not show the corresponding peaks in the XRD pattern. | ||
| Fig. 1 (a) XRD patterns of SNCF, ER-SNCF-20s and carbon cloth. (b) SEM image of ER-SNCF-20s. (c) TEM, (d) HRTEM images and (e) SAED pattern of nano-needles on the ER-SNCF-20s surface. (f) HRTEM image of ER-SNCF-20s. (g) Elemental mappings of nano-needles on the ER-SNCF-20s surface. | ||
XPS is used to measure the valence states of the surface elements in ER-SNCF-20s. As shown in Fig. 2a, the Co 2p3/2 peaks of SNCF and ER-SNCF-20s can be fitted with the Co3+ and Co2+ peaks at 780.2 and 782.1 eV, respectively, along with satellite peaks (marked as “sat”). The atomic ratio of Co3+/Co2+ can be calculated by the area covered by the corresponding fitted curves. The Co3+/Co2+ value of SNCF abruptly decreases from 2.87 to 1.94 after electrochemical reduction (Table S1†), indicating that a large amount of Co3+ converts to Co2+ on the SNCF surface. On the basis of the fitting results of Sr 3d spectra (Fig. 2b), the Sr content on the surface of SNCF increases from 35.73% to 39.58% after electrochemical reduction, which can be related to the precipitation of Sr from the bulk of the perovskite. The Fe 2p XPS spectra of SNCF and ER-SNCF show two peaks, associated with Fe 2p1/2 and 2p3/2, respectively, the XPS spectrum of Fe is almost unchanged after electrochemical reduction (Fig. S5†). As seen in Fig. 2c, the fitting peaks of O 1s at 529.2, 530.2, 531.5 and 532.8 eV can be attributed to lattice oxygen (Olatt), reactive oxygen species (O22−/O−), surface oxide (Osurf) and surface adsorbed water, respectively. Among them, the reactive oxygen species (O22−/O−) are strongly associated with the formation of oxygen vacancies.49,50 From the XPS fitting results, the content of O22−/O− of SNCF increases from 15.74% to 25.08% after electrochemical reduction (Table S1†), proving that electrochemical reduction can introduce a substantial number of oxygen vacancies. By comparing with the microstructures and the valence state surface elements of SNCF and ER-SNCF-20s, the active surface reconstruction of SNCF by electrochemical reduction is summarized in Fig. 2d: (1) precipitating Sr, eliminating the Sr rich layer and exposing more active sites and (2) introducing oxygen vacancies and increasing reactive oxygen species.
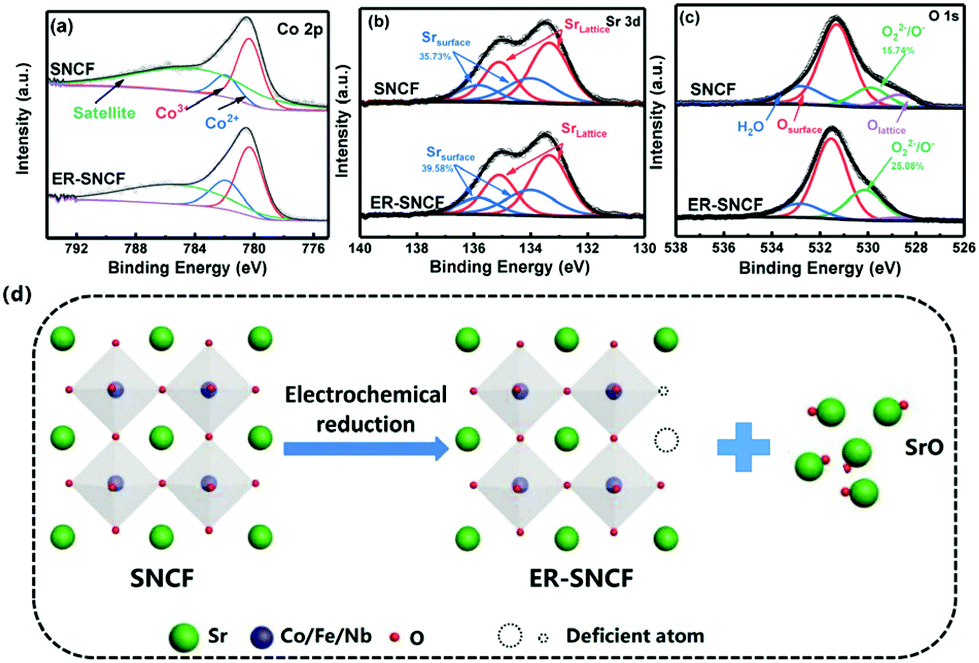 | ||
| Fig. 2 Co 2p (a), Sr 3d (b) and O 1s (c) XPS spectra of SNCF and ER-SNCF-20s. (d) Schematic illustration of the active surface reconstruction of SNCF by electrochemical reduction. | ||
The optimal electrochemical reduction parameters are first confirmed, and the OER LSV curves of SNCF before and after electrochemical reduction are shown in Fig. 3a and Fig. S6.† Obviously, the SNCF sample after electrochemical reduction at −1.0 V for 20 s (ER-SNCF-20s) has the highest OER activity. ER-SNCF-20s provides an OER overpotential of 278 mV at 10 mA cm−2 (Table S2†), which is much lower than those of the pristine SNCF (341 mV) and commercial IrO2 (344 mV) (Fig. 3b). Compared with these perovskites studied recently and some representative Co-based OER catalysts in 1 M KOH (Table S3†), the OER overpotential of ER-SNCF-20s at 10 mA cm−2 is almost the lowest. Significantly, the overpotential of ER-SNCF-20s appears to be even lower than those of SNCF and IrO2 at large current densities, and its overpotential at 100 mA cm−2 is merely 340 mV (Fig. 3b). The above results indicate that an appropriate electrochemical reduction can significantly enhance the OER activity of SNCF.
 | ||
| Fig. 3 (a) OER LSV curves of SNCF, different ER-SNCF samples, IrO2 and CC. (b) Overpotentials of SNCF and ER-SNCF-20s at 10 mA cm−2 and 100 mA cm−2. (c) Tafel plots of SNCF, different ER-SNCF samples and IrO2. Cdl values (d) and EIS spectra recorded at 1.5 V vs. RHE (e) of SNCF and different ER-SNCF samples. (f) Chronopotentiometric curves of ER-SNCF-20s and IrO2 at a constant current density of 10 mA cm−2. All the tests are performed in 1 M KOH. | ||
The OER kinetics of SNCF before and after electrochemical reduction are assessed by Tafel slopes (Fig. 3c and Fig. S6†). The Tafel slope of ER-SNCF-20s is 56 mV dec−1 which is the lowest among these SNCF samples with different electrochemical reduction treatments (different reduction times and voltages) and even lower than that of IrO2 (Table S2†). The small Tafel slope represents the fast OER kinetics. Therefore, the OER kinetics of SNCF is obviously boosted after the electrochemical reduction treatment, and ER-SNCF-20s has the fastest OER kinetics. We also calculated the double-layer capacitance (Cdl) from cyclic voltammetry (CV) curves at different scan rates (Fig. S7†) to determine the electrochemically active surface area (ECSA). As shown in Fig. 3d, the Cdl value of SNCF is improved significantly after electrochemical reduction. After 20 s of electrochemical reduction treatment, the value of Cdl reaches 18.2 mF cm−2, which is approximately 4 times higher than SNCF (4.4 mF cm−2). This indicates that the electrochemical reduction can effectively reconstruct the active surface of SNCF. The electrochemical impedance spectra (EIS) of SNCF before and after electrochemical reduction are shown in Fig. 3e. After electrochemical reduction, the arc of EIS in the high frequency zone becomes much smaller compared with that of the pristine SNCF, indicating the decreased charge transfer resistance (Rct) of ER-SNCF-20s after electrochemical reduction. Stability is an essential parameter to evaluate the actual application of an OER catalyst. As shown in Fig. 3f, the voltage of ER-SNCF-20s at 10 mA cm−2 almost remains unchanged throughout the 10 h chronoamperometry measurement, indicating a superior stability of ER-SNCF-20s. After the electrochemical reduction, Sr/O vacancies and SrO nano-needles are induced from the bulk of SNCF. By investigating in detail with separation between vacancies effects and SrO blocking effects, we can find that the Sr/O vacancies can improve the OER activity of SNCF, while SrO deteriorates the OER activity of SNCF (Fig. S8†). In addition, Sr/O vacancies induced by electrochemical reduction mainly distributed on the surface of SNCF, which reconstructs the active surface of cobalt-based perovskites for the OER.51
An Sr20Nb2Co14Fe4O52 (SNCF) (001) model (Fig. 4a) is constructed to simulate the experimentally measured SrNb0.1Co0.7Fe0.2O3 by performing DFT calculations, which is expected to offer further insight into the enhancement of OER activity in the coexistence of Sr and O vacancies (SrV and OV). The OER evolution processes on stoichiometric SNCF (Sr20Nb2Co14Fe4O52), Ov-SNCF (Sr20Nb2Co14Fe4O51), SrV-SNCF (Sr19Nb2Co14Fe4O52) and SrVOV-SNCF (Sr19Nb2Co14Fe4O51) are compared following the four-electron elementary steps shown in Fig. 4 and Fig. S9.† Surface Co is shown as a preferred adsorption site for the OER process by forming the corresponding intermediates of OH*, O* and OOH* (Fig. S10†). For all the samples, the rate-determining step (RDS) occurs at the step of *OOH → O2. The overpotential of SNCF at the RDS is as high as 1.07 V vs. NHE and introducing Sr and O defects are beneficial for decreasing the overpotential (η) of the RDS. SrVOV-SNCF needs an overpotential (η) of 0.53 V vs. NHE to trigger the step of *OOH → O2, and it is the lowest among those of the four samples. This indicates that the coexistence of Sr and O vacancies can greatly improve the OER activity of SNCF. The relationship between the adsorption free energies (ΔGOOH*) of *OOH intermediates and overpotentials (ηOER) for the RDS of different samples are shown in Fig. 4d. It is noted that the overpotential of the RDS is strongly correlated with the adsorption energy of intermediate OOH*. The fact is reasonable that the larger adsorption strength of intermediate *OOH corresponds to larger overpotentials due to more difficulty of forming O2 molecules. Therefore, the low adsorption strength of intermediate *OOH should be another critical factor for the remarkably enhanced OER activity of our ER-SNCF-20s.
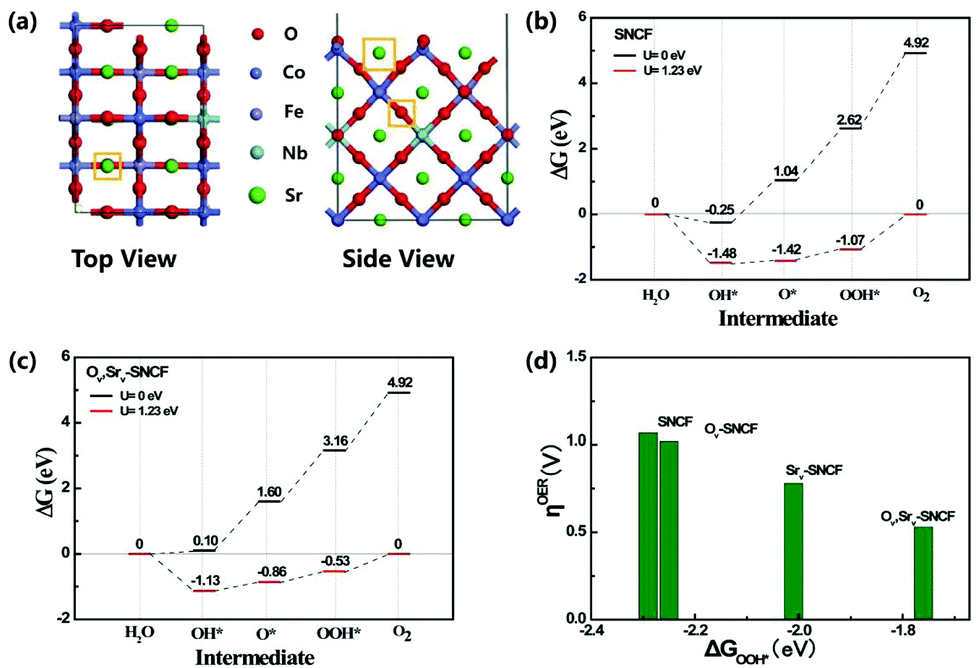 | ||
| Fig. 4 The calculated OER evolution processes of stoichiometric SNCF (001) and with the coexistence of Sr and O vacancies. (a) Structural illustration of Sr and O vacancy sites on an SNCF (001) surface model; (b) and (c) show the Gibbs free energy (ΔG) diagrams of the OER on the surface Co for stoichiometric SNCF(001) and SrVOV-SNCF(001); and (d) the relationship between overpotential (ηOER) and ΔGOOH* for different SNCF vacancy sites. | ||
To demonstrate the universality of the electrochemical reduction strategy on Co-based perovskites, Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF), La0.6Sr0.4Co0.8Fe0.2O3−δ (LSCF) and SrCo0.5Fe0.5O3−δ (SCF) as the representative Co-based perovskites are used with the same electrochemical reduction treatment as that of ER-SNCF-20s. BSCF, LSCF and SCF after electrochemical reduction are denoted as ER-BSCF, ER-LSCF and ER-SCF. As shown in the XRD patterns (Fig. S11†), the main phases of BSCF, LSCF and SCF almost remain unchanged after electrochemical reduction. Similar to ER-SNCF-20s, many SrO nano-needles appear on the surfaces of BSCF, LSCF and SCF due to the precipitation of Sr in the Sr rich layer (Fig. 5a–c). Moreover, as shown in Fig. 5d–f, the OER catalytic activities of these Co-based perovskites are significantly enhanced after electrochemical reduction, and the overpotentials of ER-BSCF, ER-SCF and ER-LSCF are 284, 265 and 306 mV at 10 mA cm−2, respectively. The electrochemical reduction can also greatly improve the OER kinetics of these Co-based perovskites, and the Tafel slopes of ER-BSCF, ER-SCF and ER-LSCF are 59.6, 45.2 and 70.4 mV dec−1, respectively (Fig. 5g–i).
 | ||
| Fig. 5 SEM images of BSCF (a), SCF (b) and LSCF (c) after electrochemical reduction. OER LSV curves of BSCF (d), SCF (e) and LSCF (f) before and after electrochemical reduction. Tafel plots of BSCF (g), SCF (h) and LSCF (i) before and after electrochemical reduction. | ||
Due to the severe oxidation and corrosion of chloride ions, OER catalysts for seawater splitting must possess excellent OER activity and robust stability.14,16 The good durability of ER-SNCF-20s can be explained by its high valence of Nb5+ cations at the B-site, which contributes to the effective phase stability.33 The OER activity of ER-SNCF-20s in alkaline seawater is evaluated by LSV measurement. In Fig. S12,† the OER activity of ER-SNCF-20s is much higher than those of RuO2 and SNCF in alkaline seawater, with an overpotential of only 283 mV at a current density of 10 mA cm−2. To evaluate the OER activity and stability of our ER-SNCF-20s for practical seawater splitting, a home-made electrolytic cell is constructed with ER-SNCF-20s and Pt/C as the anode and cathode, respectively (Fig. 6a). The OER catalytic activities of ER-SNCF-20s loaded on the different substrates (nickel foam and carbon cloth) are shown in Fig. S13,† and a negligible difference can be seen from the two electrodes. Compared with carbon cloth, nickel foam provides better hydrophilicity which can facilitate the release of oxygen bubbles. Therefore, we prepared ER-SNCF-20s on nickel foam (ER-SNCF-20s/NF) as the anode for seawater splitting. As shown in Fig. 6b and c, using ER-SNCF-20s/NF as an anode, the splitting overpotentials of alkaline water (1 M KOH), simulated alkaline seawater (1 M KOH + 0.5 NaCl) and alkaline natural seawater (1 M KOH + seawater) are 297, 299 and 327 mV at 10 mA cm−2, respectively. The seawater is collected from the East China Sea (Fig. S14†). Compared with that for simulated alkaline seawater, the larger overpotential for splitting alkaline natural seawater may be ascribed to the existence of bacteria and microorganisms as well as small particles in natural seawater. Nevertheless, our ER-SNCF-20s still shows high OER catalytic activity in alkaline natural seawater, and the overpotential at 10 mA cm−2 is much lower than the thermodynamic overpotential of the oxidation of chloride to hypochlorite (490 mV). At a high current density of 100 mA cm−2, the splitting overpotential of alkaline natural seawater using the ER-SNCF-20s anode reaches 536 mV which is slightly higher than 490 mV, whereas it is still difficult to achieve the oxidation of chlorides under this overpotential due to the fact that the oxidation of chlorides also need a certain overpotential.15 Thus, the OER remains dominant in the competition with chloride oxidation at a high current density of 100 mA cm−2 during alkaline natural seawater splitting with ER-SNCF-20s.
 | ||
| Fig. 6 (a) Schematic illustration of the overall alkaline natural seawater electrolysis using ER-SNCF-20s and Pt/C as the anode and cathode, respectively. (b) Polarization curves of an electrolyzer with ER-SNCF-20s in different electrolytes. (c) Overpotentials for driving water splitting at 10 and 100 mA cm−2 in 1 M KOH, 1 M KOH + 0.5 M NaCl and 1 M KOH + seawater. (d) Long-term stability test of an electrolyzer with ER-SNCF-20s conducted at a constant current density of 10 mA cm−2 in 1 M KOH + seawater. (e) Experimental and theoretical gaseous products (H2 and O2) obtained using a two-electrode electrolyzer at a fixed current density of 10 mA cm−2 in 1 M KOH + seawater. Photographs showing hydrogen production from seawater driven by wind (f) and solar powers (g). | ||
The stability of the anodes in seawater splitting is indispensable due to the fact that the pollutants and planktons in seawater can continuously attack the anodes.14 The long-term stability of our electrolytic cell with the ER-SNCF-20s anode is measured in alkaline natural seawater. As shown in Fig. 6d, our electrolytic cell with the ER-SNCF-20s anode can stably split the alkaline natural seawater for 300 h without degradation, proving that ER-SNCF-20s can be used as a very robust anode for seawater electrolysis. In order to verify whether the other gas products are produced during seawater electrolysis, we collected the gas products separately using the drainage method at 10 mA cm−2 at the anode and cathode sides (Fig. S15†). After calculations, the theoretical gas products and the measured gas products basically maintain the same value (Fig. 6e). Furthermore, our electrolytic cell with the ER-SNCF-20s anode could be used to generate hydrogen from renewable energy, such as wind and solar energies (Fig. 6f and g and ESI video†).
3. Conclusion
In summary, a rapid, facile and universal electrochemical reduction treatment was proposed to reconstruct the active surface of Co-based perovskites in a few seconds. After the electrochemical reduction, the Sr-rich layer on the Co-based perovskite surface could be eliminated by Sr precipitation along with the formation of A-site defects and the introduction of a substantial number of oxygen vacancies. As a result of surface reconstruction, the ECSA of Co-based perovskite SNCF could increase by about 4 times, and then the OER activity was significantly enhanced. After surface reconstruction, ER-SNCF-20s exhibited a low overpotential of merely 278 mV at 10 mA cm−2. This can be related to the significant decrease of the overpotential at the rate-determining step of *OOH → O2 by introducing Sr and O vacancies. Using ER-SNCF-20s as an anode for alkaline natural seawater splitting, a low overpotential of 327 mV could be achieved at 10 mA cm−2. More importantly, the ER-SNCF-20s anode was very robust during natural seawater splitting, and it operated continuously for 300 h without any decay.4. Experimental section
4.1 Chemicals
La(NO3)3, Ba(NO3)2, Fe(NO3)3·9H2O, Co(NO3)2·6H2O, Sr(NO3)2, C10H5NbO20, EDTA (C10H16N2O8), citric acid (C6H8O7), ethanol, KOH, IrO2 and Pt/C were purchased from Aladdin Ltd. Conductive carbon (VXC-72) was purchased from Cabot Ltd, Nafion solution (5 wt%, 80 μL) was obtained from DuPont Ltd. Carbon cloth was obtained from CeTech Co., Ltd. Ni foam was purchased from Sinero Co., Ltd. All chemical reagents in the Experimental section were used directly.4.2 Electrode preparation
Co-based perovskites were synthesized by the sol–gel method (see the ESI†) followed by ball milling for 1 h. 5 mg of the Co-based perovskites, 5 mg of conductive carbon were spread in 2 mL of ethanol with 80 μL of Nafion solution. The dispersion was added dropwise on carbon cloth or nickel foam (1 × 1 cm−2), and then dried on a heating plate at 80 °C. Then, the electrode with the different Co-based perovskites was used as the working electrode, and 1 M KOH alkaline solution was used as the electrolyte. The reference electrode is an Ag/AgCl electrode (CHI 111) with a saturated 3.5 M KCl solution as the salt bridge and the counter electrode is a graphite rod. Negative voltages from −0.8 to −1.4 V were applied on the working electrode for different times (10–60 s). The obtained samples were denoted as ER-SNCF-x s, where x is the electrochemical reduction time. The loading amount of the above-mentioned catalyst was 2 mg cm−2. The preparation procedures of ER-BSCF, ER-SCF and ER-LSCF electrodes were almost the same as that of the ER-SNCF-20s electrode, except for the replacement of SNCF with BSCF, SCF and LSCF, respectively. The electrochemical reduction time and voltage of these three samples are fixed as 20 s and −1 V, respectively.To prepare an IrO2 electrode, 5 mg of IrO2 was dispersed in 2 ml of ethanol with 80 μL of Nafion solution. Then the dispersion was dropped on carbon cloth (1 × 1 cm−2) and dried on a heating plate at 80 °C. A Pt/C electrode was prepared with the similar method. The loading amount of the above-mentioned catalyst was 2 mg cm−2.
4.3 Characterization
X-ray diffraction was carried out with a Bruker D8 Advance X-ray diffract meter (Cu Kα = 1.5418 Å) at a scan rate of 0.02° s−1. Micromorphologies were observed by scanning electron microscopy (SEM, Hitachi S4800), energy dispersive X-ray spectroscopy (EDS, Tecnai F20), transmission electron microscopy (TEM, Tecnai F20), and selected area electron diffraction (SAED, Tecnai F20). The elemental information on the surface was obtained by X-ray photoelectron spectroscopy (XPS, AXIS ULTAR).4.4 Electrochemical characterization
All the electrochemical measurements were conducted on an electrochemical workstation (CHI Instruments 760E) in a standard three-electrode system at room temperature. 1 M KOH alkaline solution was used as the electrolyte. The reference electrode is an Ag/AgCl electrode and the counter electrode is a graphite rod. Before the linear sweep voltammetry (LSV) test, O2 was pumped into the electrolyte for 30 min, and the electrodes are activated by CV scanning (10 mV s−1, 0.2–1.0 V). The OER LSV curve was recorded at 5 mV s−1 within a scanning potential of 0.2 to 1.0 V (vs. Ag/AgCl). Then, the OER LSV data were obtained with IR correction. All the potentials were calibrated to RHE: ERHE = EAg/AgCl + 0.1976 + 0.0592 × pH. The catalyst stability was measured by chronopotentiometry at 10 mA cm−2. Electrochemical impedance spectroscopy (EIS) was carried out in a frequency range of 0.1 Hz–106 Hz. The electrochemical double-layer capacitance (Cdl) was measured by CV scans at different scan rates of 0.132–0.232 V vs. Ag|AgCl (3.5 M KCl). By drawing the curve of Δj (Δj = (janode − jcathode) × 0.5) against the scanning rate, the slope of the line indicates Cdl.4.5 Water splitting test
Overall alkaline water (1 M KOH), simulated alkaline seawater (1 M KOH + 0.5 M NaCl) and alkaline natural seawater (1 M KOH + seawater) were used to obtain the polarization curve in a homemade two-electrode cell using ER-SNCF-20s as the anode and Pt/C as the cathode. The natural seawater was collected from the East China Sea. The cell stability was measured by chronopotentiometry at 10 mA cm−2 in alkaline natural seawater.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (Grant No. 51871126), the Ningbo major special projects of the Plan “Science and Technology Innovation 2025” (Grant No. 2020Z107), the Zhejiang Provincial Natural Science Foundation of China Grant (Grant No. LY21E010002) and the K. C. Wong Magna Fund in Ningbo University.Notes and references
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC
.
- W.-Z. Chen, P.-Y. Liu, L. Zhang, Y. Liu, Z. Liu, J. He and Y.-Q. Wang, Chem. Eng. J., 2021, 424, 130434 CrossRef CAS
- S. Yuan, M. Xia, Z. Liu, K. Wang, L. Xiang, G. Huang, J. Zhang and N. Li, Chem. Eng. J., 2022, 430, 132697 CrossRef CAS
- W. Gan, L. Wu, Y. Wang, H. Gao, L. Gao, S. Xiao, J. Liu, Y. Xie, T. Li and J. Li, Adv. Funct. Mater., 2021, 31, 2010951 CrossRef CAS
- Y. Jiang, S. Gao, J. Liu, G. Xu, Q. Jia, F. Chen and X. Song, Nanoscale, 2020, 12, 11573–11581 RSC
- S. Ye, Y. Zhang, W. Xiong, T. Xu, P. Liao, P. Zhang, X. Ren, C. He, L. Zheng and X. Ouyang, Nanoscale, 2020, 12, 11079–11087 RSC
- T. E. Mallouk, Nat. Chem., 2013, 5, 362–363 CrossRef CAS PubMed
- J. Zhou, L. Yu, Q. Zhou, C. Huang, Y. Zhang, B. Yu and Y. Yu, Appl. Catal., B, 2021, 288, 120002 CrossRef CAS
- D. T. Tran, S. Prabhakaran, D. H. Kim, N. Hameed, H. Wang, N. H. Kim and J. H. Lee, Nano Energy, 2021, 88, 106277 CrossRef
- W. Tong, M. Forster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Nat. Energy, 2020, 5, 367–377 CrossRef CAS
- S. Dresp, F. Dionigi, M. Klingenhof and P. Strasser, ACS Energy Lett., 2019, 4, 933–942 CrossRef CAS
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Nat. Commun., 2019, 10, 5106 CrossRef
- S. Wang, P. Yang, X. Sun, H. Xing, J. Hu, P. Chen, Z. Cui, W. Zhu and Z. Ma, Appl. Catal., B, 2021, 297, 120386 CrossRef CAS
- Y. Kuang, M. J. Kenney, Y. Meng, W. H. Hung, Y. Liu, J. E. Huang, R. Prasanna, P. Li, Y. Li, L. Wang, M. C. Lin, M. D. McGehee, X. Sun and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6624–6629 CrossRef CAS PubMed
- L. Yu, L. Wu, B. McElhenny, S. Song, D. Luo, F. Zhang, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2020, 13, 3439–3446 RSC
- L. Wu, L. Yu, F. Zhang, B. McElhenny, D. Luo, A. Karim, S. Chen and Z. Ren, Adv. Funct. Mater., 2021, 31, 2006484 CrossRef CAS
- T. Ma, W. Xu, B. Li, X. Chen, J. Zhao, S. Wan, K. Jiang, S. Zhang, Z. Wang and Z. Tian, Angew. Chem., 2021, 133, 22922–22926 CrossRef
- G. Liu, Y. Xu, T. Yang and L. Jiang, Nano Mater. Sci., 2020 DOI:10.1016/j.nanoms.2020.12.003
- Y. Duan, Z. Y. Yu, S. J. Hu, X. S. Zheng, C. T. Zhang, H. H. Ding, B. C. Hu, Q. Q. Fu, Z. L. Yu, X. Zheng, J. F. Zhu, M. R. Gao and S. H. Yu, Angew. Chem., Int. Ed., 2019, 58, 15772–15777 CrossRef CAS PubMed
- T. Wu, S. Sun, J. Song, S. Xi, Y. Du, B. Chen, W. A. Sasangka, H. Liao, C. L. Gan, G. G. Scherer, L. Zeng, H. Wang, H. Li, A. Grimaud and Z. J. Xu, Nat. Catal., 2019, 2, 763–772 CrossRef CAS
- Y. Dou, L. Zhang, J. Xu, C. T. He, X. Xu, Z. Sun, T. Liao, B. Nagy, P. Liu and S. X. Dou, ACS Nano, 2018, 12, 1878–1886 CrossRef CAS PubMed
- T. Kou, S. Wang, J. L. Hauser, M. Chen, S. R. J. Oliver, Y. Ye, J. Guo and Y. Li, ACS Energy Lett., 2019, 4, 622–628 CrossRef CAS
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS
- P. Chen, K. Xu, Z. Fang, Y. Tong, J. Wu, X. Lu, X. Peng, H. Ding, C. Wu and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 14710–14714 CrossRef CAS PubMed
- X. Jia, Y. Zhao, G. Chen, L. Shang, R. Shi, X. Kang, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Energy Mater., 2016, 6, 1502585 CrossRef
- H. Ge, G. Li, J. Shen, W. Ma, X. Meng and L. Xu, Appl. Catal., B, 2020, 275, 119104 CrossRef CAS
- L. Cai, B. Qiu, Z. Lin, Y. Wang, S. Ma, M. Wang, Y. H. Tsang and Y. Chai, J. Mater. Chem. A, 2018, 6, 21445–21451 RSC
- F. Yu, H. Zhou, Y. Huang, J. Sun, F. Qin, J. Bao, W. A. Goddard, 3rd, S. Chen and Z. Ren, Nat. Commun., 2018, 9, 2551 CrossRef
- M. Cui, C. Yang, B. Li, Q. Dong, M. Wu, S. Hwang, H. Xie, X. Wang, G. Wang and L. Hu, Adv. Energy Mater., 2020, 11, 2002887 CrossRef
- J. Hou, B. Zhang, Z. Li, S. Cao, Y. Sun, Y. Wu, Z. Gao and L. Sun, ACS Catal., 2018, 8, 4612–4621 CrossRef CAS
- J. G. Li, H. Sun, L. Lv, Z. Li, X. Ao, C. Xu, Y. Li and C. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 8106–8114 CrossRef CAS
- Y. Zuo, D. Rao, S. Ma, T. Li, Y. H. Tsang, S. Kment and Y. Chai, ACS Nano, 2019, 13, 11469–11476 CrossRef CAS PubMed
- Y. Zhu, W. Zhou, Z. G. Chen, Y. Chen, C. Su, M. O. Tade and Z. Shao, Angew. Chem., Int. Ed., 2015, 54, 3897–3901 CrossRef CAS PubMed
- Y. Zhu, W. Zhou, J. Sunarso, Y. Zhong and Z. Shao, Adv. Funct. Mater., 2016, 26, 5862–5872 CrossRef CAS
- Y. Zhu, W. Zhou, Y. Zhong, Y. Bu, X. Chen, Q. Zhong, M. Liu and Z. Shao, Adv. Energy Mater., 2017, 7, 1602122 CrossRef
- F. Dong, L. Li, Z. Kong, X. Xu, Y. Zhang, Z. Gao, B. Dongyang, M. Ni, Q. Liu and Z. Lin, Small, 2021, 17, e2006638 CrossRef PubMed
- Y. Bu, O. Gwon, G. Nam, H. Jang, S. Kim, Q. Zhong, J. Cho and G. Kim, ACS Nano, 2017, 11, 11594–11601 CrossRef CAS PubMed
- B. Zhao, L. Zhang, D. Zhen, S. Yoo, Y. Ding, D. Chen, Y. Chen, Q. Zhang, B. Doyle, X. Xiong and M. Liu, Nat. Commun., 2017, 8, 14586 CrossRef CAS PubMed
- Q. Lin, Y. Zhu, Z. Hu, Y. Yin, H.-J. Lin, C.-T. Chen, X. Zhang, Z. Shao and H. Wang, J. Mater. Chem. A, 2020, 8, 6480–6486 RSC
- G. Ou, C. Yang, Y. Liang, N. Hussain, B. Ge, K. Huang, Y. Xu, H. Wei, R. Zhang and H. Wu, Small Methods, 2019, 3, 1800279 CrossRef
- C. Zhao, N. Li, R. Zhang, Z. Zhu, J. Lin, K. Zhang and C. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 47858–47867 CrossRef CAS PubMed
- L. Yu, H. Zhou, J. Sun, F. Qin, F. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2017, 10, 1820–1827 RSC
- Y. Lin, H. Wang, C. K. Peng, L. Bu, C. L. Chiang, K. Tian, Y. Zhao, J. Zhao, Y. G. Lin, J. M. Lee and L. Gao, Small, 2020, 16, e2002426 CrossRef PubMed
- B. Wang, X. Han, C. Guo, J. Jing, C. Yang, Y. Li, A. Han, D. Wang and J. Liu, Appl. Catal., B, 2021, 298, 120580 CrossRef CAS
- B. Koo, K. Kim, J. K. Kim, H. Kwon, J. W. Han and W. Jung, Joule, 2018, 2, 1476–1499 CrossRef CAS
- W. Guo, L. Cui, H. Xu and C. Gong, Appl. Surf. Sci., 2020, 529, 147165 CrossRef CAS
- Y.-Q. Zhang, H.-B. Tao, Z. Chen, M. Li, Y.-F. Sun, B. Hua and J.-L. Luo, J. Mater. Chem. A, 2019, 7, 26607–26617 RSC
- Z. Teng, Z. Xiao, G. Yang, L. Guo, X. Yang, R. Ran, W. Wang, W. Zhou and Z. Shao, Mater. Today Energy, 2020, 17, 100458 CrossRef
- N. A. Merino, B. P. Barbero, P. Eloy and L. E. Cadús, Appl. Surf. Sci., 2006, 253, 1489–1493 CrossRef CAS
- J. Zhu, H. Li, L. Zhong, P. Xiao, X. Xu, X. Yang, Z. Zhao and J. Li, ACS Catal., 2014, 4, 2917–2940 CrossRef CAS
- Y. Zhu, L. Zhang, B. Zhao, H. Chen, X. Liu, R. Zhao, X. Wang, J. Liu, Y. Chen and M. Liu, Adv. Funct. Mater., 2019, 29, 1901783 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr01516a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |