
A cell-free artificial anabolic pathway for direct conversion of CO2 to ethanol†
Wanrong
Dong‡
ab,
Xiuling
Ji‡
a,
Yuhong
Huang
 *a,
Yaju
Xue
a,
Boxia
Guo
a,
Dongbo
Cai
b,
Shouwen
Chen
*b and
Suojiang
Zhang
*a
*a,
Yaju
Xue
a,
Boxia
Guo
a,
Dongbo
Cai
b,
Shouwen
Chen
*b and
Suojiang
Zhang
*a
aBeijing Key Laboratory of Ionic Liquids Clean Process, CAS Key Laboratory of Green Process and Engineering, State Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China. E-mail: yhhuang@ipe.ac.cn; sjzhang@ipe.ac.cn
bState Key Laboratory of Biocatalysis and Enzyme Engineering, Environmental Microbial Technology Center of Hubei Province, College of Life Sciences, Hubei University, Wuhan 430062, China. E-mail: chenshouwen@hubu.edu.cn
First published on 17th October 2023
Abstract
Biological CO2 activation and conversion to high-value ethanol are a feasible and green strategy to close the carbon cycle. However, naturally evolved CO2 utilization pathways involve carbon loss or ATP consumption. Herein, we report a complete anabolic pathway for direct conversion of CO2 to ethanol by constructing and assembling three functional modules including CO2 activation, formaldehyde → acetyl-CoA, and ethanol synthesis in a carbon-conserved and ATP-independent system. These artificial ethanol metabolic pathway fluxes were strengthened by screening efficient key enzymes for CO2 activation, promoting formaldehyde assimilation, and employing a two-step reaction. 13C-labeled CO2 demonstrated the feasibility of the pathway converting CO2 into ethanol in vitro by detecting the carbon flow. With this anabolic pathway, we obtained an ethanol concentration of 0.37 mM at a conversion rate of 4.33 nmol CO2 min−1 mg−1 enzyme and 0.5 mM R5P supply. This modularization strategy provides a new avenue in the construction of artificial metabolic pathways for ethanol synthesis from CO2.
Biological activation and conversion of CO2 into high-value ethanol are a win–win strategy towards both controlling CO2 emissions and easing energy crisis. Various natural CO2 utilization pathways have been identified, including the Wood–Ljungdahl (WL) pathway, the 3-HP bicycle, the 3-hydroxypropionate/4-hydroxybutyrate (HP/HB) cycle, the dicarboxylate/4-hydroxybutyrate (DC/HB) cycle, the reductive glycine pathway, and the reductive tricarboxylic acid (TCA) cycle.1 These pathways fix CO2 or its bicarbonate carrier (HCO3−) to produce the ethanol precursor acetyl coenzyme A (acetyl-CoA) via acetyl-CoA synthase (ACS)-mediated condensation of CO2, thiamine diphosphate (ThDP)-mediated umpolung activation, pyruvate dehydrogenase-mediated pyruvate decarboxylation, or reversal of the oxidative TCA cycle.2 However, there is no biosynthetic pathway precedent for the direct conversion of CO2 to ethanol and redesigning the natural metabolic pathways to accommodate such CO2 utilization is still challenging owing to the inherent limitations of carbon loss, ATP requirement or oxygen sensitivity.
In an effort to convert CO2 to ethanol in a fully carbon-conserved and/or ATP-independent manner, recent research studies have first shifted focus to the construction of artificial anabolic pathways for the precursor acetyl-CoA.3 For example, the methanol condensation cycle (MCC) condensed two molecules of formaldehyde into one acetyl-CoA without carbon loss, ATP requirement and anaerobic conditions by the combination of the ribulose monophosphate pathway with the nonoxidative glycolysis pathway.4 Similarly, the synthetic acetyl-CoA (SACA) pathway achieved the carbon-conserved, ATP-independent and oxygen-insensitive synthesis of acetyl-CoA by combining ThDP-dependent glycolaldehyde synthase design and pathway construction.5 Thanks to these efforts, we can achieve a complete anabolic pathway for ethanol synthesis from CO2 by including enzymes from CO2 to formaldehyde and from acetyl-CoA to ethanol to extend the MCC. However, the equilibrium direction of multi-enzyme reaction remains difficult to control.6 All reactions are reversible except the MCC, and in particular, the CO2 activation to formaldehyde is thermodynamically unfavorable due to the low solubility of CO2 and the inefficiency of activation enzymes.7 Thus, the net concentration of ethanol is primarily a matter of maintaining the formaldehyde intermediate converted from CO2 at a relatively high concentration.
To address this, we introduced a modularization strategy based on the enzymatic activation of CO2, formaldehyde assimilation into acetyl-CoA, and ethanol synthesis modules to regulate the metabolic fluxes for direct conversion of CO2 to ethanol. The efficient enzymatic CO2 activation to formaldehyde was achieved by the high substrate binding affinity and catalytic efficiency of both formate dehydrogenase mutant (ΔPaFDH48)8 and formaldehyde dehydrogenase (SzFaldDH)9 from Paracoccus sp. MKU1 and Streptomyces zinciresistens, thereby providing an attractive starting point for the artificial CO2 utilization pathway. This activation module can ensure sufficient formaldehyde concentrations for further conversion during the catalytic process, allowing the construction of a complete biosynthetic pathway using CO2 as a feedstock. By assembling three functional modules, we first constructed a carbon-conserved and ATP-independent artificial ethanol pathway from CO2, proposed as CO2 To Ethanol (CTE) (Scheme 1). 13C-labeled CO2 and gas chromatography-tandem mass spectrometry (GC-MS) revealed that modular optimization can strengthen the metabolic flux of the CTE pathway, resulting in an ethanol concentration of 0.37 mM at a conversion rate of 4.33 nmol CO2 min−1 mg−1 enzymes and 0.5 mM R5P supply.
 | ||
| Scheme 1 Construction and modular assembly of a complete anabolic pathway for direct conversion of CO2 to ethanol. | ||
To determine the thermodynamic and kinetic feasibility of the CTE pathway constructed from CO2 activation, formaldehyde assimilation, and ethanol synthesis modules, we first calculated its Gibbs energy change for each reaction and max–min driving force (MDF) during the conversion of CO2 to ethanol. The entire reaction from CO2 to ethanol was thermodynamically favorable with a total Gibbs energy change (ΔrG′m) of −25.8 kJ mol−1, except for the initial CO2 activation, which had a thermodynamically unfavorable cascade reaction (Fig. 1). The large negative values of ΔrG′m for hexulose-6-phosphate synthase (Hps), phosphohexulose isomerase (Phi) and phosphoketolase (Fpk) (−9.7, −10.5, and −53.5 kJ mol−1) in the formaldehyde assimilation module provided important driving forces for the final formation of ethanol. Meanwhile, the CTE pathway also achieved a positive MDF value of 0.21 kJ mol−1 at allowable reduced nicotinamide adenine dinucleotide (NADH) concentrations (Fig. S1†), further indicating its feasibility of thermodynamic operation. Combining the results of ΔrG′m and MDF, we concluded that the CTE pathway was theoretically functional for the biosynthesis of ethanol from CO2in vitro.
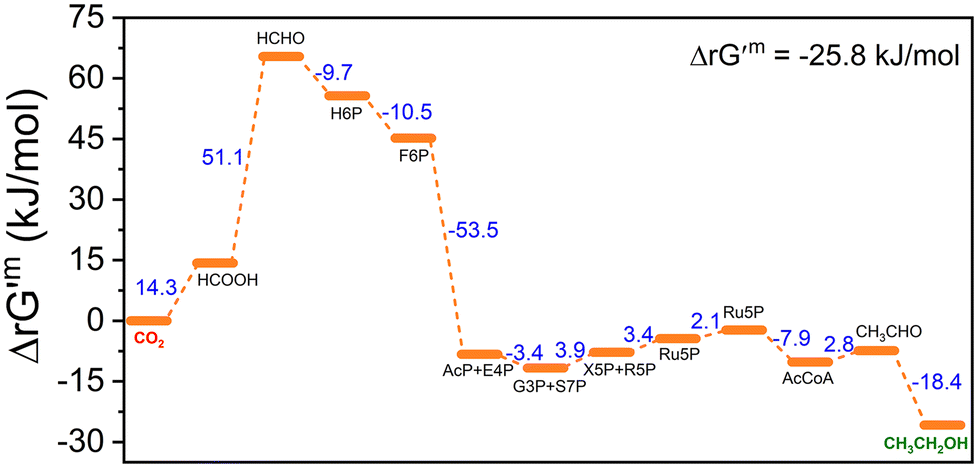 | ||
| Fig. 1 Thermodynamic data of CTE. ΔrG′m: the Gibbs energy change in a physiological standard with a 1 mM concentration. | ||
Since the bioactivation of CO2 to formaldehyde is thermodynamically unfavorable, there exists no complete biosynthetic pathway for multicarbon molecules from CO2.10 The high reductive activity of FDH and FaldDH in the activation module is a key factor in supplying sufficient formaldehyde for reactions of the other two modules. However, commercially available dehydrogenases yielded only 0.013 mM formaldehyde after 5 h, which was far from efficient activation.11 We thus constructed four alternative CO2 activation modules using PaFDH or its mutant ΔPaFDH48 and SzFaldDH or the previously reported efficient FaldDH from Burkholderia multivorans (BmFaldDH) in tandem7,12 (Fig. 2A and B). After expression and purification, the reductive activity of all candidate activation assays was investigated using CO2 as the substrate. The activation modules containing ΔPaFDH48 displayed higher reductive activities compared to PaFDH and commercial FDH from C. boidinii (CbFDH). A significant increase in the reductive activity was only observed in the activation module containing ΔPaFDH48 and SzFaldDH acting in tandem, which was 10.68-fold higher than that achieved by the commercial dehydrogenase activation module. Thus, efficient ΔPaFDH48 and SzFaldDH were chosen to construct the CO2 activation module.
 | ||
| Fig. 2 Resolving main bottlenecks in the ethanol synthesis from CO2. (A) Pathway for CO2 activation assays. (B) Comparison of the relative activity of FDH and FaldDH acting in tandem during CO2 activation at 25 °C for 1 h. (C) Pathway for Hps activity assays. (D) Screening highly active Hps from different strains. (E) Concentration optimization of Fpk to avoid the kinetic trap. (F) The synthesis of ethanol from formaldehyde by assembling formaldehyde assimilation and ethanol synthesis modules. | ||
The formaldehyde assimilation into acetyl-CoA and ethanol synthesis modules was further established by integrating the previously reported MCC4a and the aldehyde dehydrogenase (AlDH) and alcohol dehydrogenase (ADH) to reduce acetyl-CoA.13 With the exception of commercial ADH, the catalytic activities of all corresponding enzymes in both modules were demonstrated in individual assays after gene synthesis and enzyme expression and purification (Fig. S2–S7†). As the initial step in the MCC, Hps irreversibly catalyzed formaldehyde and ribulose-5 phosphate (Ru5P) to hexulose-6-phosphate (H6P), which realized the formaldehyde assimilation and in turn provided the driving force for CO2 activation.14 To find highly active Hps, we purified enzymes from three different strains including Bacillus subtilis, Methylococcus capsulatus and Bacillus methanolicus (BsHps, McHps, and BmHps)4a,15 (Fig. 2C). The relative activity of McHps has been shown to be the highest compared to those of BmHps and the inactive BsHps, as determined by measuring generated NADPH at 340 nm (Fig. 2D). However, its low enzyme activity under the optimized buffer conditions still led to small amounts of formaldehyde assimilation. Meanwhile, we discovered and screened two Hps using peptide pattern recognition (PPR).16 Unfortunately, both purified enzymes from Methylocaldum marinum demonstrated relatively lower activity than McHps (Fig. S8†). Taken together, McHps is chosen to accomplish formaldehyde assimilation into H6P, which is isomerized to fructose-6-phosphate (F6P) by Phi. F6P can be cleaved by Fpk to acetylphosphate (AcP) and erythrose 4-phosphate (E4P). Subsequently, the generated AcP and E4P were converted to acetyl-CoA by phosphate acetyltransferase (Pta) and regenerated Ru5P via a carbon rearrangement, respectively. During this process, the Fpk concentration was optimized because the inherent kinetic trap of Fpk would inevitably accumulate E4P at the expense of decreasing AcP yield.4a As the Fpk concentration increased, the ethanol yield in the system initially increased and then decreased (Fig. 2E). The maximum yield of ethanol reached 2.51 mM, when the Fpk concentration was 160 μg mL−1. Based on this formaldehyde assimilation module, we further added another two known dehydrogenases, AlDH and ADH, which would convert acetyl-CoA into ethanol. Consequently, 1.83 mM ethanol was produced at a formaldehyde concentration of 6.0 mM (Fig. 2F). Our results indicated the feasibility of the ethanol synthesis from formaldehyde by assembling formaldehyde assimilation and ethanol synthesis modules in vitro.
On establishing individual modules for the conversion of CO2 into ethanol, we constructed the entire pathway by assembling three modules and achieved a detectable ethanol concentration of 0.10 mM. Since CO2 and initially added 0.5 mM ribose 5-phosphate (R5P) were employed as feedstock, 13C-labeled experiments to detect the carbon flux were thus performed to verify the actual source of carbon in ethanol. By comparing with “unlabeled” controls, we tested the ethanol synthesis from the CTE pathway using 13C-labeled CO2 and HCO3−, respectively. GC-MS showed that the main fragmentation patterns (i.e. [M − 1]+ and [M]+) of ethanol (molecular weight = 46) achieved in the control group were 45 and 46 ions, corresponding to unlabeled carbon in ethanol (Fig. 3A and S9A†). In contrast, another 47 ions were detected by 13C-labeled CO2 along with 45 and 46 ions, while both 47 and 48 ions were detected by 13C-labeled HCO3− (Fig. 3B and S9B†). The emerging ions were designated as single labeled [2-13C]-ethanol and fully labeled [1,2-13C]-ethanol. These undetectable 48 ions from 13C-labeled CO2 led to speculation that the low formaldehyde concentration from the CO2 activation module may not be able to supply multiple passes of CO2 conversion. Our results confirmed that some of the carbons in ethanol came from CO2, verifying the full functionality of ethanol synthesis from CO2 in CTE.
 | ||
| Fig. 3 13C-labeled CO2 analysis in the CTE pathway. (A) Mass spectrum of ethanol with unlabeled CO2via the CTE pathway. (B) Mass spectrum of ethanol with 13C-labeled CO2via the CTE pathway. (C) Schematic illustration of 13C tracing from 13C-labeled CO2 to ethanol. | ||
We further proposed a carbon flux network to understand the metabolic logic of the CTE pathway. As shown in Fig. 3C, the 13C-labeled CO2 was first converted to 13C-labeled formaldehyde through a cascade reaction. Then the resulting 13C-labeled formaldehyde and Ru5P, which was isomerized from the initial added R5P, entered the formaldehyde assimilation reaction and successively produced 13C-labeled H6P and F6P by Hps and Phi. Finally, 13C-labeled F6P was cleaved to E4P and 13C-labeled AcP, which was further used to produce single labeled [2-13C]-ethanol (black line). Meanwhile, 13C-labeled F6P and E4P were used to regenerate unlabeled R5P and synthesize 13C-labeled xylulose 5-phosphate (X5P), which was then isomerized to 13C-labeled Ru5P. Similarly, the 13C-labeled Ru5P and the newly generated 13C-labeled formaldehyde would also enter the following formaldehyde assimilation and ethanol synthesis reactions to produce fully labeled [1,2-13C]-ethanol (orange lines). In addition, some fully labeled [1,2-13C]-ethanol can be formed from 13C-labeled formaldehyde and double 13C-labeled Ru5P produced from multiple passes (violet line). Therefore, we concluded that a single pass of CO2 conversion in CTE mainly synthesized single labeled [2-13C]-ethanol, while [1,2-13C]-ethanol can be synthesized by the assistance of the Ru5P regeneration cycle.
After validating the full function for ethanol synthesis from CO2 in CTE, we proceeded towards improving this pathway by addressing the bottleneck of the inadequate supply of formaldehyde. Due to the unfavorably reversible cascade reaction conditions for CO2 activation, we met the need for relatively high formaldehyde concentrations in the formaldehyde assimilation module with a two-step enzymatic cascade reaction in CTE. In the first hour, CO2 was first activated to formaldehyde by ΔPaFDH48 and SzFaldDH in tandem, and the resulting formaldehyde with the precursor Ru5P was irreversibly converted to H6P and then to F6P by Hps and Phi. In the following 1 h, the accumulated F6P was further cleaved to AcP and E4P by supplementing the remaining 7 core enzymes and CoA, which were used to produce ethanol and for carbon rearrangement, respectively (Fig. 4A). The final concentration of ethanol reached 0.30 mM, which was almost 3-fold higher than that achieved using the one-pot strategy (0.10 mM) (Fig. 4B). Such yield improvement demonstrates the effectiveness of the two-step reaction.
 | ||
| Fig. 4 Ethanol synthesis from CO2via the CTE pathway. (A) Two-step strategy for ethanol synthesis from CO2. (B) Ethanol concentration via the two-step reaction. (C) Time profile of ethanol synthesis from CO2 over 2.5 h with the first 1 h of F6P accumulation. | ||
With the successful improvement of the CTE pathway with the two-step reaction, we intended to convert CO2 into ethanol over a 2.5 h time course. Starting with 12 mL min−1 of CO2 and 1 h of F6P accumulation, the CTE pathway using ΔPaFDH48 and SzFaldDH achieved the highest ethanol concentration of 0.37 mM in the following 30 min (Fig. 4C). The ethanol synthesis rate reached 4.33 nmol CO2 min−1 mg−1 enzymes, which is 1.37-fold higher than those of PaFDH and BmFaldDH. After 30 min, the ethanol concentration decreased slightly, which may be due to the instability of intermediates like acetyl-CoA and AcP.4a Our result demonstrated the feasibility of continuous ethanol synthesis from CO2 in the CTE pathway.
Conclusions
In summary, a carbon-conserved and ATP-independent anabolic pathway for direct conversion of CO2 to ethanol was presented. Thanks to the modularization strategy based on the enzymatic activation of CO2, formaldehyde assimilation into acetyl-CoA, and ethanol synthesis modules, this complete biosynthetic pathway achieves not only the efficient activation of CO2 into formaldehyde but also the carbon rearrangement with high carbon and energy efficiency from formaldehyde. As a result, CO2 was converted into ethanol with a concentration of 0.37 mM at a conversion rate of 4.33 nmol CO2 min−1 mg−1 enzymes. Our study presents a green and attractive avenue for the direct conversion of CO2 into versatile biofuels, providing a feasible strategy and valuable inspiration for constructing a complete biosynthetic pathway from CO2.Author contributions
Y. Huang, S. Chen and S. Zhang supervised the project. Y. Huang conceived the project and performed the PPR prediction. W. Dong performed the experimental work. X. Ji discussed and wrote the manuscript. Y. Xue and B. Guo worked on the purification and protein quantification of key enzymes for CO2 activation. D. Cai discussed the manuscript. X. Ji, W. Dong and Y. Huang revised the manuscript with support from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key Research and Development Program (2021YFC2104200), the “Transformational Technologies for Clean Energy and Demonstration” Strategic Priority Research Program of the Chinese Academy of Sciences (XDA21000000), and the CAS Project for Young Scientists in Basic Research (YSBR-072). This work was also partially supported by the Open Funding Project of the State Key Laboratory of Biocatalysis and Enzyme Engineering (SKLBEE2021001).References
-
(a) Z. Liu, K. Wang, Y. Chen, T. Tan and J. Nielsen, Nat. Catal., 2020, 3, 274–288 CrossRef CAS
; (b) M. Kopke, C. Held, S. Hujer, H. Liesegang, A. Wiezer, A. Wollherr, A. Ehrenreich, W. Liebl, G. Gottschalk and P. Durre, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13087–13092 CrossRef CAS PubMed
- S. Bierbaumer, M. Nattermann, L. Schulz, R. Zschoche, T. J. Erb, C. K. Winkler, M. Tinzl and S. M. Glueck, Chem. Rev., 2023, 123, 5702–5754 CrossRef CAS PubMed
-
(a) I. W. Bogorad, T. S. Lin and J. C. Liao, Nature, 2013, 502, 693–697 CrossRef CAS PubMed
-
(a) I. W. Bogorad, C. T. Chen, M. K. Theisen, T. Y. Wu, A. R. Schlenz, A. T. Lam and J. C. Liao, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15928–15933 CrossRef CAS PubMed
- X. Lu, Y. Liu, Y. Yang, S. Wang, Q. Wang, X. Wang, Z. Yan, J. Cheng, C. Liu, X. Yang, H. Luo, S. Yang, J. Gou, L. Ye, L. Lu, Z. Zhang, Y. Guo, Y. Nie, J. Lin, S. Li, C. Tian, T. Cai, B. Zhuo, H. Ma, W. Wang, Y. Ma, Y. Liu, Y. Li and H. Jiang, Nat. Commun., 2019, 10, 1378 CrossRef PubMed
- W. Liu, Y. Hou, B. Hou and Z. Zhao, Chin. J. Chem. Eng., 2014, 22, 1328–1332 CrossRef CAS
- X. Ji, Y. Xue, Z. Li, Y. Liu, L. Liu, P. K. Busk, L. Lange, Y. Huang and S. Zhang, Green Chem., 2021, 23, 6990–7000 RSC
-
Y. Huang, Y. Xue, X. Ji and S. Zhang, CN2022115986319, 2022
-
Y. Huang, X. Ji, Y. Xue, B. Guo and S. Zhang, CN2022115994885, 2022
-
(a) T. Zheng, M. Zhang, L. Wu, S. Guo, X. Liu, J. Zhao, W. Xue, J. Li, C. Liu, X. Li, Q. Jiang, J. Bao, J. Zeng, T. Yu and C. Xia, Nat. Catal., 2022, 5, 388–396 CrossRef CAS
- M. Guo, F. Gu, L. Meng, Q. Liao, Z. Meng and W. Liu, Sep. Purif. Technol., 2022, 286, 120480 CrossRef CAS
- R. K. Singh, R. Singh, D. Sivakumar, S. Kondaveeti, T. Kim, J. Li, B. H. Sung, B.-K. Cho, D. R. Kim, S. C. Kim, V. C. Kalia, Y.-H. P. J. Zhang, H. Zhao, Y. C. Kang and J.-K. Lee, ACS Catal., 2018, 8, 11085–11093 CrossRef CAS
- Q. Wang, C. Sha, H. Wang, K. Ma, J. Wiegle, A. E. Abomohra and W. Shao, Sci. Rep., 2021, 11, 1050 CrossRef CAS PubMed
- I. Orita, N. Sakamoto, N. Kato, H. Yurimoto and Y. Sakai, Appl. Microbiol. Biotechnol., 2007, 76, 439–445 CrossRef CAS PubMed
- J. E. N. Muller, F. Meyer, B. Litsanov, P. Kiefer, E. Potthoff, S. Heux, W. J. Quax, V. F. Wendisch, T. Brautaset, J. C. Portais and J. A. Vorholt, Metab. Eng., 2015, 28, 190–201 CrossRef CAS PubMed
-
(a) P. K. Busk and L. Lange, Appl. Environ. Microbiol., 2013, 79, 3380–3391 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03159d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |