
Modular construction of N-arylated amino acid esters enabled by a photoredox-catalyzed multicomponent reaction†
Qing
Li‡
,
Hanhan
Sun‡
,
Fengying
Yan
,
Yuanyuan
Zhao
,
Yicheng
Zhang
,
Chao
Zhou
,
Man-yi
Han
 ,
Hongji
Li
and
Xianwei
Sui
*
,
Hongji
Li
and
Xianwei
Sui
*
Key Laboratory of Green and Precise Synthetic Chemistry and Application, Ministry of Education, School of Chemistry and Materials Science, Huaibei Normal University, Huaibei, Anhui 235000, P. R. China. E-mail: suixw@chnu.edu.cn
First published on 27th July 2023
Abstract
An efficient photoredox-catalyzed three-component coupling reaction of aryl amine, glyoxalate and alkyltrifluoroborates has been reported. A variety of N-arylated amino esters were constructed rapidly using an organic photocatalyst under mild conditions. Broad substrate scope and good functional group tolerance are the key features of this strategy.
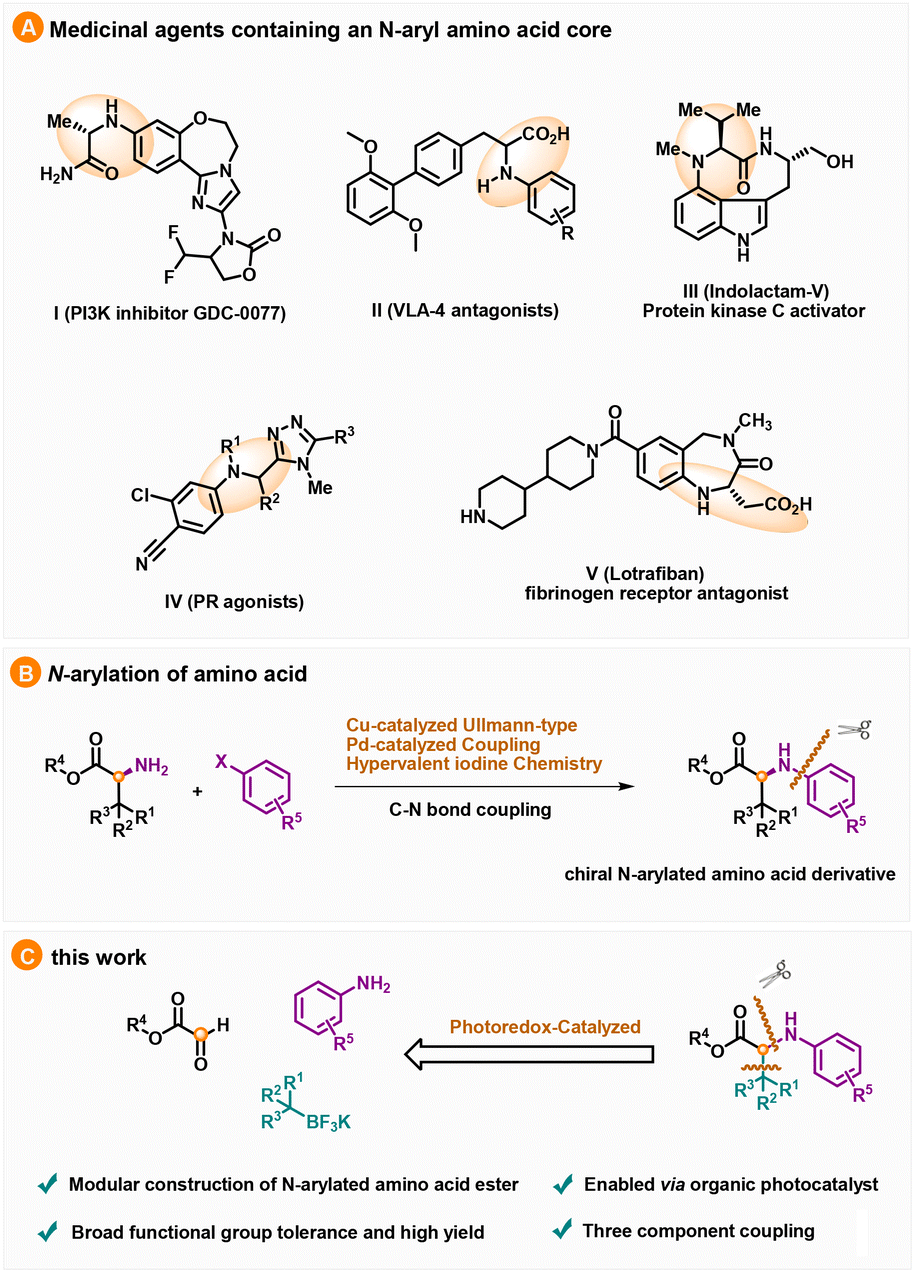
Amino acid derivatives can be employed as inexpensive building blocks in organic syntheses as well as in pharmaceuticals, agrochemicals and materials science.1 Moreover, the incorporation of functionalized amino acids in peptides and proteins allows the development of new strategies to study protein structures and functions and thus has made great advances in chemical biology.2 Among them, N-arylated α-amino acids and their esters are part of the core structures of many medicinally important agents, such as the PI3K inhibitor GDC-0077 (1),3 VLA-4 antagonists (2),4 protein kinase C (PKC) activator indolactam-V (3),5 PP agonists (4)6 and lotrafiban (5), which can act as a fibrinogen receptor antagonist (Scheme 1A).7
 | ||
| Scheme 1 Photoredox-catalyzed multicomponent reactions for the synthesis of N-arylated amino acid esters. | ||
Given the broad applications of N-arylated α-amino acids, much effort has been devoted to the development of an efficient method to access this framework. Particularly, the synthesis of enantioenriched N-arylated α-amino esters is mainly based on the direct arylation of the existing free amino group in chiral α-amino acid derivatives through Cu-catalyzed Ullmann-type N-arylation reactions,8 Pd-catalyzed coupling reactions,9 and hypervalent iodine chemistry,10 as well as other methodologies (Scheme 1B).11 Despite these advances, the development of diverse strategies based on new retro-synthesis analysis to prepare N-arylated α-amino esters is still highly desirable.
Photoredox-catalyzed reactions have drawn great attention owing to their high reaction selectivity, broad substrate tolerance and mild reaction conditions, and many significant motifs can be accessed easily from inexpensive feedstocks via a radical process.12 In 2016, the Molander group realized visible-light induced photoredox-catalyzed three-component coupling via an Ir photocatalyst, and a variety of secondary amines was delivered through the addition of alkyl radicals generated from alkyltrifluoroborates to aldimines formed in situ from aldehydes and anilines.13 Soon thereafter, the radical precursor scope of this step-economical multicomponent reaction was expanded to organo(tristrimethylsilyl)silanes,14 alkanes,15 alkyl iodides,16 alkyl carboxylic acids17 and 4-substituted-DHPs.18 Thanks to the great advances in boron compound preparation achieved in recent years,19 alkyltrifluoroborates have become readily available radical precursors that have broad substrate scope and sustainability. Given our interest in developing efficient and economical synthetic methodologies for amino acid derivatives, we envisioned that N-arylated amino esters could be constructed through a visible-light induced organic photocatalyst-mediated three-component reaction employing alkyltrifluoroborates, aryl amines and glyoxalate as substrates, which would serve as a step-economical transformation (Scheme 1C). Herein, we report a photoredox-catalyzed modular construction of racemic N-arylated amino acid esters catalyzed by the organic photocatalyst 9-mesityl-10-methylacridinium perchlorate, and alkyltrifluoroborates were employed as the radical precursor.
Our investigation commenced with ethyl glyoxalate (1), p-anisidine (2) and cyclopentyltrifluoroborate (3) as the model substrates for reaction condition optimization. After exploring various reaction parameters in the presence of a blue light-emitting diode (LED), we were delighted to find that the use of a catalytic combination of 9-mesityl-10-methylacridinium perchlorate (PC-I), Cu(MeCN)BF4, and p-tolylsulfonic acid (PTSA) delivered the desired N-arylated amino acid ester (4) in an 87% yield employing MeCN as the solvent at room temperature for 18 h (entry 1, Table 1). No desired product was formed when the reaction was performed in the dark and in the absence of the photocatalyst (entries 2 and 5, Table 1). The employment of dioxane instead of MeCN as the solvent furnished 4 in 25% yield while the use of DCM led to only trace desired products (entries 6 and 7, Table 1). Other photocatalysts such as Ir[dF(CF3)ppy]2(ppy)PF6 (PC-II), Ru(bpy)3Cl2 (PC-III) and 4-CZIPN were also investigated in our system only to find the aldimines as the major product by GC-MS (entries 8–10, Table 1). The yield of the N-arylated amino acid ester decreased to 55% when no PTSA was used in our reaction, probably owing to the slow aldimine formation rate (entry 4, Table 1). It should be noted that the copper catalyst was also necessary in the transformation, and only 39% yield of 4 was generated in the absence of Cu(MeCN)BF4, while 43% yield was obtained by using Cu(MeCN)PF6 as the copper source.
|
|
||
|---|---|---|
| Entry | Variation from the “standard conditions” | Yield (%) |
| a Reaction conditions: 2 (0.2 mmol), 1 (1.2 equiv.), 3 (1.4 equiv.), PC-I (5 mol%), Cu(MeCN)BF4 (5 mol%), and PTSA (5 mol%) in MeCN (2.0 mL) at room temperature under Ar and blue LED irradiation for 18 h. Isolated yields. PTSA = p-tolylsulfonic acid. | ||
| 1 | None | 87 |
| 2 | No PC-I | 0 |
| 3 | No Cu(MeCN)BF4 | 39 |
| 4 | No PTSA | 55 |
| 5 | No visible light | 0 |
| 6 | DCM instead of MeCN | 5 |
| 7 | Dioxane instead of MeCN | 25 |
| 8 | PC-II instead of PC-I | 3 |
| 9 | PC-III instead of PC-I | 0 |
| 10 | 4CZIPN instead of PC-I | 0 |
| 11 | Cu(MeCN)PF6 instead of Cu(MeCN)BF4 | 43 |
| 12 | 1.2 equiv. 3 instead of 1.4 equiv. 3 | 71 |

|
||

With the optimized conditions in hand, we then investigated the scope of aniline using 1 and 3 as the reaction partners. As illustrated in Table 2, both electron-withdrawing and electron-donating substituents worked well in this reaction providing N-arylated amino acid esters in moderate to excellent yields (4–19). Functional groups such as methoxyl, fluoride, methyl, t-butyl, amine, OCF3, trifluoromethyl, ester and amide groups at the para-position of aniline were all tolerated providing the corresponding products in 68–94% yields (4–15). It is worth noting that the chloride functionality was compatible with this reaction delivering 9 in 94% yield, which could be used for further modification. Anilines bearing ortho-substituents (12 and 13) or meta-substituents (14, 15, and 16) were suitable substrates producing the desired products in good yields. Moreover, the heteroaryl anilines gave the corresponding target compounds in 77%, 67% and 89% yields, respectively (17–19).
| a Reaction conditions: anilines (0.2 mmol), 1 (1.2 equiv.), 3 (1.4 equiv.), PC-I (5 mol%), PTSA (5 mol%), and Cu(MeCN)BF4 (5 mol%) in MeCN (2.0 mL) at room temperature under Ar and blue LED irradiation for 18 h. Isolated yields. |
|---|

|
We next proceeded to study the performance of various alkyltrifluoroborates (Table 3). The reaction was compatible with both secondary and tertiary alkyltrifluoroborates providing the products in 63–99% yields. Surprisingly, primary alkyltrifluoroborates, with a markedly higher oxidation potential (Ered1/2 = +1.90 V vs. SCE), worked well under the reaction conditions (20–25). The reaction of bromide and ester-containing substrates proceeded smoothly delivering 22 and 25 in 72% and 67% yield, respectively, providing ample opportunity for further elaborations. Moreover, functional groups like allylic, ether and amide groups were also tolerated in this transformation affording the N-arylated amino acid derivatives in 80–91% yields (23, 29 and 30). In addition, a derivative of gemfibrozil was prepared using this protocol (34). Benzyl glyoxalate was also tested under the standard conditions providing the corresponding amino acid ester in 88% yield. When methylglyoxal was used instead of glyoxalate as the substrate, the reaction worked well to afford the α-amino ketone derivative in good yield (36).
| a Reaction conditions: 2 (0.2 mmol), 1 (1.2 equiv.), alkyl trifluoroborate (1.4 equiv.), PC-I (5 mol%), PTSA (5 mol%), and Cu(MeCN)BF4 (5 mol%) in MeCN (2.0 mL) at room temperature under Ar and blue LED irradiation for 18 h. Isolated yields. |
|---|

|
A gram-scale reaction of 5.5 mmol of 1, 2 and 3 was performed to deliver 4 in 78% yield (Scheme 2A). The scalability of this method demonstrates great value in the practical synthesis of N-arylated amino acid esters. The synthetic utility of this reaction was highlighted by its use in the total synthesis of a VLA-4 antagonist (Scheme 2B). The first step was a Suzuki coupling using a procedure reported by the Cetinkaya group.20 Borylation of 37 occurred smoothly to form 38 in 66% yield, which was treated with KHF2 and used for the three-component coupling without further purification. N-Arylated amino ester 39 was delivered in 71% yield after two steps, and subsequent hydrolysis gave the target molecule 4 in 90% yield. In order to elucidate the reaction mechanism, 2,2,6,6-tetramethyl-1-piperidinoxyl (TEMPO) was added to the reaction system and the yield of 4 was completely inhibited, whereby the TEMPO-radical adduct was detected by HR-MS (Scheme 2C). In addition, the imine intermediate was isolated under different conditions for use in the radical addition step. It should be noted that both PTSA and copper can promote the formation of 38 and PTSA played a more important role compared to the copper catalyst; meanwhile, both PTSA and copper catalyst can promote either the radical addition step or the reduction of the N-centered radical cation, where copper seems more effective compared to PTSA (Scheme 2D). Based on the literature precedents,13–19 a plausible mechanism for the developed transformation was proposed (Scheme 2D). Initially, the acridium photocatalyst [Mes-Acr-Me]+(Acr+) is excited by visible light to a highly oxidizing excited state Acr+*. In this state, the photocatalyst can abstract an electron from the alkyltrifluoroborate via a single-electron transfer (SET) event to generate an alkyl radical while being reduced to the acridinium radical Acr˙. The alkyl radical undergoes addition to the in situ generated N-arylated imine to form an N-centered radical cation. Finally, the N-centered radical cation is reduced by the acridium radical, closing the photocatalytic cycle and furnishing the desired N-arylated amino ester product.
 | ||
| Scheme 2 Application and mechanistic studies. | ||
Conclusions
We have reported a photoredox-catalyzed three-component coupling of aryl amines, glyoxalate and alkyltrifluoroborates. A variety of N-arylated amino esters can be synthesized rapidly using an organic photocatalyst under mild conditions. This method has the advantage of broad substrate scope and good functional group tolerance and was highlighted in the synthesis of a VLA-4 antagonist.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Huaibei Normal University and the University Natural Science Research Project of Anhui Province of China (KJ2020A1199) are gratefully acknowledged.References
- A. F. M. Noisier and M. A. Brimble, Chem. Rev., 2014, 114, 8775 CrossRef CAS PubMed
.
-
(a) D. A. Dougherty, Curr. Opin. Chem. Biol., 2000, 4, 645 CrossRef CAS PubMed
- C. Han, S. M. Kelly, T. Cravillion, S. J. Savage, T. Nguyen and F. Gosselin, Tetrahedron, 2019, 75, 4351 CrossRef CAS
- G. A. Doherty, T. Kamenecka, E. McCauley, G. Van Riper, R. A. Mumford, S. Tong and W. K. Hagmann, Bioorg. Med. Chem. Lett., 2002, 12, 729 CrossRef CAS PubMed
-
(a) A. P. Kozikowski, S. Wang, D. Ma, J. Yao, S. Ahmad, R. I. Glazer, K. Bogi, P. Acs, S. Modarres, N. E. Lewin and P. M. Blumberg, J. Med. Chem., 1997, 40, 1316 CrossRef CAS PubMed
- M. Hammond, J. R. Patterson, S. Manns, T. H. Hoang, D. G. Washburn, W. Trizna, L. Glace, E. T. Grygielko, R. Nagilla, M. Nord, H. E. Fries, D. J. Minick, N. J. Laping, J. D. Bray and S. K. Thompson, Bioorg. Med. Chem. Lett., 2009, 19, 2637 CrossRef CAS PubMed
- W. H. Miller, T. W. Ku, F. E. Ali, W. al. E. Bondinell, R. R. Calvo, L. D. Davis, K. F. Erhard, L. B. Hall, W. F. Huffman, R. M. Keenan, C. Kwon, K. A. Newlander, S. T. Ross, J. M. Samanen, D. T. Takata and C.-K. Yuan, Tetrahedron Lett., 1995, 36, 9433 CrossRef CAS
-
(a) K. K. Sharma, S. Sharma, A. Kudwal and R. Jain, Org. Biomol. Chem., 2015, 13, 4637 RSC
-
(a) A. Dominguez-Huerta, I. Perepichka and C.-J. Li, Commun. Chem., 2018, 1, 45 CrossRef
-
(a) J. D. Mckerrow, J. M. A. Al-Rawi and P. Brooks, Synth. Commun., 2010, 40, 1161 CrossRef CAS
-
(a) H. Hammoud, M. Schmitt, E. Blaise, F. Bihel and J.-J. Bourguignon, J. Org. Chem., 2013, 78, 7930 CrossRef CAS PubMed
- For reviews, see:
(a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed
- J. Yi, S. O. Badir, R. Alam and G. A. Molander, Org. Lett., 2019, 21, 4853 CrossRef CAS PubMed
- X. Yu, C. G. Daniliuc, F. A. Alasmary and A. Studer, Angew. Chem., Int. Ed., 2021, 60, 23335 CrossRef CAS PubMed
-
(a) S. Pillitteri, P. Ranjan, L. G. Voskressensky, E. V. Van der Eycken and U. K. Sharma, Mol. Catal., 2021, 514, 111841 CrossRef CAS
- X. Wang, B. Zhu, J. Dong, H. Tian, Y. Liu, H. Song and Q. Wang, Chem. Commun., 2021, 57, 5028 RSC
- M. Xu, Y. Hua, X. Fu and J. Liu, Chem. – Eur. J., 2022, 28, e202104394 CrossRef CAS PubMed
- Z. Yao, J. Yang, Z. Luo, H. Wang, X. Zhang, J. Ye, L. Xu and Q. Shi, Green Chem., 2022, 24, 7968 RSC
-
(a) M. W. Campbell, J. S. Compton, C. B. Kelly and G. A. Molander, J. Am. Chem. Soc., 2019, 141, 20069 CrossRef CAS PubMed
- N. Gurbuz, I. Ozdemir, S. Demir and B. Cetinkaya, J. Mol. Catal. A: Chem., 2004, 209, 23 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01633a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |