
α-C–H functionalization of glycine derivatives under mechanochemical accelerated aging en route the synthesis of 1,4-dihydropyridines and α-substituted glycine esters†
Keyu
Xiang‡
 a,
Ping
Ying‡
ab,
Tao
Ying
a,
Weike
Su
a and
Jingbo
Yu
*a
a,
Ping
Ying‡
ab,
Tao
Ying
a,
Weike
Su
a and
Jingbo
Yu
*a
aNational Engineering Research Center for Process Development of Active Pharmaceutical Ingredients, Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology. Hangzhou. 310014, P. R. China. E-mail: yjb@zjut.edu.cn
bCollege of Ecology, Lishui University, Lishui, 323000, P.R. China
First published on 7th March 2023
Abstract
The emergence of accelerated aging reaction provided a safer, cleaner, and more sustainable technology for material manufacturing and biomass treatment but still underexploited in organic synthesis and medicinal chemistry. We report the first mechanochemical accelerated aging strategy for solvent-minimal (cascade) cross dehydrogenative coupling (CDC) reactions between glycine esters/amides and a range of nucleophiles, which features clean and convenient setup, ambient temperature, atmospheric oxidation, and feasibility, for multigram-scale synthesis. By virtue of these facts, the present method provided an expedient and sustainable alternative to synthesize biologically important α-glycine derivatives and functionalized 1,4-dihydropyridines including the precursor of the antioxidant AV-154 and calcium channel blocker analogs. Mechanistically, a pre-grinding of the reactants and silica gel/NaCl facilitated spontaneous oxidation of glycine esters/amides under open air without continuous energy input followed by a coupling reaction (and sequential transformations). Multiform green metrics calculation demonstrates that the current accelerated aging protocol meets many of the principles of green chemistry such as waste prevention, high atom economy, unnecessary solvent, and good energy efficiency.
Introduction
Mechanochemistry has experienced dramatic development over the past decade and enabled many chemical transformations in a sustainable way.1 The use of mechanical agitation to cause chemical reactions by direct absorption of mechanical energy has many practical advantages such as short reaction time, reliable energy and mass transfer, minimal solvents/reagents consumption, and even lead to better reactivity and selectivity.2 Interestingly, some mechanochemical reactions can occur without continuous mechanical agitation, being accelerated or directed by mild and short-lasting low-energy impetus. Such transformations are considered accelerated aging (AA)3 (Fig. 1A), a specific diffusion-based process with inherently simple, low-cost, safe, and energy-saving properties, especially for expandable chemical production. The recent outstanding research by groups of Friščić, Schurko, Jayasankar, and Moores showcased a big breakthrough of AA in the synthesis of metal–organic framework (MOFs),4 nanoparticles,5 co-crystals,6 and polymers.7 The organic accelerated aging reactions date back more than thirty years, pioneered by Toda8 but rarely investigated in recent 15 years. Representative works are Štrukil's,9 Baltas, and Calvet's10 independent works of vapor digestion accelerated amination of thiocarbamoyl benzotriazoles and pre-grinding facilitated Diels–Alder reaction. Besides, accelerated aging was found beneficial to explain the mechanism of solid-state diaza-Cope rearrangement but continuous ball milling was still necessary for high conversion.11 In view of the notable advantages of accelerated aging, we envision that this technique is possible to provide new opportunities to increase the efficiency and sustainability of exquisite organic synthetic procedures for manufacturing high-value chemicals.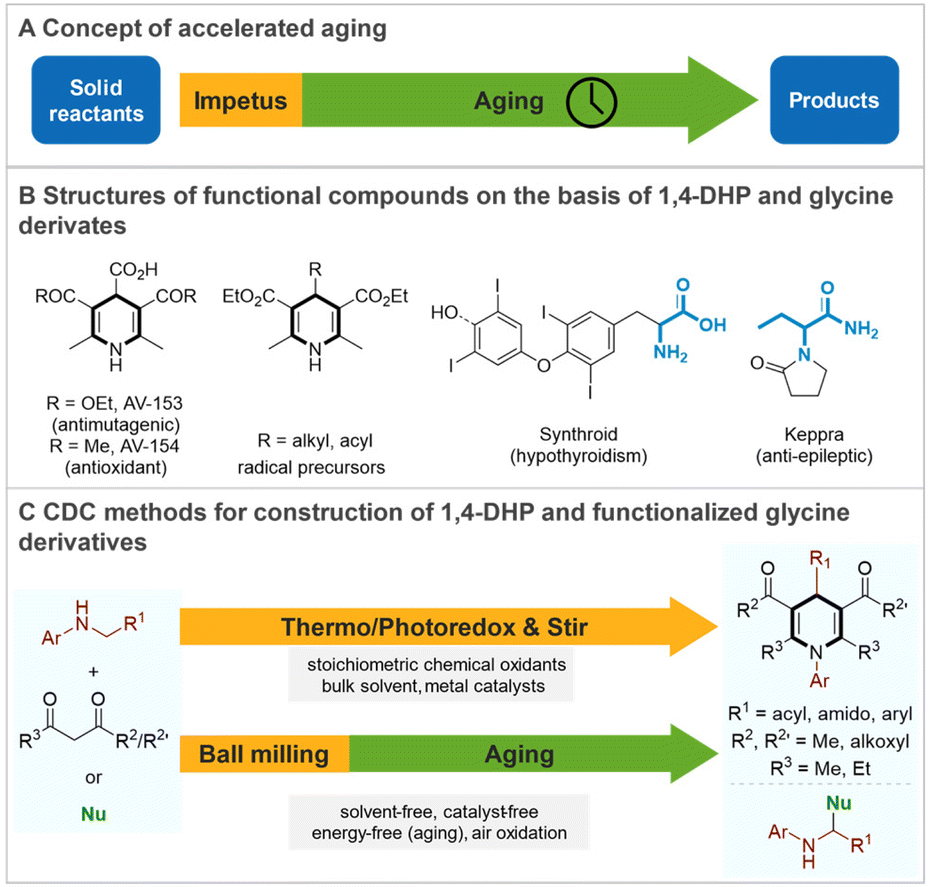 | ||
| Fig. 1 (A) Concept of accelerated aging. (B) Structure of functional compounds on the basis of 1,4-DHP and glycine derivates. (C) CDC method for construction of 1,4-DHP and functionalized glycine derivatives. | ||
The 1,4-dihydropyridine (1,4-DHP) is a prevalent chemical core found in a multitude of compounds ranging from calcium channel blockers, anticonvulsants, and antimutagenic bioactive molecules, to radical precursors (Fig. 1B).12 Their synthesis mostly relies on the multicomponent reactions of aldehydes, β-ketoesters, and specific amine sources, which is known as Hantzsch-type synthesis.12a,c,13 Recently, the development of cascade cross dehydrogenative coupling (CDC)14 -cyclization strategy15 has provided an efficient alternative for the synthesis of 1,4-DHPs using glycines as accessible building blocks. For example, Jia15a developed the first Fe/TMSCl-TBPA+/O2 promoted cascade CDC between glycinates and β-ketoesters in acetonitrile solvent to achieve 1,4-DHPs, where the cleavage of the C–N bonding and the reassembly of amine fragments led to the bond formation in an atom- and step-economic fashion. Subsequent works of Zhu and Le showed that similar reactions could be carried out successfully in toluene by both Cu catalysis15b and photocatalysis15c (with Ir(ppy)3 as a sensitizer). On the other hand, non-cascade CDC-type functionalization of α-C–H bonds of glycines16 also attracted considerable interest due to the abundance and prevalence of α-glycine derivatives in natural products and bioactive molecules (Fig. 1B). Despite the enormous advances that have been made, the use of bulk toxic solvents, metal/(photo) catalysts, external oxidative additives, and/or heating greatly depreciates the sustainability and practicality of these strategies, which calls for continuous improvement. Based on the practical requirements of green and effective synthesis of the privileged frameworks of 1,4-DHPs as well as the α-functionalized glycines, and our research interests in mechanochemistry,17 herein, we demonstrate a novel AA facilitated (cascade) CDC of glycinates and various nucleophiles under metal-free and solvent-minimal conditions. (Fig. 1C). This mechanochemical accelerated aging protocol is not only a method without a catalyst, bulky solvent, and additional strong oxidant, but also a design for safer and simpler chemical synthesis that requires less energy input.
Results and discussion
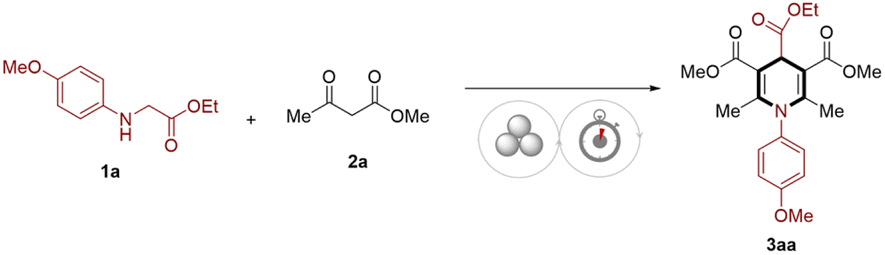
During the optimization of the reaction conditions, ethyl N-(4-methoxyphenyl)glycinate (1a) and methyl acetoacetate (2a) were used as the model substrates under the mechanochemical accelerated aging conditions for the synthesis of 1,4-DHP. The desired product 3aa was obtained in 67% yield after 30 min pre-grinding at 30 Hz, followed by aging at 40 °C for 24 h. For the initial screen, we selected several auxiliaries as shown in Table 1 and Table S1,† among which silica gel was found to be the most effective, while other salts or mineral alternatives all depreciated the product yield (Table 1, entries 1–4). As the grinding-aid agent and absorbent in the reaction, silica gel could mix substrates efficiently, whereas, in the absence of the auxiliary, only a trace product was formed because of the agglomeration of the reactants (Tables S1 and S2†), this phenomenon also occurs when less silica gel was used (Fig. S3†). In addition, silica gel serving as a slightly acidic medium has a positive effect due to the increase in acidity of the reaction environment2h,n which could promote this transformation. Thus, we continued with the optimization by using 600 mg silica gel (details see Table S3†) and found that the yield of 3aa was decreased by reducing the milling frequency and milling time (Table 1, entries 5 and 6). However, prolonging the milling time could not significantly improve the reaction yield (Table 1, entry 6), which implied that the milling frequency acted as a key parameter for the following aging transformations (see also Fig. S5†). After the preliminary tuning of the stoichiometry, 2a was used in the reaction methodology, and 3.0 eq. of 2a was found to be the optimal balance (Table 1, entry 7 vs. 1). Reducing the amount of 2a gave rise to a depreciated product yield, even with prolonged milling time (Table 1, entry 8).|
|
||||
|---|---|---|---|---|
| Entry | Variation from “standard conditions” | Yield of 3aa/% | ||
| a Reaction conditions: 1a (0.5 mmol), 2a (1.5 mmol) and silica gel (0.6 g)/auxiliaries (with same volume) were pre-grinded for 30 min at 30 Hz, using two stainless-steel balls (dMB = 1.4 cm) in a 25 mL stainless-steel jar, then aging in an opened flask for 24 h at 40 °C. Yield of isolated product. b The reaction were carried out in a 25 mL glass flask in the presence of 1a (0.5 mmol), 2a (1.5 mmol) and CH3CN (5 mL). c The reaction were carried out in a 25 mL glass flask in the presence of 1a (0.5 mmol), 2a (1.5 mmol), TFA (0.125 mmol) and CH3CN (5 mL). d Neat stirring of 1a (0.5 mmol), 2a (1.5 mmol) and 0.6 g silica gel in a 25 mL glass flask. e Neat stirring of 1a (0.5 mmol), 2a (1.5 mmol), TFA (0.125 mmol) and 0.6 g silica gel in a 25 mL glass flask (rt = room temperature, n.d. = not detected). | ||||
| 1 | None | 67 | ||
| 2 | Without silica gel | N.d. | ||
| 3 | Auxiliary: NaCl/KBr/Na2SO4 | 59/31/37 | ||
| 4 | Auxiliary: kieselguhr/Al2O3 | N.d. | ||
| 5 | Milling frequency: 25 Hz/20 Hz/15 Hz | 53/48/35 | ||
| 6 | Milling time: 20 min/60 min | 55/68 | ||
| 7 | 2a (4.0 eq.)/2a (2.2 eq.) | 68/54 | ||
| 8 | 2a (2.2 eq.), milling time: 60 min | 57 | ||
| 9 | 2a (2.2 eq.), aging time: 48 h | 65 | ||
| 10 | 2a (2.2 eq.), aging at rt | 50 | ||
| 11 | 2a (2.2 eq.), aging at rt, aging time: 48 h/84 h | 61/71 | ||
| 12 | 2a (2.2 eq.), aging under O2, 48 h/84 h | 64/72 | ||
| 13 | 2a (2.2 eq.), aging under N2, 84 h | Trace | ||
| 14b |

|
CH3CN | Air | N.d. |
| 15b | 40 °C | O2 | N.d. | |
| 16c | CH3CN | Air | 15 | |
| 17c | 40 °C, TFA | O2 | 36 | |
| 18d |

|
Stirred neat | Air | N.d. |
| 19d | 40 °C | O2 | N.d. | |
| 20e | Stirred neat | Air | N.d. | |
| 21e | 40 °C, TFA | O2 | N.d. | |
For sustainability considerations, we wondered if the product yield could be improved by prolonging the aging time in the presence of less amount of 2a and under lower aging temperature, offering a good balance between the cost of production, environmental impact, and efficacy. To our delight, varying the aging time from 24 h to 48 h showed a significant effect on the product yield even at a reduced aging temperature (Table 1, entry 9 vs. 7, entry 11 vs. 10). The best result (71% of 3aa) could be finally achieved in the presence of 2.2 eq. of 2a after aging for 84 h at room temperature (Table S3,† entry 14). It should be noted that replacing the aging atmosphere with oxygen had little effect on the yield (Table 1, entry 12), while the nitrogen atmosphere suppressed the reaction completely (Table 1, entry 13). Considering that column chromatography purification is time-consuming and energy-demanding and could be a significant obstacle, particularly working at a larger scale, a simple work-up procedure and purification method were then developed to produce 3aa. According to the dissolution difference between the products and the undesired substances, the target product can be separated from the reaction mixture by simply rinsing with cyclohexane,18 a usable non-polar solvent, without leaching any side products (Fig. S8†).
A comparison study of the reactors was also been made for this cascade CDC reaction. Running the model reaction system in 5 mL CH3CN gave no target product (Table 1, entry 14), while an auto-oxidation side-product (ethyl 2-((4-methoxyphenyl)amino)-2-oxoacetate)16b of 1a was generated (Scheme S2†) under the oxygen atmosphere (Table 1, entry 15). Further study showed that acidic additive (TFA) could increase the solution reaction yield to 15% (under air) and 36% (under oxygen), respectively (Table 1, entries 16 and 17), however, neat solid stirring at 40 °C under both air and oxygen atmosphere gave very poor results with or without TFA (Table 1, entries 18–21). In sharp contrast, the pre-grinding greatly facilitated the reaction and produced 3aa in 67% yield (Table 1 entry1), highlighting the outstanding performance of the mechanical force-accelerated aging strategy. Besides, the possibility of catalytic behavior of some leaching metals such as Fe, Co, and Ni during the milling process19 was excluded by carrying out the reaction using Teflon jars and balls (Table S3,† entry 16).
With the optimal conditions, we started by experimenting with different N-PMP glycine esters 1a–1e with methyl acetoacetate (2a), which could be effectively converted into products 3aa–3ea in 55–81% yields, amongst the bulky ester group (OtBu) lowered the transformation. Nevertheless, a slight loss of efficiency was observed for 3na carrying a congested steric environment on the phenyl ring. The experimental results also indicated that the electronic variation on the benzene rings had a remarkable impact on the reaction yields, the electron-donating substituents such as OMe and Me were favorable to the transformation, while electron-withdrawing groups (4-F, 4-Cl, 4-Br) led to a depreciation in the conversion and a small amount of liquid additive20 (LAGs = TFA, η = 0.009) was required to improve the yields of 3ia–3ka (46–64%). It should be noted that the presence of terminal double bonds in glycine ester was adaptable to give the target product 3mb, and the allyl group could be retained for further derivatization. For β-carbonyl esters, different ester groups such as –OMe, –OEt, –OiPr, –OBn, and –OAd (2a–2f) were tolerated under the mechanochemical aging conditions, giving corresponding products in desired yields (up to 85%). Satisfactorily, methyl 3-oxopentanoate (2f) and acetylacetone (2g) could be applied in this transformation with no obvious suppression in the products yields (3af), especially in the presence of trace amounts of liquid additive (3ag–3cg, 3fg, and 3hg). In addition, N-arylglycine esters 1a and 1l were able to achieve the transformation smoothly with two different β-carbonyl esters/acetylacetone (2a, 2b, 2c, 2g) to furnish unsymmetrical products albeit with relatively low yields. Unfortunately, neither N-alkylglycine ester nor N-arylglycine esters with strong electron-withdrawing groups (nitro-, cyano-, and carbonyl) on the benzene rings were competent in the reaction, which was probably due to their low reactivity or the formation of a less stable intermediate during the C–H activation21 (Fig. S6†).
It is noteworthy to mention that the cascade CDC was not restricted to N-arylglycine esters, a series of N-arylglycine amides, including aliphatic primary amide, aliphatic secondary amide and aromatic amide were also suitable for this transformation (Table 3). The linear amides (4a and 4b) seem to be more beneficial to the reaction compared to the branched ones (4c and 4d). When secondary amides 4e and 4f were subjected to the optimized reaction conditions, the expected 1,4-dihydropyridines 5ea, 5fa were not obtained by neat pre-grinding, but instead, produced in very good yields under LAGs promotion. The scope was further extended to aromatic amides, where N-phenylacetamide 4g was perfectly compatible with the mechanochemical AA conditions to afford 5ga in satisfactory yield through easy workup and purification. Notably, the use of amine derivative as substrate makes the cascade strategy applicable for the late-stage modification of API. Indeed, an aesthetic benzocaine derivative of 4h reacted smoothly to form the corresponding 1,4-DHP product in a moderate yield. Moreover, it should be highlighted that this solid-state transformation is particularly useful for poorly soluble substrate22 such as N-arylglycine amide, which is decorated with quinoline moiety 4i.
After giving more attention to details about the structure-reactivity relationship, we observed that benzyl β-carbonyl esters 6b and β-keto esters bearing tertiary α-carbon 6c–6d were adaptable to our accelerated aging conditions, but only gave CDC products 7aa–7ad without any cascade proceedings, which is considerably influenced by the steric and electronic nature of the substituent groups. The utility of this AA-facilitated CDC reaction was further illustrated by using other kinds of nucleophiles (Table 4). Aliphatic nitromethane 6a reacted smoothly with 1a, giving 7aa in satisfactory yield. Aromatic nucleophiles including mesitylene, N,N-dimethylaniline, thiophene, and indole were completely compatible with this reaction, delivering the target products 7af–7fi in good yields ranging from 62% to 83%. Besides, the C–N CDC reaction (7le) was also achievable under the standard conditions. However, the reaction with cyclic β-keto esters (6j–6o) failed to produce the coupling products under neat conditions. The rigid steric hindrance of α-carbon may retard their reactivity and hence trace amounts of catalyst were required. In these cases, five-membered-ring substrates (7aj, 7an and 7co) were more reactive than the six-membered-ring ones (7ak–7al and 7dm), while the steric hindrance of alkyl at the ester terminus had an inappreciable effect on the yield (7dm).
Besides, we investigated the recycling of the grinding auxiliary (silica gel) in order to improve the environmental performance of the whole process, unfortunately, the yield diminished significantly in the 2nd cycle (Fig. 2). Inspired by our previous work17g that NaCl has a satisfying performance in the recovery process as well as the comparable results obtained using NaCl as an alternative grinding auxiliary in this cascade CDC reaction (see 3ga, 3gb, 3af, and 3cg in Table 2, 5ea in Table 3, 7ac and 7ah in Table 4, and also Fig. S7 in ESI†), we assessed the recyclability of this grinding auxiliary and found it could maintain efficiency after 5 cycles.
 | ||
| Fig. 2 Recycling and recovery of the grinding auxiliary (isolated yields obtained without column chromatography). | ||
| a Reaction conditions:1 (0.5 mmol), 2 (1.1 mmol), and silica gel (0.6 g) were pre-grinded for 30 min at 30 Hz, using two stainless-steel balls (dMB = 1.4 cm) in a 25 mL stainless-steel jar, then aging in an opened flask for 84 h at rt. Isolated yields without column chromatography were shown in parentheses. b LAGs (TFA, η = 0.009) was added. c NaCl (1.5 g) was used instead of silica gel. d aging for 96 h. |
|---|

|
| a Reaction conditions: 4 (0.5 mmol), 2 (1.1 mmol) and silica gel (0.6 g) were pre-grinded for 30 min at 30 Hz, using two stainless-steel balls (dMB = 1.4 cm) in a 25 mL stainless-steel jar, then aging in an opened flask for 84 h at rt. Isolated yields without column chromatography were shown in parentheses. b LAGs (TFA, η = 0.009) was added. c NaCl (1.5 g) was used instead of silica gel. (PMP = para methoxy phenyl). |
|---|

|
| a Reaction conditions:1 (0.5 mmol), 6 (0.5 mmol) and silica gel (0.6 g) were pre-grinded for 30 min at 30 Hz, using two stainless-steel balls (dMB = 1.4 cm) in a 25 mL stainless-steel jar, then aging in an opened flask for 96 h at rt. Isolated yields without column chromatography were shown in parentheses. b LAGs (TFA, η = 0.01) was added. c NaCl (1.5 g) was used instead of silica gel. d 1 mol% Cu(OTf)2 was added, aging for 24 h at rt. |
|---|

|
To further manifest the practicality of this new accelerated aging protocol, the cascade reaction of N-arylglycine esters/amides and 1,3-dicarbonyl compounds have been conducted on a multigram scale. As illustrated in Scheme 1, 3aa, 3ag, and 5cb were isolated in 64% (2.58 g), 61% (2.26 g), and 57% (2.74 g) yields, respectively. Notably, the target product 3ag could be readily transformed into an antioxidant 9 (AV-15423). Further demonstration of the synthetic utility of the cascade CDC reaction was via the expedient synthesis of calcium channel blocker (nitrendipine) analogs (11) using N-benzyl-4-methoxyaniline (10) and 2a as substrates, recoverable NaCl as grinding auxiliary (details see ESI, sections 3.8 and 4.4†).
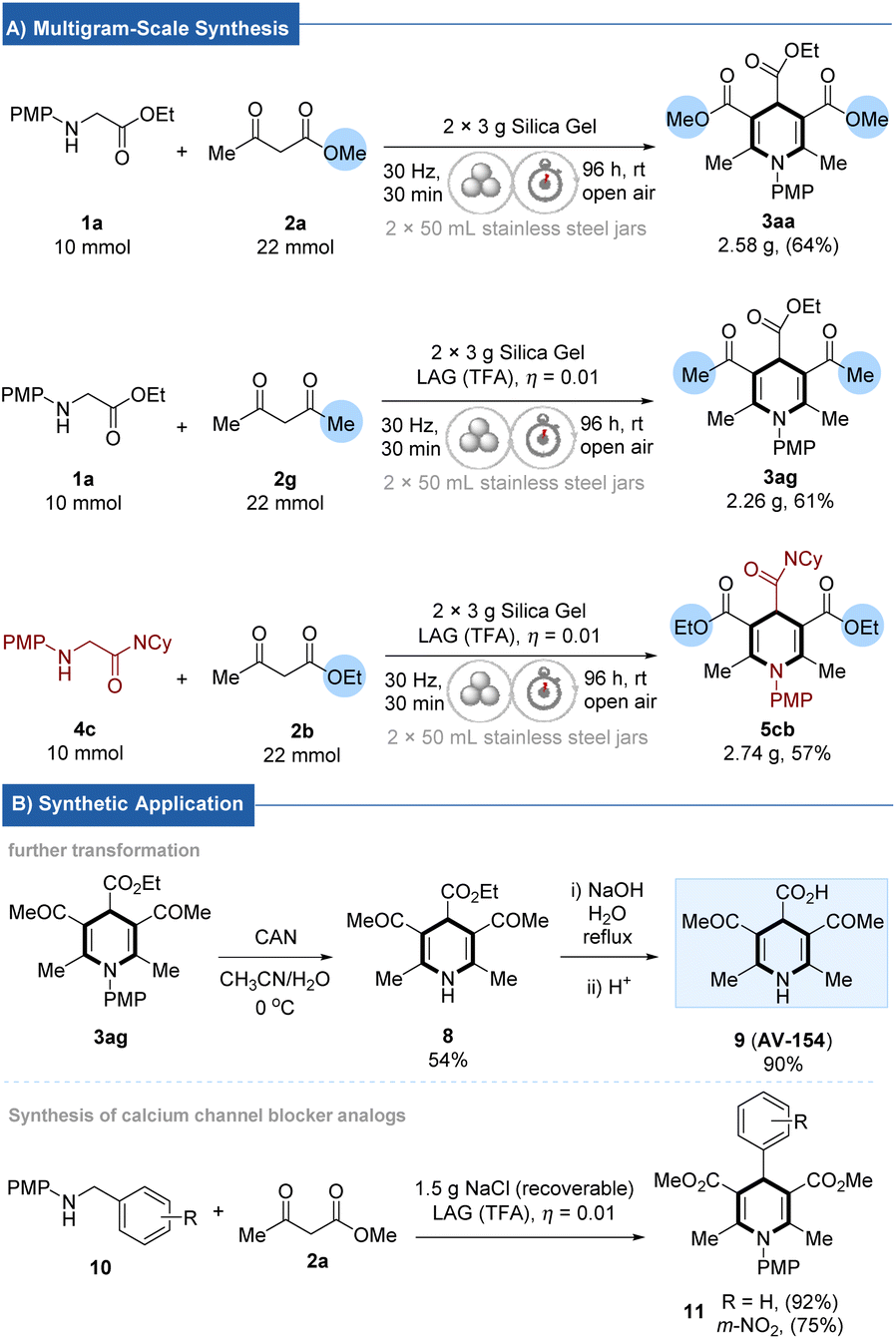 | ||
| Scheme 1 (A) Multigram-scale synthesis. (B) Synthetic application. (Isolated yields without column chromatography were shown in parentheses). | ||
To elucidate the reaction mechanism, we conducted a preliminary mechanistic investigation of the cascade process (Scheme 2). First, radical inhibition and trapping experiments were performed (Scheme 2A). The addition of 2.0 eq. of 2,6-di-tertbutyl-4-methylphenol (BHT) suppressed the cascade process of 1a and 2a, and a BHT-trapped product 12 was isolated and identified (details see Scheme S6 and Fig. S9, S10†) which strongly indicate that radical species are involved in the reaction pathway. Second, 3aa was obtained in 38% yield when N-glycine ester 1a was replaced by imine 13 under mechanical milling conditions, which implied imine as a potential intermediate of the cascade reaction (Scheme 2B). Next, a three-component reaction of 1a, 2a, and p-toluidine was performed to give 3aa (42%) along with a newly reassembled product 3fa (15%), which suggested that the transformation has undergone a C–N bond cleavage and fragment-reassembly process (Scheme 2C). Besides, the aging or neat stirring of the reaction sample from the pre-grinded 1a/silica gel (no imine was detected after pre-grinding) gave 16%/30% yields of 3aa, while physical pre-mixing of them did not afford any product, even at an elevated stirring temperature (60 °C) (Scheme 2D), demonstrating that the mechanical activation of 1a/silica gel is the driving force for the cascade CDC reaction.
 | ||
| Scheme 2 Control experiments. | ||
We hypothesize that the pre-grinding process might have some effects on the morphologies of silica gel. The microscopic differences of the commercial silica gel (C), silica gel after grinding at 30 Hz (C-30), and silica gel after ball-milling with 1a and 2a at 30 Hz (BM-30) were then investigated by laser particle analysis and N2 physical adsorption test (Table 5). From the data obtained by the measurements using a Mastersizer, the average particle size sequence of these silica gel was BM-30 (29.40 μm) < C-30 (72.08 μm) < C (86.60 μm). Notably, the neat grinding process resulted in a sharp reduction of the surface area and the total pore volume of silica gel. This phenomenon was probably due to aggregation24 (the particle size distribution was drastically changed, see Fig. S11†) and the collapse of pore structure25of silica gel after the intensive mechanical grinding. While co-grinding the silica gel with organic substrates (BM-30) could largely weaken this disruption yet lead to a small decrease in the average pore diameter. This might be attributed to the introduction of the reactants into the pores of silica gel.26 Such microscopic changes led to easy pre-association of silica gel and reactants,10 providing a high-performance dispersion of the substrates and favoring the oxidation of glycine ester with air and the subsequent transformations, which is hardly proceeding under conventional solution-based conditions.
| Type | Average particle size (μm) | S BET (m2·g−1) | Pore volume (cm3·g−1) | Pore size (nm) |
|---|---|---|---|---|
| C | 86.60 | 371.31 | 1.00 | 10.46 |
| C-30 | 72.08 | 16.57 | 0.05 | 11.18 |
| BM-30 | 29.40 | 219.55 | 0.41 | 7.17 |
On the basis of the experimental results and literature reports,15a a possible mechanism for this transformation is proposed, as shown in Scheme 3. Glycine ester 1a gives a single electron to oxygen (in the air) to produce the radical cation A after the mechanical activation, and generate active species O2˙−, which abstracted a hydrogen atom from radical cation A to produce the iminium ion intermediate B. Then, B reacted with an ethyl acetoacetate 2a to afford intermediate C, which was nucleophilically attacked by another ethyl acetoacetate 2a and followed a subsequent C–N bond cleavage, producing 1,5-dicarbonyl intermediate D. Finally, the leaving aniline further condensed with 1,5-dicarbonyl intermediate D to afford 3aa.
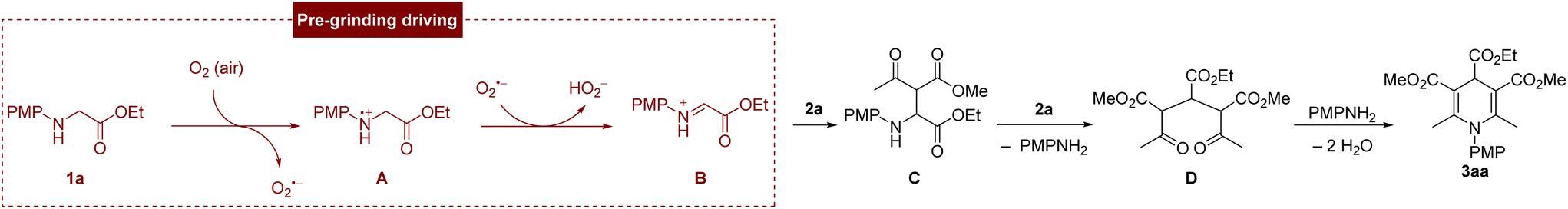 | ||
| Scheme 3 The proposed mechanism. | ||
In order to assess the green metrics of our mechanochemical AA process compared to more traditional cascade CDC approaches, the accessible green chemistry metrics such as environmental factor (E factor),27 reaction mass efficiency (RME),28 and power demand were quantified (detailed calculations see ESI†). As seen in Fig. 3A and B, our strategy gave low values of E factor (4.8 [1.1629],/5.3/6.1 kg kg−1 for 3aa/3ab/3lb), while the solution-based strategies gave much higher values of E factor (15.5–42.1/14.7–32.8/16.1–38.7 kg kg−1). These significant differences are mainly attributed to the excessive usage of solvents during the solution-based processes. While the higher RME of 17% (46%29)/15%/13% given by our AA strategy implied less waste generated. Notably, replacing the auxiliary silica gel with recoverable NaCl remarkably improved the environmental performance of the whole process (see Fig. 3 and ref. 29). Moreover, the power demand30 assessment (Fig. 3C) showed that the higher power demand of the literature methodologies compared with our strategy was mainly due to the heating and irradiation process. These results strongly display that the cascade CDC process driven by AA is an environmentally-friendly approach for the synthesis of 1,4-DHPs, even compared with modified Hantzsch processes31 (details see ESI, section 6.5†). Besides, the green metrics of the CDC reaction for the synthesis of 7ah were also evaluated to give similar results (details see ESI, section 6.6†).
 | ||
| Fig. 3 Comparison of (A) E factors (B) reaction mass efficiency and (C) power demands for the synthesis of 3aa between previous works and this work. | ||
Recently, Millipore Sigma's/Merck's Quantitative Green Chemistry Evaluator DOZN 2.0 has been applied to compare the relative greenness of similar chemicals, synthetic routes, and chemical processes, which utilizes the 12 Principles of Green Chemistry to calculate an aggregate score.32 The aggregate score is on a scale of 0–100 (0 being the most desired). The lower the score, the “greener” the procedure. Here, DOZN 2.0 was employed to evaluate different strategies for the synthesis of 1,4-dihydropyridine 3aa. As illustrated in Fig. 4, our work has the lowest DOZN 2.0 aggregate scores, indicating the greener synthesis of 3aa. The improved resource use group includes the green chemistry principles 1, 2, 7, 8, 9, and 11 (Table S4†), and the reduced human and environmental hazard group includes the principles 3, 4, 5, 10, and 12 (Table S4†) scores, the lowest values for our AA process, as there are no hazardous solvents and catalysts used during the process. Moreover, the lowest value of our increased energy efficiency group (principle 6, Table S4†) means the least energy is used in the production, which is mainly due to the avoidance of heating and solvent removal process as well as the employment of the ambient pressure. By these quantitative calculations, it is obvious that our strategy is the “greenest” one compared with previous strategies.
 | ||
| Fig. 4 (A) Comparisons of the greenness of a mechanochemical AA process (this work) versus previous solution-based processes and their respective aggregated scores assessed by the DOZN 2.0 tool for the preparation of 3aa. (B) Against the 12 Principles of Green Chemistry (indicated by #) and assessed by DOZN 2.0 for the preparation of 3aa. | ||
Conclusions
In conclusion, an operationally simple yet highly practical (cascade) CDC was developed. This protocol avoids the requirement for strong oxidants, (metal-based catalysts), bulk solvents, and continuous agitation conditions, enabling the formation of 1,4-DHPs and α-functionalized glycines from readily accessible starting materials. Mechanistic studies demonstrate that a brief mechanical pre-grinding of the reactants with silica gel can lead to an efficient pre-association of them, facilitating facile oxidation of glycine esters/amides by air to give an iminium intermediate and favouring the subsequent coupling reaction. Moreover, the recycling of NaCl as an alternative grinding auxiliary, the scaling-up studies, the synthetic application in drug-like molecules, and the multiform green metrics calculation demonstrate the practicality and greenness of the developed methodology, which will have a great impact on organic transformation, especially on solid–gas reactions in the near future.Author contributions
KY and JB suggested the idea; KY and P performed the experiments and the greenness evaluation. T modified the starting material synthesis and conducted data analysis. WK and JB supervised the project. Writing – review, and editing were conducted by KY, P, and JB. All authors have read and approved the final manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We gratefully acknowledge the National Natural Science Foundation of China (No. 21978270), Zhejiang Provincial Natural Science Foundation of China (No. LY23B060005) and National Key R&D Program of China 2021YFC2101000 for financial support.References
-
(a) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC
; (b) C. Bolm and J. G. Hernández, ChemSusChem, 2018, 11, 1410–1420 CrossRef CAS PubMed
-
(a) L. Konnert, F. Lamaty, J. Martinez and E. Colacino, J. Org. Chem., 2014, 79, 4008–4017 CrossRef CAS PubMed
- I. Huskić, C. B. Lennox and T. Friščić, Green Chem., 2020, 22, 5881–5901 RSC
- C. A. O'Keefe, C. Mottillo, J. Vainauskas, L. Fábián, T. Friščić and R. W. Schurko, Chem. Mater., 2020, 32, 4273–4281 CrossRef
-
(a) A. Moores, Curr. Opin. Green Sustainable Chem., 2018, 12, 33–37 CrossRef
- A. Jayasankar, D. J. Good and N. Rodríguez-Hornedo, Mol. Pharmaceutics, 2007, 4, 360–372 CrossRef CAS PubMed
- T. D. Nardo, C. Hadad, A. N. V. Nhien and A. Moores, Green Chem., 2019, 21, 3276–3285 RSC
-
(a) F. Toda, M. Yagi and K. Kiyoshige, J. Chem. Soc., Chem. Commun., 1988, 958–959 RSC
- M. Đud, O. V. Magdysyuk, D. Margetića and V. Štrukil, Green Chem., 2016, 18, 2666–2674 RSC
- L. Gonnet, A. Chamayou, C. André-Barrès, J. Micheau, B. Guidetti, T. Sato, M. Baron, M. Baltas and R. Calvet, ACS Sustainable Chem. Eng., 2021, 9, 4453–4462 CrossRef CAS
- W. Ma, Y. Liu, N. Yu and K. Yan, ACS Sustainable Chem. Eng., 2021, 9, 16092–16102 CrossRef CAS
-
(a)
L. Yet, Privileged Structures in Drug Discovery, John Wiley & Sons, Inc, Hoboken, NJ, USA, 2018, vol. 3, pp. 59–82 Search PubMed
-
(a) R. Lavilla, J. Chem. Soc., Perkin Trans. 1, 2002, 1141–1156 RSC
-
(a) C. Huang, H. Kang, J. Li and C. Li, J. Org. Chem., 2019, 84, 12705–12721 CrossRef CAS PubMed
-
(a) X. Jia, Y. Wang, F. Peng, C. Huo, L. Yu, J. Liu and X. Wang, Adv. Synth. Catal., 2014, 356, 1210–1216 CrossRef CAS
-
(a) G. Zhang, Y. Zhang and R. Wang, Angew. Chem., Int. Ed., 2011, 50, 10429–10432 CrossRef CAS PubMed
-
(a) J. Yu, C. Zhang, X. Yang and W. Su, Org. Biomol. Chem., 2019, 17, 4446–4451 RSC
-
Francisco G. Calvo-Flores, in Comprehensive Foodomics, ed. Alejandro Cifuentes, Elsevier, Oxford, 2021, vol. 2, pp. 690-709 Search PubMed
- K. Martina, F. Baricco, S. Tagliapietra, M. J. Moran, G. Cravotto and P. Cintas, New J. Chem., 2018, 42, 18881–18888 RSC
-
(a) G. A. Bowmaker, Chem. Commun., 2013, 49, 334–348 RSC
- J. Xie and Z. Huang, Angew. Chem., Int. Ed., 2010, 49, 10181–10185 CrossRef CAS PubMed
-
(a) R. Takahashi, T. Seo, K. Kubota and H. Ito, ACS Catal., 2021, 11, 14803–14810 CrossRef CAS
- L. Milkovic, T. Vukovic, N. Zarkovic, F. Tatzber, E. Bisenieks, Z. Kalme, I. Bruvere, Z. Ogle, J. Poikans, A. Velena and G. Duburs, Antioxidative, 2018, 7, 123 CrossRef PubMed
- C. Chen, F. Mentink-Vigier, J. Trébosc, I. Goldberga, P. Gaveau, E. Thomassot, D. Iuga, M. Smith, K. Chen, Z. Gan, N. Fabrègue, T. Métro, B. Alonso and D. Laurencin, Chem. – Eur. J., 2021, 27, 1–16 CrossRef
- C. Li, X. Wang, A. Yang, P. Chen, T. Zhao and F. Liu, ACS Omega, 2021, 6, 35389–35397 CrossRef CAS PubMed
- R. Zhao, J. Li, B. Huang and M. Cai, Catal. Lett., 2023, 153, 178–187 CrossRef CAS
- R. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC
- J. Andraos and M. Sayed, J. Chem. Educ., 2007, 84, 1004–1010 CrossRef CAS
- The calculation was based on the use of NaCl as grinding auxiliary with 5 times recycling and recovery. Details see ESI, section 6.1.†.
-
(a) F. Schneider, T. Szuppa, A. Stolle, B. Ondruschka and H. Hopf, Green Chem., 2009, 11, 1894–1899 RSC
-
(a) T. Reddy, G. Reddy, L. Reddy, C. Meda, K. Parsa, K. Kumar, Y. Lingappa and M. Pal, Eur. J. Med. Chem., 2013, 62, 395–404 CrossRef CAS PubMed
-
(a) P. Sharma, C. Vetter, E. Ponnusamy and E. Colacino, ACS Sustainable Chem. Eng., 2022, 10, 5110–5116 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, optimization data, mechanism study, green metric calculation, characterization data and NMR spectra. See DOI: https://doi.org/10.1039/d3gc00538k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |