
Molecularly or atomically precise nanostructures for bio-applications: how far have we come?
Jie
Wang†
 a,
Ping
Li†
b,
Chao
Wang
*a,
Ning
Liu
*a and
Dongming
Xing
*a
a,
Ping
Li†
b,
Chao
Wang
*a,
Ning
Liu
*a and
Dongming
Xing
*a
aThe Affiliated Hospital of Qingdao University, Qingdao University, Qingdao 266071, China. E-mail: wangchao20086925@126.com; leirum_ln@qdu.edu.cn; xdm@qdu.edu.cn
bSchool of Rehabilitation Sciences and Engineering, University of Health and Rehabilitation Sciences, Qingdao 266071, China
First published on 9th June 2023
Abstract
A huge variety of nanostructures are promising for biomedical applications, but only a few have been practically applied. Among the various reasons, the limited structural preciseness is a critical one, as it increases the difficulty in product quality control, accurate dosing, and ensuring the repeatability of material performance. Constructing nanoparticles with molecule-like preciseness is becoming a new research field. In this review, we focus on the artificial nanomaterials that can currently be molecularly or atomically precise, including DNA nanostructures, some metallic nanoclusters, dendrimer nanoparticles and carbon nanostructures, describing their syntheses, bio-applications and limitations, in view of up-to-date studies. A perspective on their potential for clinical translation is also given. This review is expected to provide a particular rationale for the future design of nanomedicines.
Wider impactNanostructures with a molecular or atomic structural preciseness distinguish themselves from common nanomaterials. This feature will enable the comparison of material performance across different labs and manufacturers, produce highly repeatable efficacies and predictable side effects, and thereby help to realize the practical bio-application of nanomaterials. It is increasingly recognized that relatively poor structural preciseness is an important reason why the clinical translation of nanomedicines has been outmatched by small molecules and biomacromolecules. Numerous efforts have been devoted to this field, while a precise atomic/molecular arrangement is still difficult to achieve for a nanoparticle containing tens of thousands of atoms, due to the structural complexity and probably the intrinsic limitations in synthesis and characterization techniques. Fortunately, a new dawn has appeared in recent years. Here we summarize the recent advances in this research direction, with a particular emphasis on the advancement in synthesis techniques. Four kinds of nanostructures are at the forefront, each with its own limitations, and insights into the required optimizations for future clinical translation are given. |
1. Introduction
Nanotechnology has been flourishing for decades and attracting tremendous efforts to realize its bio-applications. Nanoformulations have been demonstrated preclinically and clinically to possess exceptional advantages over free molecules, including reduced toxicity and improved therapeutic efficiency, but most of the approved or clinically tested drugs are still small molecules or biomarcomolecules. Nano-drugs approved by the Food and Drug Administration (FDA) are mounting, while the majority of them are based on liposomes or the conjugation of polymers.1 Liposomes are relatively special given the extremely high biocompatibility and the versatile loading pattern for cargos;2 the conjugation of polymers such as polyethylene glycol (PEG) produces nano-sized formulations but is actually a molecule-based chemical modification method.3 Natural nanostructures, such as proteins, cell membranes and virus, have achieved great success in clinics and labs, such as the FDA-approved albumin-bound paclitaxel,4 the extensive exploration of ferritin5 and cell membrane-derived vesicles6 as drug carriers, and the use of viral vectors for vaccination, drug delivery and bio-imaging.7 Nevertheless, employing natural nanostructures may face some challenges including difficulty in large scale production, limited modifiability and potential immunogenicity. Researchers are devoting great efforts to push the clinical translation of the various artificial nanoformulations in the laboratory, which are in much greater variety than those used in clinical settings.The reasons why nanoformulations cannot match small molecules in clinical translation include a failure to bring improved efficacy or reduced side effects,8 as well as toxicity-related concerns.9 Nevertheless, another important reason is being increasingly recognized, which is the lower structural preciseness of nanoformulations compared to small molecules which have defined structures.10 Nanoformulations that contain multiple ingredients in one particle have been constructed in many studies, raising challenges in practical applications despite the encouraging treatment outcomes; reducing the number of ingredients is helpful, but the particle-to-particle variety of each ingredient is a trickier issue to address.10 Structure can be highly heterogeneous for a single-component nanomaterial. High structural heterogeneity not only increases batch-to-batch variety but also makes it difficult to study the exact pharmacodynamics and the molecular pathways of drug-body interactions. For example, multiple studies have demonstrated that the drug-to-particle stoichiometry, inter-drug spacing and surface ligand orientation, which are difficult to control for traditional nanostructures, all have a great influence on therapeutic efficiency.11,12 It is also difficult to compare the efficacy of nanostructures of “one-kind but from different labs”, thereby leading to different or even contradictory conclusions. In comparison, molecule-like structural preciseness will facilitate product quality control, yield predictable side effects, enable robust diagnostic and therapeutic performances and realize accurate dosing using a measured amount of product (Fig. 1). In this context, atomically or molecularly precise nanostructures are attracting great interest. Researchers are putting efforts toward fabricating nanomaterials with a defined number and arrangement of atoms/molecules within, by developing new synthesis routes, exploiting the highly specific recognition between chemical (e.g., bonds for click reactions)13 or biochemical (e.g., nucleic acids, biotin and avidin)14 entities, etc.
 | ||
| Fig. 1 Illustration of the importance of structural preciseness of nanomaterials for their biomedical applications. | ||
DNA nanostructures, some metallic nanoclusters, dendrimer nanoparticles and carbon nanostructures are currently typical nanostructures that can be molecularly or atomically precise. Each has its own way to achieve the high preciseness, and synthetic breakthroughs are the key to success. Therefore, the synthesis routes are fully described in this review. The advantages and limitations of each nanostructure are analyzed with particular attention on the ease of controlling structural preciseness, the facileness for synthesis and the biocompatibility. A perspective on their potential clinical translation is also given. Note that all the introduced are artificial nanostructures, while the biomacromolecule- and cell membrane-based ones are not included since they intrinsically have relatively high structural preciseness and are biomedically promising even regardless of the degree of preciseness.
2. DNA nanostructures
2.1. Origin and basic principles
DNA nanotechnology has been explored extensively.15 As a natural genetic material, DNA follows a strict A–T and G–C pairing law to form Watson–Crick helical double strands that are about 2 nm in diameter and 3.4 nm per helical turn. This provides a highly predictable intermolecular interaction and makes DNA strands natural building blocks for on-demand assembly.15,16 Seeman et al. demonstrated in the early 1980s that by breaking the sequence symmetry of Holliday junctions, one could lock the mobile junction to obtain an immobile structure.17,18 The immobile Holliday junctions could be considered as a crossover between two DNA duplexes, while stronger junctions such as double crossover,19 triple crossover,20 and six-helix bundles21 have been subsequently constructed by connecting multiple duplexes. By repeatedly assembling these junctions via the pairing of cohesive ends, DNA nanostructures will be constructed (Fig. 2a). 3D nanostructures are readily available because the duplexes in the immobile junctions can occupy unparallel planes.22 This immobile junction-based assembly forms the initial fundamental structure for the synthesis of DNA nanostructures and has inspired various synthesis techniques (Fig. 2). A large variety of DNA nanostructures have emerged with precisely controlled size and structure. Favored by the specific recognition between A–T and C–G bases, the inter-particle variation will be limited, probably at a level relevant to the occurrence rate of gene mutation for the simple DNA nanostructures. Moreover, DNA itself can be functional (e.g., therapeutic, diagnostic or recognizable by enzymes) by virtue of a specific nucleotide sequence,23 which further makes DNA nanostructures biomedically promising. | ||
| Fig. 2 Typical techniques for DNA nanostructure synthesis. (a) Oligonucleotide tile-based assembly, (b) DNA origami technique, and (c) single-stranded assembly. | ||
2.2. Synthesis methods
Seeman's method22 is a tile-based bottom-up synthesis. Four-arm immobile junctions yield 2D structures, while 3D DNA structures have been built by creating six-arm junctions. For example, Mao et al. used a tile-based synthesis for simplified preparation of DNA polyhedra, by using many copies of identical units for hierarchical self-assembly.24 The formed three-point-star tiles served as vertices to enable the formation of 3D structures; by controlling the flexibility and concentration of the tiles, DNA tetrahedra, dodecahedra and buckyballs were obtained. Tile-based synthesis was the major method during the early years of DNA nanotechnology in virtue of its powerfulness and simplicity.25,26 Using a number of identical tiles, nanostructures can be obtained after a one-pot annealing treatment. The main drawbacks of this method include the need for high-purity oligonucleotide strands for the formation of tiles, as well as the difficulty in designing the strand sequence.Nowadays multiple strategies are available, including origami assembly, single-stranded assembly, nanomaterial-templated preparation, etc. Origami assembly, which involves the folding of a long single-stranded DNA with the help of short ‘staple strands’ (Fig. 2b), is a powerful alternative to the tile-based bottom-up synthesis.29 Its advantages include: (i) the design and synthesis are convenient without the need for sequence optimization and strand purification, and the DNA origami can even be obtained with unpurified staple strands;15 (ii) the feasibility to obtain uniform products with relatively large size, generally about one order of magnitude larger in size than that obtained from tile-based bottom-up synthesis. Origami assembly readily produces 3D structures, as first demonstrated by Shih et al.30,31 By building staple crossovers to bridge different origami layers, 3D structures formed upon the stacking of the bridged layers, and advanced hierarchical assembly consisting of multiple stacked 3D structures were also obtained.31 After evolving for more than a decade, DNA origami can now produce nanostructures of almost any topology, including the already reported genie bottle,31 Möbius strip,32 spherical or ellipsoidal shells,33 polyhedral meshes,27etc. Difficulty in the origami assembly method mainly originates from the need for a large number (generally more than one hundred) of staple strands that will lower the cost-effectiveness, and the total size may be restricted by the length of the long sing-stranded scaffold DNA. Fortunately, computation-aided design and automated routing methods have relieved the difficulty. For example, Högberg et al. demonstrated the feasibility of using a routing algorithm and a relaxation simulation to facilitate the synthesis of complex 3D DNA meshes.27 In the routing algorithm, for a given 3D mesh, the odd-degree vertices were paired and the corresponding edges were assigned as double edges, and then the scaffold was routed based on A-trails (a specific Eulerian trail), followed by the routing of staple strands (Fig. 3a). Computation-aided rotational relaxation and length modification of the arris edges helped to reduce and evenly distribute the strain (Fig. 3b). Very complex structures were designed and successfully synthesized with the help of the routing algorithm and relaxation simulation (Fig. 3c).27
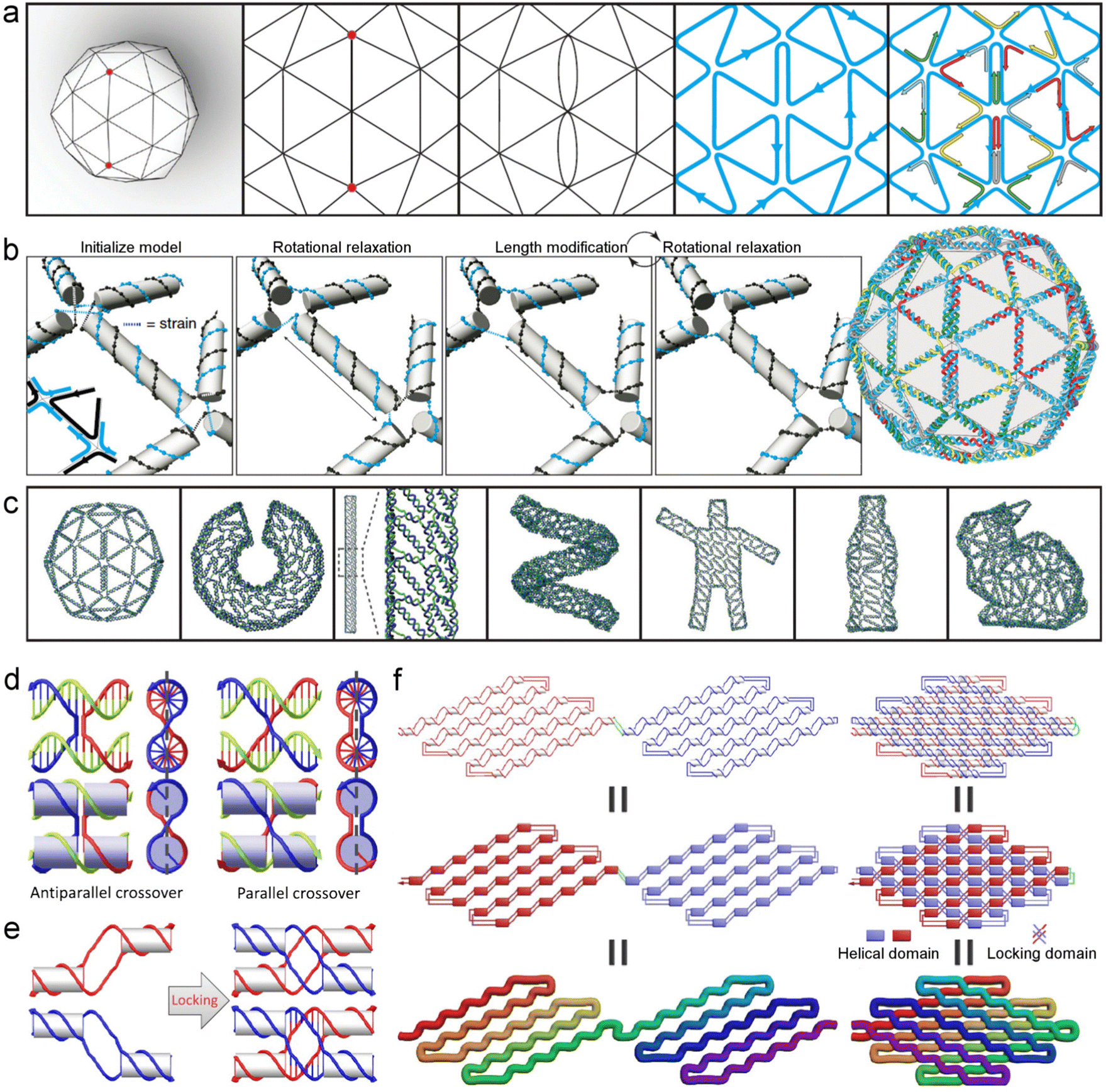 | ||
| Fig. 3 Typical synthesis strategies for DNA nanostructures. (a and b) DNA origami via a routing algorithm. (a) The algorithm process involves drawing a 3D mesh, paring the odd-degree vertices, introducing double edges, routing the scaffold according to A-trails, and then routing the staple strands. (b) Scheme of rotational relaxation and length modification for reducing the strain in the 3D mesh. (c) The multiple complex DNA meshes have been as-designed and prepared. Reproduced with permission.27 Copyright 2015, Springer Nature. (d–f) Single-stranded assembly. (d) Scheme of the (left) antiparallel and (right) parallel crossover designs. Dashed lines indicate the plane containing all DNA helical axes. (e) Scheme illustrating the formation of a locking domain. (f) Scheme of the formation of single-stranded origami by covering one folded layer with another symmetrical layer. Reproduced with permission.28 Copyright 2017, American Association for the Advancement of Science. | ||
A single-stranded assembly method (Fig. 2c), developed in 2012,37,38 greatly reduces the technological complexity for constructing DNA nanostructures. Yin et al. utilized 42-based DNA strands, which were divided into four domains with each acting as sticky ends to bind with four local neighbors, to successfully construct complex 2D patterns.37 3D structures were obtained using 32-nucleotide ‘bricks’ which bind to four local neighbors in a 3D space; each 8-base pair defines a voxel, resembling a Lego-like building block.38,39 This technique can yield large-sized structures without the limitation of the length of single-stranded DNA in the origami method. Drawbacks of this technique include the increasing number of ‘bricks’ when preparing larger structures, leading to an increased cost in sequence design; the product yield decreases sharply with the increase of structure size.15 More recently, Han et al. integrated the single-stranded assembly method with the DNA origami technique.28 By creating a parallel inter-helical cohesion that does not need the strands to cross the central plane (dashed lines in Fig. 3d), knot-free structures were obtained with minimal possibility to be kinetically trapped during the folding process. Moreover, they designed a partially complemented double-stranded DNA (dsDNA), with each paired domain located between two adjacent unpaired domains. Locking domains formed via the proximity of two adjacent parallel crossovers after covering one folded layer with another symmetrical layer (Fig. 3e–f).
Since all these strategies applied the basic laws of base-paring and immobile Holliday junction, researchers have integrated different strategies to minimize the disadvantages of each method. For example, Winfree et al.40 and Seeman et al.41 all combined the DNA origami method with tile-based assembly to construct large DNA origami-arrays. All these progresses, along with the wide exploitation of computer-aided sequence design, make DNA nanostructures increasingly easy to prepare.
2.3. DNA nanostructure-based bio-applications
DNA nanostructures possess intrinsic structural preciseness and high biocompatibility, and the on-demand fabricability in sequence, topology and porosity further make them attractive in drug delivery and bio-imaging. Fluorescence,42 computed tomography (CT),43 PET36 and magnetic resonance imaging44 are all applicable with DNA nanostructures by virtue of the high modifiability of nucleic acid, thereby providing diagnostic information with potential therapeutic value. For drug uploading, drugs can be covalently conjugated, nonspecifically adsorbed (e.g., via electrostatic attraction), spatially confined by the porous structure, and the DNA strand itself can be functional via sequence design (Fig. 4a). Particularly, doxorubicin (Dox) can be stably intercalated into a DNA double helix and will be released at acidic environments due to protonation and the structural metamorphosis of the DNA double helix.45 For porosity-enabled drug loading, many DNA polyhedra have been used including tetrahedral,46,47 hexahedral,48 octahedral,49 and icosahedral50 nanocages. Particle morphology is a powerful parameter influencing the delivery efficiency.51,52 Tetrahedral DNA nanostructures (TDNs) represent the simplest type and are relatively easy and cost-effective to fabricate. They are highly stable against deformable shear force due to the high stability of triangles, and are also proved stable against enzymatic degradation,53 for example, reported to remain intact in living HEK cells for more than 48 hours.54 Moreover, TDNs enter cells easily, reported more easily than DNA double strands,55 which facilitates intracellular drug delivery. | ||
| Fig. 4 Bio-applications of DNA nanostructures. (a) Typical drug loading modes of DNA nanostructures. (b) Usage of DNA icosahedra for antigen presentation. Antigen-to-icosahedron stoichiometry, antigen spacing and the dimensionality of carriers were effectively modulated. Reproduced with permission.11 Copyright 2020, Springer Nature. (c) Scheme of the site-specific synthesis of silica nanostructures on a DNA origami template. Reproduced with permission.34 Copyright 2018, Wiley-VCH. (d) Scheme of the rotation of dsDNA relative to a surface-bound enzyme, which was amplified using a dye-labelled DNA origami rotor and tracked via dye position trajectory. Scale bar: 100 nm. Reproduced with permission.35 Copyright 2019, Springer Nature. (e) Scheme of the preferential accumulation of DNA origami in kidneys after intravenous injection, and the protection of kidney tubules by scavenging ROS. Positron emission tomography (PET) imaging results of mice injected with either circular single-stranded DNA M13 or rectangular DNA nanostructures, are displayed. Reproduced with permission.36 Copyright 2018, Springer Nature. | ||
Functionalizing the nucleotide sequence of DNA nanostructures enables stimuli-responsive drug delivery. Gu et al. constructed a telomerase-responsive DNA icosahedron by incorporating telomerase primer and telomeric repeats into the icosahedral structure.56 Platinum nanoparticles were encapsulated as a model drug during the formation of icosahedra, and in the presence of telomerase, the primers were extended, leading to chain substitution and subsequence release of the caged nanoparticles. Douglas et al. demonstrated the use of DNA nanobarrels for logic-gated payload release.57 Each barrel consisted of two domains that were covalently attached in the rear and could be noncovalently fastened by staples modified with DNA aptamer-based locks. The locks could be opened by antigen keys via switching between an aptamer-complement duplex and an aptamer–target complex. Incorporating two different locks onto the barrel realized an ‘AND’ logic-gated drug delivery, in which the drug was released only when the two locks were both opened.57
The molecular-level preciseness of DNA nanostructures enables many applications that need fine spatiotemporal or stoichiometric control. Bathe et al. studied the role of nanoscale antigen organization in B cell activation, where antigens were conjugated onto DNA nanostructures with well-controlled stoichiometry, spacing and dimensionality (Fig. 4b).11 The results showed that an inter-antigen spacing of ∼25–30 nm and the use of a rigid nanostructure were favorable for B cell triggering. Precise synthesis of silica nanostructures were realized by using DNA nanostructures as templates.34,58 Protruding dsDNA has a stronger affinity to positively charged silica precursors than the DNA origami surface, and therefore silica nanostructures could be precisely prepared via the silicification on the designed dsDNA patterns (Fig. 4c).34 This strategy may pave the way for templated synthesis of a large variety of nanomaterials with DNA nanostructure-like preciseness. Zhuang et al. realized the rotation tracking of genome-processing enzymes using DNA origami rotors.35 dsDNA was attached onto a DNA origami rotor that was labelled with fluorescent dyes, and rotation of the dsDNA relative to a surface-bound DNA-processing enzyme was detected via dye position trajectory (Fig. 4d).
In general, DNA nanostructures are promising in multiple bio-applications. Favored by the strict pairing between complementary nucleotides, the inter-particle variation is quite limited. The number of carried ligands per particle can also be controlled very precisely, which is not easy to achieve using common nanoparticles. This enables precise drug delivery as well as quantitative evaluation about the effect of the delivered agents. Meanwhile, it was suggested by Cai et al. that DNA nanostructures (e.g., 2D origamis of around 100 nm in length) exhibited preferential renal uptake and excretion, and alleviated acute kidney injury by scavenging reactive oxygen species (ROS) (Fig. 4e),36 suggesting a particular application potential in kidney disease treatment. Nevertheless, several shortcomings still exist. First, as a genetic material, DNA is a natural cellular component and has no direct or accurate toxicity, but the long-term biosafety regarding the genetic stability is a potential issue to address.59 Secondly, large scale (e.g., milligram scale) fabrication of DNA nanostructures is still costly using current techniques, which is not very favorable for wide application. Thirdly, DNA nanostructures may be damaged in nuclease-rich environments. Although not all applications need a high nuclease resistance, this capability will improve the applicability in most scenarios including prolonged circulation in serum.60 Nevertheless, new techniques are constantly emerging to bring solutions to these issues. For example, the simplification of gene amplification procedures and computer-aided sequence design61,62 will lower the cost for producing desired nanostructures; strategies of enhancing the nuclease resistance for DNA nanostructures are available, such as using L-DNA63,64 and increasing the number of DNA crossovers.65
3. Atomically precise inorganic nanoclusters
Motivated by the small-size-related properties, synthesizing and applying metallic nanocrystals has been a major research topic in the last two decades. Size and composition are of great significance for their performances in catalysis, bio-sensing and biomedicine. Therefore, reliable preparation that enables precise control over particle characteristics and inter-particle variation is highly desirable. This has been extensively explored at the beginning stage, as an important criterion for optimizing the synthesis procedures. For example, for metal chalcogenide quantum dots (QDs) which have size- and surface-dependent fluorescence,66 considerable efforts have been devoted to preparing products with highly uniform size/shape and narrow emission width.67,68 For a nanoparticle of several nanometers in size, it will contain hundreds to thousands of atoms in the particle core and it is difficult to control the exact number of atoms or ligands, although the measured size distribution may be quite narrow. Therefore, as a further step, researchers have been trying to prepare nanoparticles with a defined number of atoms/ligands in the particle core/surface.69 The spatial arrangement or crystallinity of the atoms is also in atomic precision which finally leads to a well-defined molecule-like chemical structure. This precision is indeed important since the stability and physicochemical properties of nanocrystals are largely a function of the spatial arrangements of atoms (the crystalline phase). Meanwhile, different from common nanoparticles that rely on transmission electron microscopy (TEM) imaging for size determination, these nanoclusters can be characterized with techniques such as single crystal X-ray crystallography (SCXC) and mass spectrometry.70,713.1. Noble metal nanoclusters
An important type of material in this field is noble metal-based nanoclusters. A pioneering work was reported by Jadzinsky et al. who prepared p-mercaptobenzoic acid (p-MBA)-protected gold (Au) nanoparticles comprised of 102 Au atoms and 44 p-MBAs.72 The central Au atoms were packed in a Marks decahedron and were surrounded by additional layers of Au atoms. SCXC and the electron density map defines an unambiguous particle structure, and it could be confirmed that Au atoms up to 5.5 Å from the particle center did not contact the sulfur on p-MBA.72 Afterwards, dozens of Aux(SR)y (SR = thiolate ligand) nanoclusters were reported.73 The number of Au atoms per particle varies from several to more than one hundred. These materials typically lie in the size regime of smaller than ∼3 nm, which enables the Au nanoclusters to exhibit interesting properties different from the common Au nanoparticles (∼5–100 nm in size), such as the disappearance of surface plasmon resonance effect and the appearance of the molecule-like HOMO–LUMO electronic transition.74,75For synthesis methods, two main systematic methodologies have been established: the size focusing method75 and the ligand exchange-induced size/structure transformation (LEIST) method.76 The size focusing strategy creates a solution environment that makes the Aux(SR)y (x and y are exact numbers) particularly stable compared with particles of any other composition. The stable particles are preserved, while the unstable ones reconfigure to be stable; after a required duration (also called aging), highly uniform Aux(SR)y are obtained. Jin et al. summarized this method as ‘survival of the robustest’, just like the natural law ‘survival of the fittest’.75 The size focusing process can actually be explained by the potential energy theory which has been applied to guide nanomaterial synthesis,77 that the given synthesis condition creates a low-energy stable environment for the particles with a ‘magic’ atom-ligand number and arrangement (Fig. 5). The size focusing method has yielded many Aux(SR)y,75 as well as ‘common’ (without atomic preciseness) nanoparticles with highly uniform size.78,79 A typical synthesis process involves a preliminary reduction of Au(III) into Au(I) by a thiol-containing ligand in a selected solvent, a further reduction to Au(0) by a strong reductant (e.g., NaBH4) at low temperature, and finally an aging process that allows the size focusing transition to fully occur, during which the reaction can be monitored using techniques such as matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry.70
 | ||
| Fig. 5 Illustration of the potential energy of the reaction solution during the synthesis of M-SR NPs, the formation of atomically precise nanoclusters Mx(SR)yvia aging, and the formation of new Mx′(SR′)y′ nanoclusters by using new ligands or varying other conditions. | ||
The LEIST method, built on the basis of the size focusing process, involves the transition from one stable size to another. By replacing the original ligands of Aux(SR)yvia ligand exchange, the solution condition becomes unfavorable for the stability of Aux(SR)y but creates a new environment that favors the formation of Aux′(SR′)y′, where the SR′ is the new surface ligand. For example, Wu et al. obtained a novel Au60S6(SCH2Ph)36 nanocluster via the LEIST of molecularly pure Au38(SC2H4Ph)24, where the original and the new ligands only had a subtle difference in structure.80 Jin et al. further showcased the effectiveness of LEIST. Using the isomeric para-, meta- and ortho-methylbenzenethiol (MBT) as surface protecting ligands, Au130(p-MBT)50, Au104(m-MBT)41 and Au40(o-MBT)24 were obtained, respectively.71 The molecular purity was confirmed by electrospray ionization mass spectrometry (ESI-MS) and MALDI-TOF. Note that replacing original ligands does not guarantee size transformation and only several selected new ligands work for a given synthesis. It has also been confirmed to be feasible to switch ligands of Aux(SR)y while keeping the x and y values constant.81
The synthesis of Agx(SR)y follows similar procedures and philosophies with that of Aux(SR)y. Thiol-containing ligands are usually used due to the ease of Ag–S bond formation.82,83 Jin et al. reported a high yield, large scale preparation method for water-soluble Ag7(DMSA)4 (DMSA is 2,3-dimercaptosuccinic acid) nanoclusters.85 The purity was improved via post-synthesis precipitation and recrystallization. Other nanoclusters containing Ag atoms ranging from several to dozens have been reported, as summarized in a recent review.86 It can be noticed that the structure of Ag-based nanoclusters is relatively small (fewer Ag atoms) compared with Au-based ones, making them closer to small molecules. Bakr et al. reported a ‘golden’ Ag25(SR)18 nanocluster which shares the identical atom/ligand count, superatom electronic configuration, absorption and emission features, and atomic arrangement, with Au25(SR)18.87 2,4-dimethylbenzenethiol was used as the SR. Notably, the emission of the Ag25(SR)18 reached 850 nm, about 100 nm longer than that of Au25(SR)18, representing an advancement in red-shifting fluorescence wavelength. Strategies for enhancing the emission efficiency of Ag nanoclusters have been extensively explored.88 Generally, Ag-based nanoclusters may have lower biocompatibility compared with Au-based ones,89 but they are much more cost-effective for large-scale preparation and may achieve wider in vitro applications.
3.2. Alloyed nanoclusters and others
Substituting one or more atoms with heteroatom(s) can tune the optical and electronic properties of the original material, forming a strong rationale for the exploration of alloyed nanoclusters. Teo et al. pioneered the field by preparing a number of bimetallic (Au–Ag) and trimetallic (e.g., Au–Ag–Pt) nanoclusters.90,91 Current techniques allow us to precisely control the species, number and position of heteroatoms in one particle, as well as the alloying extent (e.g., bimetallic or trimetallic), therefore providing countless combinations of particle composition (exemplified in Fig. 6). The similar physiochemical properties of Au and Ag drove the emergence of many Au–Ag bimetal nanoclusters. For example, Bakr et al. prepared Ag24Au(SR)18 nanoclusters via a galvanic exchange strategy, using pure Au25(SR)18 as the template (SR here is 2,4-dimethylbenzenethiol).92 The position of the introduced Au was stationary (at the Ag25 center). The Galvanic exchange method was proved necessary since the direct preparation of Ag24Au led to mixed products of Ag25−xAux(SR)18, x = 1–8. More intense alloying, i.e., higher introduced/original atom ratio, can be achieved. Li et al. prepared a biicosahedral [Au13Ag12(PPh3)10Cl8]SbF6 nanocluster (PPh3 = triphenylphosphine) by simply reducing the mixture of Au and Ag precursor solutions.93 | ||
| Fig. 6 Feasibility of preparing alloyed nanoclusters via doping. Taking Au25(SR)18 nanoclusters as an example, Ag-, Cu-, Pd-, Pt-, Cd, Hg- and Ir-doped nanoclusters have been obtained with a controlled number and position of the doping atoms. Redrawn with permission.84 Copyright 2020, Royal Society of Chemistry. | ||
Other metals and metal chalcogenides can also be atomically precise, such as Cu25H22(PPh3)12,94 Ni39(SC2H4Ph)24 and Ni41(SC2H4Ph)2595 nanoclusters. Pradeep et al. reported a Cu38(PET)25 (PET here is 2-phenylethanethiol) nanocluster; interestingly, the product exhibited 615 nm photoluminescence which was not common for Cu-based nanoclusters.96 Owen et al. reported multiple atomically precise CdSe QDs, including Cd35Se20X30L30, Cd56Se35X42L42, and Cd84Se56X56L56 (X = O2CPh, L = H2N–C4H9), whose structures were confirmed via single-crystal X-ray diffraction and atomic pair distribution function analysis.97 Notably, the synthesis route enabled gram scale synthesis and all the nanoclusters were photoluminescent.
3.3. Typical bio-applications
The atomically precise nanoclusters, especially the Au-based ones, have high biocompatibility. Meanwhile, nanoclusters were mostly prepared to be oil-soluble in the earlier studies, while now water-soluble molecules, such as glutathione,98 captopril99,100 and p-MBA,72,101 have been increasingly used as capping ligands, thereby paving the way for nanoclusters to be biomedically applied.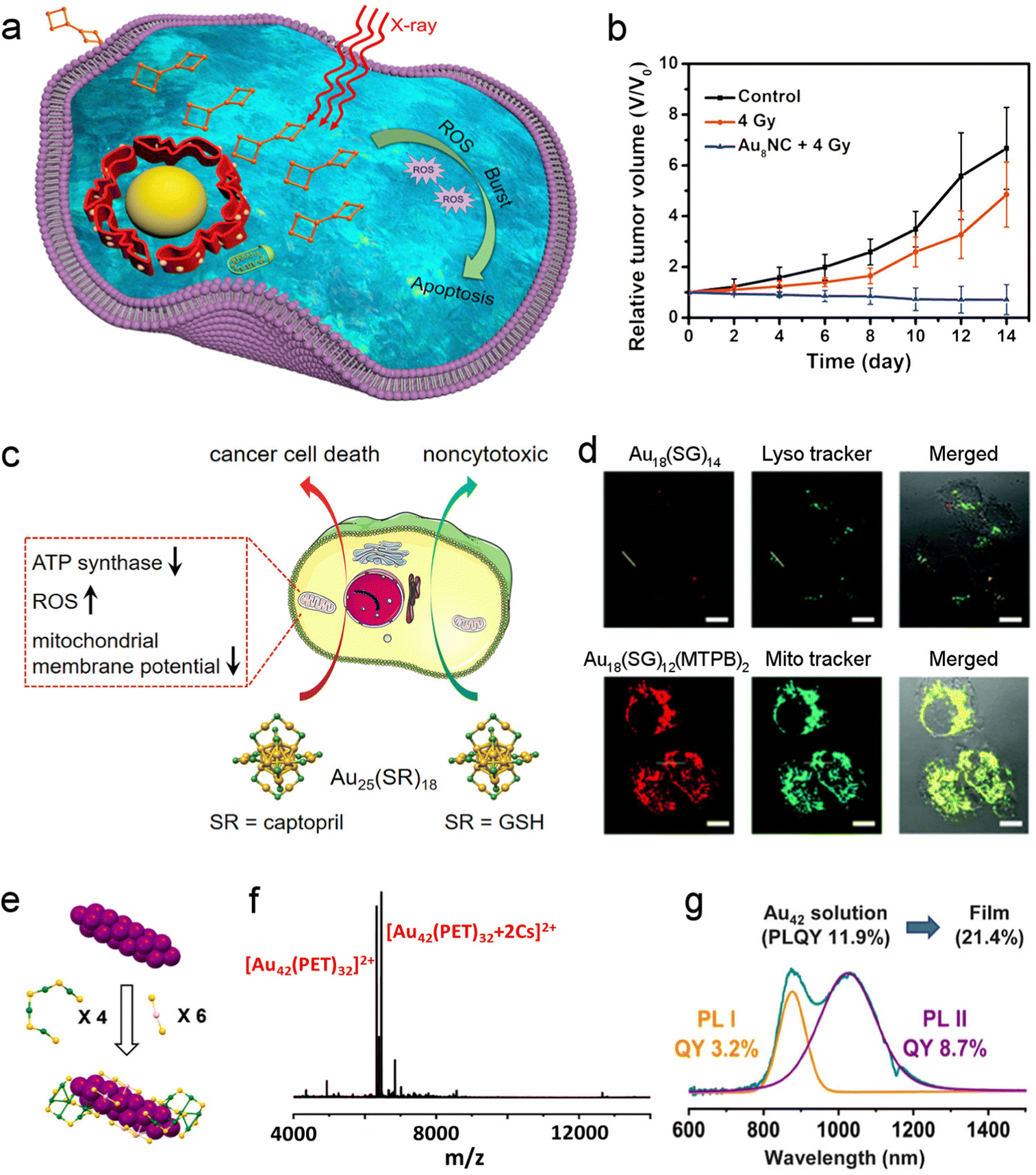 | ||
| Fig. 7 Multi-roles of nanoclusters in biomedical applications. (a) Schematic illustration of the burst ROS generation of Au8(C21H27O2) under X-ray irradiation. (b) Relative tumor volume curves of mice under the indicated treatment. Reproduced with permission.103 Copyright 2019, American Chemical Society. (c) Au25(GSH)18 were nontoxic, while Au25(Capt)18 induced cancer cell death by inhibiting ATP synthase, elevating intracellular ROS and disrupting the mitochondrial membrane potential. Reproduced with permission.100 Copyright 2022, American Chemical Society. (d) Fluorescent Au18(SG)14 served as a lysotracker, while Au18(SG)12(MTPB)2 was useful as a mitochondrion tracker. Reproduced with permission.105 Copyright 2018, Royal Society of Chemistry. (e) Structure of Au42(PET)32. Purple: core Au, green: surface Au, yellow: sulfur. (f) Mass spectrum of Au42(PET)32. (g) Emission spectrum of Au42(PET)32 and the corresponding peak fitting. Reproduced with permission.106 Copyright 2022, American Chemical Society. | ||
4. Dendrimer nanoparticles
Dendrimers are nano-sized radially symmetric molecules with well-defined tree-like branches.112 They are single-polymer nanoparticles, or particulate polymers, that generally have a spherical shape. A dendrimer commonly consists of (i) an initiator core at the center, (ii) multiple branched arms made up of monomers, and (iii) surface functional groups at the end of arms.113 Each cycle of the radial polymerization is called one generation, and the number of generations is commonly used for describing the size of a dendrimer. The high density in dens, charge or conjugation site makes them promising as drug carriers.112,114 Here we define dendrimer nanoparticles as either single-molecule dendrimers, supramolecular dendrimers that are assembled from amphiphilic molecules, or nanoparticle-cored dendrimers.4.1. Dendrimers
Like polymers, the synthesis of dendrimers involves the repetitive conjugation of monomers except that the conjugation occurred radially on the outer surface with self-limited cycles, therefore the structural preciseness of dendrimers should be at the same level as common polymers.115 The molecule structure cannot expand illimitably, because the number of conjugated monomers will increase exponentially with the increase of generation, while the overall size of the dendrimer does not increase proportionately and cannot provide enough space for further conjugation.116 As a result, structural defects (missing arms and intramolecular loops) will inevitably occur after a certain number of generations due to the increased steric hindrance. Fortunately, advances in synthetic techniques have helped to effectively overcome or suppress these limitations.Dendrimer nanoparticles can be synthesized via two main routes: divergent synthesis and convergent synthesis. The former is a radial generation-by-generation growth route from the initiator core to outer layers, while the latter needs to prepare multi-generation arms (dendrons) first, followed by coupling the dendrons with the core.113 The divergent synthesis can yield up to 10 generations or more, however, incomplete reactions occur increasingly. Moreover, the defective impurities produced in divergent synthesis are difficult to separate due to the high structural similarity. Holl et al. analyzed 5-generation (G5) poly(amido amine) (PAMAM) dendrimers via high performance liquid chromatography (HPLC), mass spectrometry, potentiometric titration, etc.117 Impurities including the generational defects (e.g., those lower than G5) and dimers of G5 could be effectively separated via HPLC, and an average of 93 arms could be identified, but the intramolecular branching defects were difficult to directly detect. In the convergent method, dendrons are built separately before the conjugation with the dendrimer core, which provides better control over defects. Moreover, the impurities generated in convergent synthesis are relatively more different with the desired dendrimers in comparison with those in the divergent synthesis, and thus can be separated more easily.118 Disadvantages of the convergent method include the reduced reactivity of dendrons with the core due to the increased steric hindrance. Notably, the divergent and convergent methods are not mutually exclusive, since a number of studies have combined the two strategies to achieve better quality control. This combined strategy involves the synthesis of a low-generation dendrimer as the supercore and low-generation dendrons as arms, which are then coupled to produce the final dendrimer.119 Higher generations can be realized via this approach.
Structural purity will be guaranteed when every reaction step proceeds with high yield. “Click” chemistry has been used to prepare dendrimers due to their high reaction specificity and yield.120 For example, Wong et al. reported an efficient approach to prepare glycerol dendrimers via thiol-yne click chemistry.121Via a simple divergent route, G3 dendrimers were obtained without transition metal catalysis or heating, and the structural purity was suggested by nuclear magnetic resonance (NMR) results, mass spectrometry as well as gel permeation chromatography.
4.2. Supramolecular dendrimers
To obtain higher generation dendrimers or simplify the synthesis procedures, researchers developed dendrimer-mimics consisting of amphiphilic dendrons, which usually bear a hydrophobic tail and multiple hydrophilic arms.122,123 The amphiphilic dendrons would self-assemble into supramolecules in an aqueous environment via noncovalent interactions. This strategy is a simplified convergent method that has removed the difficulty in conjugating the large dendrons onto the core, and makes the supramolecule resemble micelle or liposome which has achieved wide clinical applications.2 Therefore, the structural preciseness of these supramolecular dendrimers should be comparable to liposomes, with reduced structural defects compared with the same-generation single-molecule dendrimers. Peng et al. fabricated supramolecular dendrimers via the self-assembly of low-generation PAMAM dendrons, and the products exhibited a highly narrow size distribution.123 Using click chemistry to prepare the amphiphilic dendrons will further improve structural preciseness.1244.3. Nanoparticle-cored dendrimers
Nanoparticle-cored dendrimers are another category of dendrimer nanoparticles that use nanoparticles as cores for the conjugation of dendrons.126 Researchers have fabricated these dendrimers to deliver the core nanoparticle, make the dendrimers multifunctional, or to create a large number of anchoring sites for the ease of conjugation by dendrons. The high hydrophilicity of the chosen dendrons can also water-solubilize the hydrophobic nanoparticles. Nanoparticle-cored dendrimers can be prepared via replacement of original ligands by dendrons, post-synthesis conjugation of dendrons onto nanoparticles, or directly using dendrons as protecting ligands during nanoparticle synthesis.127 Obviously, the structural preciseness of nanoparticle-cored dendrimers largely depends on the used nanoparticles; using materials/molecules with well-defined structures as the core will improve the preciseness. For example, Zhou et al. fabricated molecularly precise cyanine nanodots, by using cyanine dyes as cores to prepare polylysine dendrimers (Fig. 8a–b).125 MALDI-TOF, gel permeation chromatography and HPLC demonstrated the product purity, and the nanodots were shown to be applicable for in vitro and in vivo bio-imaging (Fig. 8c). | ||
| Fig. 8 Biomedical applications of dendrimer nanoparticles. (a) Representation and some merits of cyanine-cored dendrimers. (b) Chemical structure of the cyanine-cored dendrimers. (c) Usage of cyanine-cored dendrimers for in vivo mice imaging. Reproduced with permission.125 Copyright 2022, Wiley-VCH. (d) Schematic illustration of a supramolecular dendrimer nanosystem, based on the self-assembling of 69Ga-labelled amphiphilic dendrimer, for mice PET imaging. (e) TEM image and (f) molecular-dynamics simulation model of the supramolecular dendrimers. (g) PET imaging of colorectal and pancreatic tumors on mice injected with [18F]FDG or 69Ga-labelled supramolecular dendrimers. Reproduced with permission.123 Copyright 2018, National Academy of Science. | ||
4.4. Preclinical applications
Advances in synthetic techniques led to the emergence of various dendrimers including PAMAM,118 poly(arylether),128 polylysine,129 triazine130 and phosphorus dendrimer.131 The polycationic nature of PAMAM, polylysine and phosphorus dendrimers makes them suitable for gene delivery.132 Shi et al. used phosphorus dendrimers as a nonviral vector to deliver plasmid DNA.133 Interestingly, it was found that G1 dendrimers displayed a higher delivery efficiency to HeLa cells compared with G2 and G3 counterparts, suggesting a critical role of the particle size. Self-assembled supramolecular dendrimers were also widely used. Peng et al. reported a dual-targeting dendrimer-mediated siRNA delivery system for cancer therapy.134 Amphiphilic PAMAM dendrimers self-assembled into micelle-like structures and efficiently adsorbed siRNA, while a RGDK peptide (which could bind integrin and neuropilin-1 receptor on cancer cells) was further decorated onto the micelle surface to achieve tumor-specific siRNA delivery. The system was further demonstrated to be useful for tumor PET imaging when modified with 69Ga (Fig. 8d–f).123 Compared with [18F]FDG (18F labelled 2-fluorodeoxyglucose, the clinical gold reference for PET imaging), the dendrimer-based agent showed a better detection capability for multiple cancers including colorectal cancer and pancreatic cancer (Fig. 8g), possibly due to the higher intratumoral accumulation of nanoparticles compared with small molecules.Toxicity is a critical issue determining the application potential. It has been summarized that the cytotoxicity of dendrimers mainly depends on the generation, as well as the number and nature (anionic, neutral or cationic) of the terminal moieties,135 and therefore precise control of the particle structure is needed to improve in vivo biocompatibility. High-generation cationic dendrimers are relatively cytotoxic compared with the low-generation ones. Wang et al. reported generation-, concentration- and time-dependent hemolysis of PAMAM dendrimers when incubated with red blood cells.136 The hemolysis effect followed G6-NH2 > G5-NH2 > G4-NH2 > G3-NH2, and the surface modified PAMAM-COOH and PAMAM-OH dendrimers induced much less hemolysis. Mukherjee et al. found previously that the short-term and long-term cytotoxicity of PAMAM dendrimers to both HaCaT and SW480 cells followed G6 > G5 > G4.137 A mechanism study revealed that dendrimers would be located in the mitochondria and lead to ROS production, apoptosis and DNA damage.138 Surface modification with negatively charged or neutral groups decreased cytotoxicity.139 Siegwart et al. used an alternative method to reduce toxicity by fabricating degradable dendrimers.140 Ester linkage was used to link the dendrimer core with the arms, and hepatotoxicity assays as well as liver cancer treatment outcomes suggested a balanced low toxicity and high treatment potency.
4.5. Clinical trials
There are multiple ongoing clinical trials involving the use of dendrimers, as summarized in Table 1. A majority of the ongoing trials, including several completed ones (not listed in Table 1), were launched by the Ashvattha Therapeutics, Inc., which focuses on the development of a new class of precision medicines using hydroxyl dendrimers. The hydroxyl dendrimers are used as drug carriers to minimize toxicity and maximize drug uptake in the disease tissue.141 About four dendrimer-based formulations have been developed by the company and tested in clinical trials, but it is difficult to identify their exact structures. After injection or oral dosing, the dendrimers distribute rapidly throughout the body, and the water-like surface makes them behave comparably to water without binding proteins. Enabled by their unique surface chemistry and small size, the developed dendrimers can travel across the blood–brain barrier. Moreover, they can be taken up selectively by disease cells including inflammatory cells, and as a carrier are excretable through the kidney without causing systemic toxicity. The completed trials (e.g., NCT04321980 and NCT03500627) have suggested the safety and high tolerability of the hydroxyl dendrimers. Surface conjugation with 18F makes the dendrimers a potential contrast agent.| Dendrimer | Indication | Trial purpose | Start date | Identifier and phase |
|---|---|---|---|---|
| PK: pharmacokinetics; NAC: N-acetyl-cysteine; NVAMD: neovascular age-related macular degeneration; DME: diabetic macular edema.a Estimated start date. | ||||
| 18F hydroxyl dendrimer | Amyotrophic lateral sclerosis | Study the PK, safety and distribution of the dendrimer as an imaging agent | July 2022a | NCT05395624; Phase 1 |
| NAC-conjugated G4 PAMAM146 | Severe COVID-19 | Study the efficacy and effect in reducing pro-inflammatory cytokines in severe COVID-19 | August 2020 | NCT04458298; Phase 2 |
| Hydroxyl dendrimer | NVAMD; DME | Study its safety, tolerability and PK in patients with NVAMD or DME | August 2022 | NCT05387837; Phase 2 |
| Hydroxyl dendrimer | NVAMD; DME | Study its safety, tolerability and pharmacokinetics in healthy volunteers | January 2022 | NCT05105607; Phase 1 |
| 188Re ligand-loaded G5 poly-L-lysine142 | Primary or metastatic liver cancer | Study its efficacy and safety in treating non-responding and inoperable liver cancers | March 2017 | NCT03255343; unknown |
| PEGylated poly-L-lysine dendrimer | Non-Hodgkin lymphoma | Study the safety, tolerability, PK and preliminary efficacy of the dendrimer in patients with NHL | July 2022 | NCT05205161; Phase 1 and 2 |
188Re reagent (188Re-nitro-imidazole-methyl-1,2,3-triazol-methyl-di-(2-pycolyl)amine)-loaded G5 poly-L-lysine dendrimer, also known as “[188Re]rhenium-ImDendrim” (Fig. 9), co-developed by French and Chinese researchers, is being tested as a potential radiopharmaceutical. Its anti-tumor activity has been confirmed in a liver cancer model in nude mice.142 Positive outcomes after intrahepatic injection to a human patient with adenocarcinoma and liver metastases have also been reported.143 More recently, [188Re]rhenium-ImDendrim has been trailed to treat patients with local primary and secondary lung malignancies via brachytherapy (NCT03255343).144
 | ||
| Fig. 9 Chemical structure of the [188Re]rhenium-ImDendrim. | ||
Generally, we noticed that all the trialed dendrimers were single-molecule dendrimers rather than self-assembled dendrimer supramolecules or nanoparticle-cored dendrimers. This may be due to three main reasons: (i) The development of dendrimer-based nanomedicines is still at the early stage, as all the relevant trials were launched in the last five years. (ii) Single-molecule dendrimers are more suitable for some scenarios. For example, they are generally smaller than antibodies or antibody fragments, which is important for urinary excretion and crossing tissue barriers,145 while supramolecular dendrimers will be too large to achieve the same. (iii) Inter-particle structural variation of supramolecular and nanoparticle-cored dendrimers may challenge the quality-control and the precise evaluation of pharmacokinetics and therapeutic efficacy. Nevertheless, supramolecular dendrimers possess their own advantages for bio-applications, such as higher drug loading capacity. Wider applications of dendrimers can be expected under constant efforts to improve synthetic techniques. Meanwhile, all the trialed formulations are administrated via intravenous injection, while localized delivery is also a powerful route worthy of exploration.147
5. Carbon nanostructures
Carbon nanostructures, including fullerene, carbon nanotubes, graphene nanosheets and nanoribbons, have been widely studied due to their high biocompatibility, eco-friendliness, unique optical and electronic properties and high stability.148 They can be regarded as an intermediate between small molecules and nanomaterials, and between inorganic and organic materials. Fullerene is a structurally precise molecule, while for other carbon nanostructures the synthesis techniques have been extensively evolved to enable the preparation of structurally highly uniform products.1495.1. Graphene nanosheets and nanoribbons
Graphene is a single-layered graphite material with zero bandgap, and when its lateral size decreases to the nanoscale, it becomes a graphene nanosheet (GNS) with a size-dependent bandgap. GNSs have exceptional properties such as the large edge effect and ultra-high specific surface area, and one particular merit of GNSs compared with other carbon nanostructures is the ease of exhibiting photoluminescence. A quantum yield of greater than 50% has been reported;150 for tuning PL emission wavelength, several strategies were demonstrated to be effective, such as enlarging the sp2 domain by conjugating GNSs with extra aromatic rings, and introducing an intermediate n-orbital between π and π* orbitals via conjugation with electron-donating groups.151,152 Meanwhile, GNSs are considerably biocompatible, and have been reported to be less toxic compared with microscale graphene.153GNSs can be prepared using the techniques commonly used for nanomaterial synthesis, including the top-down route by cutting, exfoliating or grinding bulk materials, and the bottom-up route by building nanostructures from atoms and molecules via hydrothermal/solvothermal treatment, microwave treatment, or traditional organic chemistry.154–157 The bottom-up route is more effective in quality control and property modulation.158 For example, incorporating heteroatoms into the basal plane of GNSs is easier through bottom-up routes compared with the top-down routes. It is currently hard to achieve high control over product quality through a simple and large-scale synthesis route (e.g., one-step hydrothermal treatment) without a separation procedure. Song et al. achieved space-confined formation of GNSs by hydrothermally treating citrate which was intercalated into the interlayer galleries of layered double hydroxides.159 The obtained GNSs had a highly narrow size distribution and was basically all single-layered due to the strict space-confinement. An aromatic C27H10N2O15 molecular structure was revealed by 13C NMR, 1H NMR, gel permeation chromatography and tandem mass spectrometry.159 This method produced much more uniform GNSs compared with the common one-step hydrothermal syntheses, but still failed in providing atomic preciseness.
Given the well-defined structure of hexagonal carbon arrangement,160,161 GNSs hold the possibility to be precisely prepared via the traditional organic synthesis route. Organic synthesis can guarantee the high structural preciseness via stepwise monitoring and purification, and has been demonstrated to be feasible for GNS synthesis.149 For example, Yan et al. reported the synthesis of colloidal GNSs through the oxidative condensation of polyphenylene dendritic precursors, yielding graphene moieties of up to 170 carbon atoms.162 The graphene was stabilized by multiple 2′,4′,6′-triakyl phenyl groups that were covalently attached to graphene edges. Nitrogen-doping and addition of functional groups were shown to be feasible to tune the bandgap and optical property of the resulting GNSs.163,164 These products represent the most uniform kind of GNSs despite the tedious synthesis procedure and the failure in providing detectable photoluminescence and being water-solubilized.
Graphene nanoribbons (GNRs) are a member of the graphene family but different from common graphene nanosheets. They have a narrow width (typically several nanometers) and large aspect ratio, resemble a linear polymer and therefore have reduced dimensionality compared with graphene sheets.167 Their remarkable mechanical, thermal, electrical and optical properties offer many opportunities in bio-applications.168 The structural variation for a GNR mainly comes from the varied length and edge structure. Top-down routes, for example cutting graphene via lithography, generate products with relatively low uniformity,169 while some established bottom-up routes have produced atomically precise GNRs. Ruffieux et al. prepared atomically precise GNRs via surface-catalyzed coupling of molecular precursors into linear polyphenylenes and subsequent cyclodehydrogenation.170 By using different monomers, the topology, width and edge periphery were effectively modulated. Raman spectroscopy and scanning tunneling microscopy demonstrated the atomic preciseness of straight, chevron-type and threefold junction GNRs. Crommie et al. further prepared atomically precise GNR heterojunctions. Fluorenone GNRs were first synthesized by annealing a molecule precursor on the Au(111) surface, and chevron GNRs were then obtained following post-growth excitation (Fig. 10a). Formation of fluorenone/pristine chevron heterojunctions was confirmed via multiple techniques including bond-resolved scanning tunneling microscopy (Fig. 10b).
 | ||
| Fig. 10 Atomically precise carbon nanostructures. (a) Synthesis route of an atomically precise GNR and the corresponding heterojunction. (b) Bond-resolved scanning tunneling microscopy image of the fluorenone/pristine chevron heterojunction. Reproduced with permission.165 Copyright 2017, Springer Nature. (c) Synthesis route of singly-capped CNTs using a cap-molecule (precursor C96H54). Illustration of the conversion of (d) cap-molecule to (e) the ultrashort singly capped CNT seed via thermal induced surface catalyzed cyclodehydrogenation. The corresponding scanning tunneling microscopy images of (f) cap-molecules and (g) CNT seeds on the Pt(111) surface. Reproduced with permission.166 Copyright 2014, Springer Nature. | ||
An important issue restricting the bio-applications of these atomically precise GNRs is the limited water-solubility. Graphene oxide nanoribbons, in which the carbon atoms are partially sp3-hybridized by bearing oxygen-containing functional groups, may be more applicable due to their higher water-solubility and ease of functionalization.171 Nevertheless, the introduction of oxygen-containing groups inevitably decreases the structural preciseness since the extent and position of oxidation are difficult to control.
5.2. Carbon nanotubes
Carbon nanotubes (CNTs) are cylindrical molecules that are composed of rolled-up single-layer carbon atoms (graphene). Since the first discovery in the early 1990s,172 numerous studies have explored their synthesis, property and application. The diameter of CNTs is typically measured in nanometers, while the length can reach millimeters. CNTs are mostly prepared via chemical vapor deposition, which involves the splitting of a carbon-containing precursor gas (e.g., CO, methane) to deposit carbon atoms on catalyst nanoparticles (e.g., Fe, Co) to generate CNTs.173 Visually uniform products have been produced in good controllability and scalability, but atomic preciseness is barely achieved. The floating catalyst chemical vapor deposition, featured by the in situ formation of catalyst nanoparticles, is to some extent helpful to improve structural preciseness,176 but it raises another problem, i.e., the collected CNTs will be attached with a large number of catalyst nanoparticles.177 Generally, the diameter of CTNs prepared via this route is relevant with the size of the catalyst nanoparticles which are heterogeneous, while CTNs with close diameters are hard to separate. Meanwhile, multiple-walled CTNs may be simultaneously generated, and structural defects on the wall plane may emerge.178 Other synthesis methods including arc discharge and laser ablation are also bothered by these limitations. Fortunately, integrating organic synthesis with the surface-catalyzed carbon atom deposition method is applicable to greatly improve the structural preciseness.By vertically ‘stacking’ carbon atoms on some already-existing carbon nanorings (e.g., cycloparaphenylenes, cyclacene), we can obtain open-ended CTNs with a tuned structure: the CTN diameter could be tuned by selecting differently sized carbon nanorings, while the use of chiral carbon nanorings led to chiral CTNs.179 For example, Itami et al. prepared structurally uniform CTNs by simply heating cycloparaphenylenes (as templates) with ethanol (as the carbon source), realizing a ‘growth-from-template’ route.180 Cycloparaphenylenes with the number of phenylenes from 9 to 16 were used to effectively tune the CTN diameter. The same group also prepared a chiral carbon nanoring, cyclo[13]paraphenylene-2,6-naphthylene, which represented a novel chiral template.181 Using cycloparaphenylenes as templates leads to ‘armchair’ edges, while using cyclacene will yield ‘zigzag’ edges, although the synthesis of cyclacene has virtually not been realized.149 Meanwhile, besides open-ended CTNs, capped CTNs can be prepared by selecting some capping molecules as templates. Fasel et al. achieved controlled synthesis of singly-capped CTNs by first synthesizing an elaborately designed cap-molecule and then converting the cap-molecule into an ultrashort singly capped nanotube seed, followed by nanotube growth via epitaxial elongation (Fig. 10c–g).166 The cap-molecule was deposited onto a Pt(111) surface and then annealed to induce cyclodehydrogenation reaction to form the singly caped seed, and epitaxial elongation was achieved by incorporating carbon atoms decomposed from ethylene or ethanol.
Generally, organic synthesis of template molecules greatly improves the structural preciseness of CTNs, especially in the control of the diameter, chirality and edge structure. Nevertheless, it is technically difficult to realize full synthesis using organic chemistry, and surface-catalyzed carbon deposition at a high temperature is needed to construct the main body of CTNs. Consequently, strict atomic preciseness is difficult to achieve. What might relieve this limitation is that in bio-applications such as drug delivery, a relatively short length is needed for CTNs, which will decrease the reaction duration of surface catalyzed carbon deposition and therefore decrease the structural variation. Moreover, using large carbon nanorings (e.g., cycloparaphenylene and cyclacene) as templates will make it easier to yield a large diameter, which is desirable for the uploading of biomacromolecules.
5.3. Carbon quantum dots
Carbon quantum dots (CQDs) are zero-dimensional quasi-spherical carbon nanomaterials. Synthesis of CQDs is facile, usually via solvothermal treatment of small molecules or biomasses. The precursors in the high temperature and high pressure environment will undergo some ‘miraculous’ reactions that are not be easy to realize via common organic synthesis, leading to the formation of crystalline structures that are often fluorescent.182 It is currently very difficult to prepare atomically precise CQDs, despite the fine control of synthesis procedures and the use of simple molecular precursors. Given the ultra-small size, researchers are binding synthesis optimization with purification techniques such as liquid chromatography to improve the product uniformity. Emission width has been widely employed to assess the size uniformity of CQDs.183 Yang et al. prepared triangular CQDs with unprecedented narrow emission bandwidth.174 Using phloroglucinol as the precursor, highly luminescent CQDs were obtained after solvothermal treatment in ethanol at 200 °C, and the products was further purified via silica column chromatography. The high color purity made the CQDs comparable with traditional dyes in providing non-overlapping multicolor fluorescence signals (Fig. 11a–d).174 Generally, the structural preciseness of CQDs may be at a similar level as other common nanomaterials such as CdSe QDs and gold nanoparticles. Emission efficiency of CQDs has been improved to a level comparable to organic dyes, such as those prepared by solvothermally treating 1,3-dihydroxynaphthalene and KIO4 (Fig. 11e).175 The CDs had a quantum yield of 53% at a red emission wavelength, thereby showing great potential in in vivo bio-imaging. Still, column chromatography was needed to improve the product purity.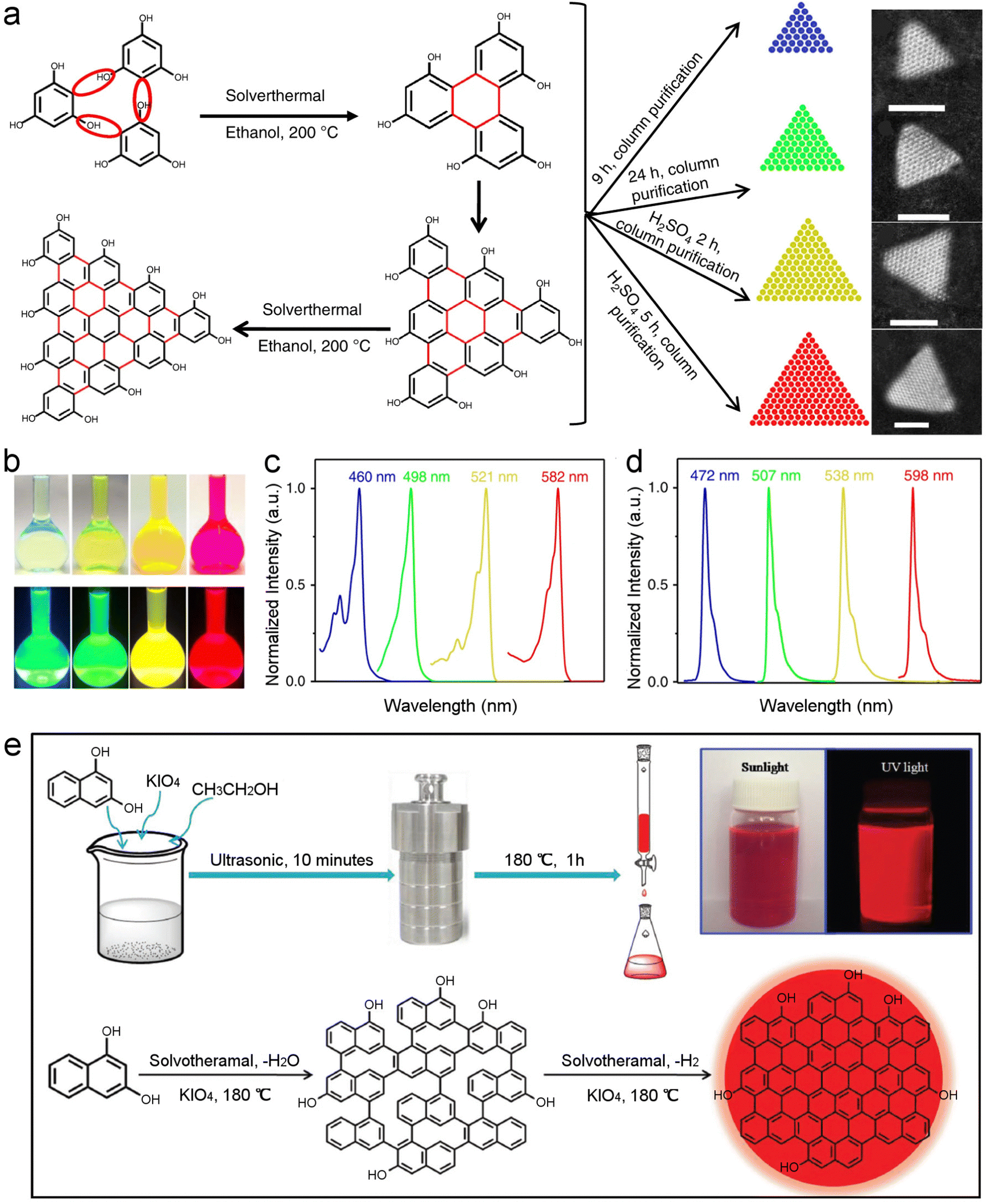 | ||
| Fig. 11 Structurally uniform CQDs. (a) Synthesis route of triangular CQDs via solvothermal treatment of phloroglucinol, and the dark-field scanning transmission electron microscopy images of differently sized triangular CQDs. Scale bar, 2 nm. (b) Photographs of the solutions of blue-, green-, yellow- and red-emissive CQDs under daylight and UV light. (c) Normalized absorption spectra and (d) normalized PL spectra of the corresponding CQDs. Reproduced with permission.174 Copyright 2018, Springer Nature. (e) Synthesis route and growth mechanism of the 53% efficient red emissive CQDs. Reproduced with permission.175 Copyright 2017, Wiley-VCH. | ||
6. Perspective remarks and conclusion
Molecule-like structural preciseness is a large challenge for nanomaterials on the way towards practical bio-applications, as it ensures product quality control and repeatable interaction with the human body. The efforts devoted to synthesis method exploration have yielded great uniformity in size, composition and surface chemistry, as often confirmed by microscopic observation, hydrodynamic diameter measurement, or the consistency and uniformity in properties. Nevertheless, greater preciseness is expected for bio-applications and the typical characterization methods for nanomaterials may not guarantee a molecule-level preciseness (Fig. 12). For example, for crystalline nanostructures that are characterized to be highly uniform, the particle size and shape may be variable; the number, position and species of lattice defects will greatly differ among particles; the location and conformation (protruding or prostrating) of surface ligands are difficult to identify under common characterizations. Particle size, shape and lattice defects determine the electronic and optical performances, while the density and orientation of ligands have a great impact on in vivo particle behaviors such as the affinity toward the cell surface and circulation time in blood. | ||
| Fig. 12 Limitations of characterization methods for nanostructures. For a batch of crystalline nanostructures that are characterized uniformly by common methods such as TEM imaging, hydrodynamic analysis, photospectrometry test and X-ray diffraction analysis (a), the (b) particle size and shape, (c) species and location of lattice defects, and (d) the density, location and conformation of surface ligands may differ greatly. | ||
Therefore, molecule-like structural preciseness may be difficult to realize via regular synthesis-characterization-optimization cycles. Sharp evolvement in synthesis strategies will be needed. DNA nanostructures, atomically precise nanoclusters, dendrimer nanoparticles and carbon nanostructures are currently representative materials that can reach molecule-like preciseness, and each has its own way to ensure a highly precise structure, and each has its own potential in specific bio-applications. For example, dendrimer nanoparticles are promising in both drug delivery and bio-imaging after the required modification, as evidenced by multiple clinical trials; the potential of Au-based nanoclusters in fluorescence imaging may be less pronounced due to the relatively low emission efficiency, but a particular promise is held in CT imaging due to the low toxicity and high X-ray attenuation efficiency of Au. Nevertheless, practical bio-applications are currently rarely realized, suggesting the need for further progress in synthesis methods and property optimization.
(i) DNA nanostructures are composed of molecularly precise nucleic acids that are organized by the strict pairing law between complementary bases, and therefore can achieve high structural preciseness despite the relatively sophisticated structure. Synthesis methods have evolved to be increasingly facile and robust, including the computation-aided sequence design61 and the non-necessity of staple strands in the newly-developed single-stranded assembly method.39 Nevertheless, for advanced 3D structures it is still hard for the structural uniformity to reach a similar level as that for simple structures such as DNA tetrahedra and small 2D structures. Meanwhile, two other limitations are restricting the wide application of DNA nanostructures, which are the high cost for large scale production, and the long-term biosafety regarding generic stability.
(ii) The atomically precise Mx(SR)y nanoclusters are prepared via reduction of metal precursors in the presence of surface ligands, followed by an aging process during which the particle size focuses. Pure MS spectra and the feasibility for SCXC measurement suggest the high structural preciseness. The nanoclusters are promising as imaging contrast agents. Meanwhile, the SR-to-M ratio is defined for a species, meaning that the drug (SR) content is constant for a specific weight of particles, which is essential for accurate dosing. The SR-to-M ratio in nanoclusters is also much higher compared with that in nanoparticles of larger size, since the particle volume increases more rapidly than surface area with increasing diameter. Therefore, the drug loading efficiency (the drug-to-carrier weight ratio) is greatly improved. Important concerns regarding the use of nanoclusters include the toxicity and the versatility for uploading drugs, since only a small range of molecules can serve as the nanocluster ligands without covalent conjugation.
(iii) Dendrimer nanoparticles may be the closest to clinical applications, as suggested by the number of relevant clinical trials. Dendrimers can be viewed as a special type of polymers but are particulate. The structural defects of dendrimers mainly come from the inter-branch linkage and the failed elongation of some branches due to increased steric hindrance. Like linear polymers that are clinically applicable despite the difficulty in precisely controlling the polymer length, dendrimers may achieve practical applications despite the presence of certain structural defects, because minor defects will influence their property but probably to a less degree than that for crystalline materials whose property heavily depends on particle size and composition.66,184 For the supramolecular dendrimer nanoparticles, the structural uniformity should be close to that of liposomes (self-assembled from amphiphilic phospholipids) that have achieved clinical applications. The toxicity of dendrimers varies greatly with chemical structure and some cationic ones may cause inflammation;185 on the other hand, some cationic dendrimers themselves are useful as immunostimulators.186 The completed and ongoing clinical trials have demonstrated the low toxicity of the tested dendrimers.
(iv) Structural preciseness of carbon nanotubes largely depends on synthesis strategy. Bottom-up routes produce relatively uniform materials compared with the top-down routes, while high structural preciseness is still difficult to achieve using common bottom-up routes, such as solvothermal treatment and chemical vapor deposition. The use of traditional organic synthesis will guarantee a high preciseness. Small-sized GNSs and GNRs can be fully prepared via organic synthesis, while for CNTs this is currently impracticable. The use of carbon nanorings as templates directs the growth of CTNs with uniform diameters, although the subsequent epitaxial elongation may increase the structural heterogeneity. Conjugation of hydrophilic groups or oxidation of nanostructures (e.g., producing graphene oxide) is often involved to improve water-solubility but will attenuate the structural preciseness. Therefore, carbon nanostructures may need further development to meet the criterion for bio-applications.
In summary, although other properties, including toxicity and the robustness of performances, are also critical aspects determining whether a material could realize clinical applications, high structural preciseness is considered as a prerequisite. This will help us to understand the material property on a single-particle level, instead of the average property of particles with widely varying structures. Tremendous progress has been made in improving the structural preciseness of nanomaterials in virtue of breakthroughs in synthetic techniques. Currently the above-discussed four types of nanomaterials currently can be molecularly or atomically precise and dendrimer nanoparticles have reached multiple clinical trials, probably due to the high biocompatibility and ease of production. It can be envisioned that with constant progress in the synthesis method, more materials will be prepared precisely and more applications will be realized.
Author contributions
Conceptualization: Jie Wang and Ping Li. Writing original draft: Jie Wang and Ping Li. Review & editing: Chao Wang and Ning Liu. Supervision: Dongming Xing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the China Postdoctoral Science Foundation (2021T140355) and the Postdoctoral Innovation Project of Shandong Province (202002025).Notes and references
- S. R. D'Mello, C. N. Cruz, M.-L. Chen, M. Kapoor, S. L. Lee and K. M. Tyner, Nat. Nanotechnol., 2017, 12, 523–529 CrossRef PubMed
.
- H. Nsairat, D. Khater, U. Sayed, F. Odeh, A. Al Bawab and W. Alshaer, Heliyon, 2022, 8, e09394 CrossRef CAS PubMed
- P. Mishra, B. Nayak and R. K. Dey, Asian J. Pharm. Sci., 2016, 11, 337–348 CrossRef
- Z. Tian and W. Yao, Front. Oncol., 2022, 12, 815900 CrossRef PubMed
- N. Song, J. Zhang, J. Zhai, J. Hong, C. Yuan and M. Liang, Acc. Chem. Res., 2021, 54, 3313–3325 CrossRef CAS PubMed
- P. Dash, A. M. Piras and M. Dash, J. Controlled Release, 2020, 327, 546–570 CrossRef CAS PubMed
- Y. H. Chung, H. Cai and N. F. Steinmetz, Adv. Drug Delivery Rev., 2020, 156, 214–235 CrossRef CAS PubMed
- S. Dordevic, M. M. Gonzalez, I. Conejos-Sanchez, B. Carreira, S. Pozzi, R. C. Acurcio, R. Satchi-Fainaro, H. F. Florindo and M. J. Vicent, Drug Delivery Transl. Res., 2022, 12, 500–525 CrossRef PubMed
- C. Egbuna, V. K. Parmar, J. Jeevanandam, S. M. Ezzat, K. C. Patrick-Iwuanyanwu, C. O. Adetunji, J. Khan, E. N. Onyeike, C. Z. Uche, M. Akram, M. S. Ibrahim, N. M. El Mahdy, C. G. Awuchi, K. Saravanan, H. Tijjani, U. E. Odoh, M. Messaoudi, J. C. Ifemeje, M. C. Olisah, N. J. Ezeofor, C. J. Chikwendu and C. G. Ibeabuchi, J. Toxicol., 2021, 9954443 CAS
- J. M. Rabanel, V. Adibnia, S. F. Tehrani, S. Sanche, P. Hildgen, X. Banquy and C. Ramassamy, Nanoscale, 2019, 11, 383–406 RSC
- R. Veneziano, T. J. Moyer, M. B. Stone, E. C. Wamhoff, B. J. Read, S. Mukherjee, T. R. Shepherd, J. Das, W. R. Schief, D. J. Irvine and M. Bathe, Nat. Nanotechnol., 2020, 15, 716–723 CrossRef CAS PubMed
- H. Lee, A. K. Lytton-Jean, Y. Chen, K. T. Love, A. I. Park, E. D. Karagiannis, A. Sehgal, W. Querbes, C. S. Zurenko, M. Jayaraman, C. G. Peng, K. Charisse, A. Borodovsky, M. Manoharan, J. S. Donahoe, J. Truelove, M. Nahrendorf, R. Langer and D. G. Anderson, Nat. Nanotechnol., 2012, 7, 389–393 CrossRef CAS PubMed
- G. Yi, J. Son, J. Yoo, C. Park and H. Koo, Biomater. Res., 2018, 22, 13 CrossRef PubMed
- S. B. van der Meer, T. Knuschke, A. Frede, N. Schulze, A. M. Westendorf and M. Epple, Acta Biomater., 2017, 57, 414–425 CrossRef CAS PubMed
- Q. Hu, H. Li, L. Wang, H. Gu and C. Fan, Chem. Rev., 2019, 119, 6459–6506 CrossRef CAS PubMed
- J. Huang, S. Gambietz and B. Sacca, Small, 2022, e2202253, DOI:10.1002/smll.202202253
- N. C. Seeman, J. Theor. Biol., 1982, 99, 237–247 CrossRef CAS PubMed
- N. R. Kallenbach, R.-I. Ma and N. C. Seeman, Nature, 1983, 305, 829–831 CrossRef CAS
- T. J. Fu and N. C. Seeman, Biochemistry, 1993, 32, 3211–3220 CrossRef CAS PubMed
- T. H. Labean, H. Yan, J. Kopatsch, F. Liu, E. Winfree, J. H. Reif and N. C. Seeman, J. Am. Chem. Soc., 2000, 122, 1848–1860 CrossRef CAS
- F. Mathieu, S. Liao, J. Kopatsch, T. Wang, C. Mao and N. C. Seeman, Nano Lett., 2005, 5, 661–665 CrossRef CAS PubMed
- J. Zheng, J. J. Birktoft, Y. Chen, T. Wang, R. Sha, P. E. Constantinou, S. L. Ginell, C. Mao and N. C. Seeman, Nature, 2009, 461, 74–77 CrossRef CAS PubMed
- L. Lin, Y. Pei, Z. Li and D. Luo, Interdiscip. Med., 2022, 1, e20220008 Search PubMed
- Y. He, T. Ye, M. Su, C. Zhang, A. E. Ribbe, W. Jiang and C. Mao, Nature, 2008, 452, 198–201 CrossRef CAS
- C. Zhang, S. H. Ko, M. Su, Y. Leng, A. E. Ribbe, W. Jiang and C. Mao, J. Am. Chem. Soc., 2009, 131, 1413–1415 CrossRef CAS PubMed
- C. Zhang, M. Su, Y. He, X. Zhao, P.-A. Fang, A. E. Ribbe, W. Jiang and C. Mao, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10665–10669 CrossRef CAS PubMed
- E. Benson, A. Mohammed, J. Gardell, S. Masich, E. Czeizler, P. Orponen and B. Hogberg, Nature, 2015, 523, 441 CrossRef CAS PubMed
- D. Han, X. Qi, C. Myhrvold, B. Wang, M. Dai, S. Jiang, M. Bates, Y. Liu, B. An, F. Zhang, H. Yan and P. Yin, Science, 2017, 358, eaao2648 CrossRef PubMed
- P. W. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed
- H. Dietz, S. M. Douglas and W. M. Shih, Science, 2009, 325, 725–730 CrossRef CAS PubMed
- S. M. Douglas, H. Dietz, T. Liedl, B. Hogberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS PubMed
- D. Han, S. Pal, Y. Liu and H. Yan, Nat. Nanotechnol., 2010, 5, 712–717 CrossRef CAS PubMed
- D. Han, S. Pal, J. Nangreave, Z. Deng, Y. Liu and H. Yan, Science, 2011, 332, 342–346 CrossRef CAS PubMed
- Y. Shang, N. Li, S. Liu, L. Wang, Z. G. Wang, Z. Zhang and B. Ding, Adv. Mater., 2020, 32, e2000294 CrossRef PubMed
- P. Kosuri, B. D. Altheimer, M. Dai, P. Yin and X. Zhuang, Nature, 2019, 572, 136–140 CrossRef CAS PubMed
- D. Jiang, Z. Ge, H. J. Im, C. G. England, D. Ni, J. Hou, L. Zhang, C. J. Kutyreff, Y. Yan, Y. Liu, S. Y. Cho, J. W. Engle, J. Shi, P. Huang, C. Fan, H. Yan and W. Cai, Nat. Biomed. Eng., 2018, 2, 865–877 CrossRef CAS PubMed
- B. Wei, M. Dai and P. Yin, Nature, 2012, 485, 623–626 CrossRef CAS PubMed
- Y. Ke, L. L. Ong, W. M. Shih and P. Yin, Science, 2012, 338, 1177–1183 CrossRef CAS PubMed
- Y. Ke, L. L. Ong, W. Sun, J. Song, M. Dong, W. M. Shih and P. Yin, Nat. Chem., 2014, 6, 994–1002 CrossRef CAS PubMed
- R. D. Barish, R. Schulman, P. W. K. Rothemund and E. Winfree, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6054–6059 CrossRef CAS PubMed
- W. Liu, H. Zhong, R. Wang and N. C. Seeman, Angew. Chem., Int. Ed., 2011, 50, 264–267 CrossRef CAS PubMed
- B. Zhang, T. Tian, D. Xiao, S. Gao, X. Cai and Y. Lin, Adv. Funct. Mater., 2022, 32, 2109728 CrossRef CAS
- S. Li, L. Xu, M. Sun, X. Wu, L. Liu, H. Kuang and C. Xu, Adv. Mater., 2017, 29, 1606086 CrossRef PubMed
- Y. He, C. Lv, X. Hou and L. Wu, Anal. Chim. Acta, 2020, 1138, 141–149 CrossRef CAS PubMed
- J. Yan, J. Chen, N. Zhang, Y. Yang, W. Zhu, L. Li and B. He, J. Mater. Chem. B, 2020, 8, 492–503 RSC
- T. Wang, Y. Liu, Q. Wu, B. Lou and Z. Liu, Smart Mater. Med., 2022, 3, 66–84 CrossRef
- T. Zhang, T. Tian, R. Zhou, S. Li, W. Ma, Y. Zhang, N. Liu, S. Shi, Q. Li, X. Xie, Y. Ge, M. Liu, Q. Zhang, S. Lin, X. Cai and Y. Lin, Nat. Protoc., 2020, 15, 2728–2757 CrossRef CAS PubMed
- W. Zhou, F. Yang, S. Li, R. Yuan and Y. Xiang, Chem. Sci., 2022, 13, 11132–11139 RSC
- L. Zhong, S. Cai, Y. Huang, L. Yin, Y. Yang, C. Lu and H. Yang, Anal. Chem., 2018, 90, 12059–12066 CrossRef CAS PubMed
- D. Bhatia, S. Arumugam, M. Nasilowski, H. Joshi, C. Wunder, V. Chambon, V. Prakash, C. Grazon, B. Nadal, P. K. Maiti, L. Johannes, B. Dubertret and Y. Krishnan, Nat. Nanotechnol., 2016, 11, 1112–1119 CrossRef CAS PubMed
- J. Wang, Q. Li, J. Xue, W. Chen, R. Zhang and D. Xing, Chem. Eng. J., 2021, 410, 127849 CrossRef CAS
- J. Wang, J. Zhou, D. Xu, J. Li and D. Deng, ACS Appl. Mater. Interfaces, 2020, 12, 47197–47207 CrossRef CAS PubMed
- R. Duangrat, A. Udomprasert and T. Kangsamaksin, Cancer Sci., 2020, 111, 3164–3173 CrossRef CAS PubMed
- A. S. Walsh, H. Yin, C. M. Erben, M. J. A. Wood and A. J. Turberfield, ACS Nano, 2011, 5, 5427–5432 CrossRef CAS PubMed
- Z. Xia, P. Wang, X. Liu, T. Liu, Y. Yan, J. Yan, J. Zhong, G. Sun and D. He, Biochemistry, 2016, 55, 1326–1331 CrossRef CAS
- Y. Ma, Z. Wang, Y. Ma, Z. Han, M. Zhang, H. Chen and Y. Gu, Angew. Chem., Int. Ed., 2018, 57, 5389–5393 CrossRef CAS PubMed
- S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831–834 CrossRef CAS PubMed
- X. Liu, F. Zhang, X. Jing, M. Pan, P. Liu, W. Li, B. Zhu, J. Li, H. Chen, L. Wang, J. Lin, Y. Liu, D. Zhao, H. Yan and C. Fan, Nature, 2018, 559, 593–598 CrossRef CAS PubMed
- T. Tian, Y. Li and Y. Lin, Bone Res., 2022, 10, 40 CrossRef CAS PubMed
- A. R. Chandrasekaran, Nat. Rev. Chem., 2021, 5, 225–239 CrossRef CAS PubMed
- H. Jun, X. Wang, W. P. Bricker and M. Bathe, Nat. Commun., 2019, 10, 5419 CrossRef PubMed
- M. Glaser, S. Deb, F. Seier, A. Agrawal, T. Liedl, S. Douglas, M. K. Gupta and D. M. Smith, Molecules, 2021, 26, 2287 CrossRef CAS PubMed
- C. Lin, Y. Ke, Z. Li, J. H. Wang, Y. Liu and H. Yan, Nano Lett., 2009, 9, 433–436 CrossRef CAS PubMed
- K. R. Kim, H. Y. Kim, Y. D. Lee, J. S. Ha, J. H. Kang, H. Jeong, D. Bang, Y. T. Ko, S. Kim, H. Lee and D. R. Ahn, J. Controlled Release, 2016, 243, 121–131 CrossRef CAS PubMed
- A. R. Chandrasekaran, J. Vilcapoma, P. Dey, S. W. Wong-Deyrup, B. K. Dey and K. Halvorsen, J. Am. Chem. Soc., 2020, 142, 6814–6821 CrossRef CAS PubMed
- D. Deng, Y. Chen, J. Cao, J. Tian, Z. Qian, S. Achilefu and Y. Gu, Chem. Mater., 2012, 24, 3029–3037 CrossRef CAS
- L. Qu and X. Peng, J. Am. Chem. Soc., 2002, 124, 2049–2055 CrossRef CAS PubMed
- C. Pu, H. Qin, Y. Gao, J. Zhou, P. Wang and X. Peng, J. Am. Chem. Soc., 2017, 139, 3302–3311 CrossRef CAS PubMed
- C. Femoni, M. C. Iapalucci, S. Ruggieri and S. Zacchini, Acc. Chem. Res., 2018, 51, 2748–2755 CrossRef CAS PubMed
- A. C. Dharmaratne, T. Krick and A. Dass, J. Am. Chem. Soc., 2009, 131, 13604–13605 CrossRef CAS PubMed
- Y. Chen, C. Zeng, D. R. Kauffman and R. Jin, Nano Lett., 2015, 15, 3603–3609 CrossRef CAS PubMed
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC
- R. Jin, H. Qian, Z. Wu, Y. Zhu, M. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS
- C. Zeng, Y. Chen, A. Das and R. Jin, J. Phys. Chem. Lett., 2015, 6, 2976–2986 CrossRef CAS PubMed
- D. C. Gary, M. W. Terban, S. J. L. Billinge and B. M. Cossairt, Chem. Mater., 2015, 27, 1432–1441 CrossRef CAS
- J. Zhang, R. W. Crisp, J. Gao, D. M. Kroupa, M. C. Beard and J. M. Luther, J. Phys. Chem. Lett., 2015, 6, 1830–1833 CrossRef CAS PubMed
- H. Liu, Z. Liu and C. Pu, Nano Res., 2022, 15, 7622–7630 CrossRef CAS
- Z. Gan, J. Chen, J. Wang, C. Wang, M. B. Li, C. Yao, S. Zhuang, A. Xu, L. Li and Z. Wu, Nat. Commun., 2017, 8, 14739 CrossRef CAS PubMed
- Z. Gan, Y. Lin, L. Luo, G. Han, W. Liu, Z. Liu, C. Yao, L. Weng, L. Liao, J. Chen, X. Liu, Y. Luo, C. Wang, S. Wei and Z. Wu, Angew. Chem., Int. Ed., 2016, 55, 11567–11571 CrossRef CAS PubMed
- N. B. Luque and E. Santos, Langmuir, 2012, 28, 11472–11480 CrossRef CAS PubMed
- A. H. Pakiari and Z. Jamsshidi, J. Phys. Chem. A, 2010, 114, 9212–9221 CrossRef CAS PubMed
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC
- Z. Wu, E. Lanni, W. Chen, M. E. Bier, D. Ly and R. Jin, J. Am. Chem. Soc., 2009, 131, 16672–16674 CrossRef CAS PubMed
- J. Yang and R. Jin, ACS Mater. Lett., 2019, 1, 482–489 CrossRef CAS
- C. P. Joshi, M. S. Bootharaju, M. J. Alhilaly and O. M. Bakr, J. Am. Chem. Soc., 2015, 137, 11578–11581 CrossRef CAS PubMed
- J. Yang and R. Jin, J. Phys. Chem. C, 2021, 125, 2619–2625 CrossRef CAS
- T. L. Botha, E. E. Elemike, S. Horn, D. C. Onwudiwe, J. P. Giesy and V. Wepener, Sci. Rep., 2019, 9, 4169 CrossRef PubMed
- B. K. Teo and H. Zhang, Inorg. Chem., 1991, 30, 3115–3116 CrossRef CAS
- B. K. Teo, Inorg. Chem., 1994, 33, 4086–4097 CrossRef CAS
- M. S. Bootharaju, C. P. Joshi, M. R. Parida, O. F. Mohammed and O. M. Bakr, Angew. Chem., Int. Ed., 2016, 55, 922–926 CrossRef CAS PubMed
- Z. Qin, J. Zhang, C. Wan, S. Liu, H. Abroshan, R. Jin and G. Li, Nat. Commun., 2020, 11, 6019 CrossRef CAS PubMed
- T.-A. D. Nguyen, Z. R. Jones, B. R. Goldsmith, W. R. Buratto, G. Wu, S. L. Scott and T. W. Hayton, J. Am. Chem. Soc., 2015, 137, 13319–13324 CrossRef CAS PubMed
- J. Ji, G. Wang, T. Wang, X. You and X. Xu, Nanoscale, 2014, 6, 9185–9191 RSC
- A. Ganguly, I. Chakraborty, T. Udayabhaskararao and T. Pradeep, J. Nanopart. Res., 2013, 15, 1522 CrossRef
- A. N. Beecher, X. Yang, J. H. Palmer, A. L. LaGrassa, P. Juhas, S. J. Billinge and J. S. Owen, J. Am. Chem. Soc., 2014, 136, 10645–10653 CrossRef CAS PubMed
- X. D. Zhang, Z. Luo, J. Chen, X. Shen, S. Song, Y. Sun, S. Fan, F. Fan, D. T. Leong and J. Xie, Adv. Mater., 2014, 26, 4565–4568 CrossRef CAS
- S. Kumar and R. Jin, Nanoscale, 2012, 4, 4222–4227 RSC
- S. R. Bhattacharya, K. Bhattacharya, V. J. Xavier, A. Ziarati, D. Picard and T. Burgi, ACS Appl. Mater. Interfaces, 2022, 14, 29521–29536 CrossRef CAS PubMed
- L. M. Tvedte and C. J. Ackerson, J. Phys. Chem. A, 2014, 118, 8124–8128 CrossRef PubMed
- X. D. Zhang, Z. Luo, J. Chen, S. Song, X. Yuan, X. Shen, H. Wang, Y. Sun, K. Gao, L. Zhang, S. Fan, D. T. Leong, M. Guo and J. Xie, Sci. Rep., 2015, 5, 8669 CrossRef CAS PubMed
- T. T. Jia, G. Yang, S. J. Mo, Z. Y. Wang, B. J. Li, W. Ma, Y. X. Guo, X. Chen, X. Zhao, J. Q. Liu and S. Q. Zang, ACS Nano, 2019, 13, 8320–8328 CrossRef CAS PubMed
- H. Kawasaki, S. Kumar, G. Li, C. Zeng, D. R. Kauffman, J. Yoshimoto, Y. Iwasaki and R. Jin, Chem. Mater., 2014, 26, 2777–2788 CrossRef CAS
- Y. Yang, S. Wang, S. Chen, Y. Shen and M. Zhu, Chem. Commun., 2018, 54, 9222–9225 RSC
- L. Luo, Z. Liu, X. Du and R. Jin, J. Am. Chem. Soc., 2022, 144, 19243–19247 CrossRef CAS PubMed
- W. T. Chang, P. Y. Lee, J. H. Liao, K. K. Chakrahari, S. Kahlal, Y. C. Liu, M. H. Chiang, J. Y. Saillard and C. W. Liu, Angew. Chem., Int. Ed., 2017, 56, 10178–10182 CrossRef CAS PubMed
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC
- S. Li, X. S. Du, B. Li, J. Y. Wang, G. P. Li, G. G. Gao and S. Q. Zang, J. Am. Chem. Soc., 2018, 140, 594–597 CrossRef CAS PubMed
- X. Y. Xie, P. Xiao, X. Cao, W. H. Fang, G. Cui and M. Dolg, Angew. Chem., Int. Ed., 2018, 57, 9965–9969 CrossRef CAS PubMed
- X. Jiang, X. Wang, C. Yao, S. Zhu, L. Liu, R. Liu and L. Li, J. Phys. Chem. Lett., 2019, 10, 5237–5243 CrossRef CAS PubMed
- E. Abbasi, S. F. Aval, A. Akbarzadeh, M. Milani, H. T. Nasrabadi, S. W. Joo, Y. Hanifehpour, K. Nejati-Koshki and R. Pashaei-Asl, Nanoscale Res. Lett., 2014, 9, 247 CrossRef
- P. Patel, V. Patel and P. M. Patel, J. Indian Chem. Soc., 2022, 99, 100514 CrossRef CAS
- J. Bugno, H.-J. Hsu and S. Hong, J. Drug Targeting, 2015, 23, 642–650 CrossRef CAS PubMed
- R. J. Smith, C. Gorman and S. Menegatti, J. Polym. Sci., 2020, 59, 10–28 CrossRef
- E. N. Augustus, E. T. Allen, A. Nimibofa and W. Donbebe, Am. J. Polym. Sci., 2017, 7, 8–14 CAS
- M. A. van Dongen, A. Desai, B. G. Orr, J. R. Baker Jr. and M. M. Holl, Polymer, 2013, 54, 4126–4133 CrossRef CAS PubMed
- Z. Lyu, L. Ding, A. Y. T. Huang, C. L. Kao and L. Peng, Mater. Today Chem., 2019, 13, 34–48 CrossRef CAS
- M. V. Walter and M. Malkoch, Chem. Soc. Rev., 2012, 41, 4593–4609 RSC
- M. Arseneault, C. Wafer and J. F. Morin, Molecules, 2015, 20, 9263–9294 CrossRef CAS PubMed
- N. Li, T.-H. Tsoi, W.-S. Lo, Y.-J. Gu, H.-Y. Wan and W.-T. Wong, Polym. Chem., 2017, 8, 6989–6996 RSC
- C. Chen, P. Posocco, X. Liu, Q. Cheng, E. Laurini, J. Zhou, C. Liu, Y. Wang, J. Tang, V. D. Col, T. Yu, S. Giorgio, M. Fermeglia, F. Qu, Z. Liang, J. J. Rossi, M. Liu, P. Rocchi, S. Pricl and L. Peng, Small, 2016, 12, 3667–3676 CrossRef CAS PubMed
- P. Garrigue, J. Tang, L. Ding, A. Bouhlel, A. Tintaru, E. Laurini, Y. Huang, Z. Lyu, M. Zhang, S. Fernandez, L. Balasse, W. Lan, E. Mas, D. Marson, Y. Weng, X. Liu, S. Giorgio, J. Iovanna, S. Pricl, B. Guillet and L. Peng, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11454–11459 CrossRef CAS PubMed
- J. Chen, A. Ellert-Miklaszewska, S. Garofalo, A. K. Dey, J. Tang, Y. Jiang, F. Clement, P. N. Marche, X. Liu, B. Kaminska, A. Santoni, C. Limatola, J. J. Rossi, J. Zhou and L. Peng, Nat. Protoc., 2021, 16, 327–351 CrossRef CAS PubMed
- J. Yang, K. Wang, Y. Zheng, Y. Piao, J. Wang, J. Tang, Y. Shen and Z. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202202128 CAS
- B. Basly, G. Popa, S. Fleutot, B. P. Pichon, A. Garofalo, C. Ghobril, C. Billotey, A. Berniard, P. Bonazza, H. Martinez, D. Felder-Flesch and S. Begin-Colin, Dalton Trans., 2013, 42, 2146–2157 RSC
- V. Brunetti, L. M. Bouchet and M. C. Strumia, Nanoscale, 2015, 7, 3808–3816 RSC
- J. Nithyanandhan and N. Jayaraman, J. Org. Chem., 2002, 67, 6282–6285 CrossRef CAS PubMed
- C. Wu, C. Gao, S. Lu, X. Xu, N. Wen, S. Zhang and M. Liu, J. Biomed. Mater. Res., Part A, 2018, 106, 440–449 CrossRef CAS PubMed
- E. E. Simanek, Molecules, 2021, 26, 4774 CrossRef CAS PubMed
- A. M. Caminade and J. P. Majoral, Molecules, 2016, 21, 538 CrossRef PubMed
- M. Nikzamir, Y. Hanifehpour, A. Akbarzadeh and Y. Panahi, J. Inorg. Organomet. Polym., 2021, 31, 2246–2261 CrossRef CAS
- L. Chen, J. Li, Y. Fan, J. Qiu, L. Cao, R. Laurent, S. Mignani, A. M. Caminade, J. P. Majoral and X. Shi, Biomacromolecules, 2020, 21, 2502–2511 CrossRef CAS PubMed
- Y. Dong, T. Yu, L. Ding, E. Laurini, Y. Huang, M. Zhang, Y. Weng, S. Lin, P. Chen, D. Marson, Y. Jiang, S. Giorgio, S. Pricl, X. Liu, P. Rocchi and L. Peng, J. Am. Chem. Soc., 2018, 140, 16264–16274 CrossRef CAS PubMed
- A. Janaszewska, J. Lazniewska, P. Trzepinski, M. Marcinkowska and B. Klajnert-Maculewicz, Biomolecules, 2019, 9, 330 CrossRef CAS PubMed
- M. H. Han, J. Chen, J. Wang, S. L. Chen and X. T. Wang, J. Biomed. Nanotechnol., 2010, 6, 82–92 CrossRef CAS PubMed
- S. P. Mukherjee, M. Davoren and H. J. Byrne, Toxicol. In Vitro, 2010, 24, 169–177 CrossRef CAS PubMed
- S. P. Mukherjee, F. M. Lyng, A. Garcia, M. Davoren and H. J. Byrne, Toxicol. Appl. Pharmacol., 2010, 248, 259–268 CrossRef CAS PubMed
- M. Ciolkowski, J. F. Petersen, M. Ficker, A. Janaszewska, J. B. Christensen, B. Klajnert and M. Bryszewska, Nanomedicine, 2012, 8, 815–817 CrossRef CAS PubMed
- K. Zhou, L. H. Nguyen, J. B. Miller, Y. Yan, P. Kos, H. Xiong, L. Li, J. Hao, J. T. Minnig, H. Zhu and D. J. Siegwart, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 520–525 CrossRef CAS PubMed
- D. A. Moreira, S. D. Santos, V. Leiro and A. P. Pego, Pharmaceutics, 2023, 15, 1054 CrossRef CAS PubMed
- G. Yang, N. Sadeg and H. Belhadj-Tahar, Drug Des., 2017, 6, 1000144 Search PubMed
- H. Belhadj-Tahar, A. Chen, Y. Jia, S. Wu, N. Sadeg, H. Zhao, C. Li, G. Gu, Y. Gao and G. Yang, J. Clin. Oncol., 2018, 36, e15569 Search PubMed
- H. Belhadj-Tahar, J. Chen, P. Song, J. Zhao, M. Quan, C. Li, X. Gu, G. Yang and Y. Gao, J. Global Oncol., 2019, 5, 96 Search PubMed
- L. M. Kaminskas, D. E. V. Pires and D. B. Ascher, Sci. Rep., 2019, 9, 15465 CrossRef PubMed
- R. Sharma, A. Sharma, S. P. Kambhampati, R. R. Reddy, Z. Zhang, J. L. Cleland, S. Kannan and R. M. Kannan, Bioeng. Transl. Med., 2018, 3, 87–101 CrossRef CAS PubMed
- J. Wang, Y. Zhang, J. Pi, D. Xing and C. Wang, J. Controlled Release, 2021, 340, 149–167 CrossRef CAS PubMed
- G. Speranza, Nanomaterials, 2021, 11, 967 CrossRef CAS PubMed
- Y. Segawa, H. Ito and K. Itami, Nat. Rev. Mater., 2016, 1, 15002 CrossRef CAS
- Y.-X. Chen, D. Lu, G.-G. Wang, J. Huangfu, Q.-B. Wu, X.-F. Wang, L.-F. Liu, D.-M. Ye, B. Yan and J. Han, ACS Sustainable Chem. Eng., 2020, 8, 6657–6666 CrossRef CAS
- B. P. Qi, H. Hu, L. Bao, Z. L. Zhang, B. Tang, Y. Peng, B. S. Wang and D. W. Pang, Nanoscale, 2015, 7, 5969–5973 RSC
- Y. Yan, J. Chen, N. Li, J. Tian, K. Li, J. Jiang, J. Liu, Q. Tian and P. Chen, ACS Nano, 2018, 12, 3523–3532 CrossRef CAS PubMed
- J. Li, X. Zhang, J. Jiang, Y. Wang, H. Jiang, J. Zhang, X. Nie and B. Liu, Toxicol. Sci, 2019, 167, 269–281 CrossRef CAS PubMed
- W. Chen, G. Lv, W. Hu, D. Li, S. Chen and Z. Dai, Nanotechnol. Rev., 2018, 7, 157–185 CrossRef CAS
- C. Zhao, X. Song, Y. Liu, Y. Fu, L. Ye, N. Wang, F. Wang, L. Li, M. Mohammadniaei, M. Zhang, Q. Zhang and J. Liu, J. Nanobiotechnol., 2020, 18, 142 CrossRef CAS PubMed
- A. Ghaffarkhah, E. Hosseini, M. Kamkar, A. A. Sehat, S. Dordanihaghighi, A. Allahbakhsh, C. van der Kuur and M. Arjmand, Small, 2022, 18, e2102683 CrossRef PubMed
- S. Chung, R. A. Revia and M. Zhang, Adv. Mater., 2021, 33, e1904362 CrossRef PubMed
- N. Abid, A. M. Khan, S. Shujait, K. Chaudhary, M. Ikram, M. Imran, J. Haider, M. Khan, Q. Khan and M. Maqbool, Adv. Colloid Interface Sci., 2022, 300, 102597 CrossRef CAS PubMed
- L. Song, J. Shi, J. Lu and C. Lu, Chem. Sci., 2015, 6, 4846–4850 RSC
- W. Su, H. Wu, H. Xu, Y. Zhang, Y. Li, X. Li and L. Fan, Mater. Chem. Front., 2020, 4, 821–836 RSC
- A. Kalluri, D. Debnath, B. Dharmadhikari and P. Patra, Method. Enzymol., 2018, 609, 335–354 CrossRef CAS PubMed
- X. Yan, X. Cui and L.-S. Li, J. Am. Chem. Soc., 2010, 132, 5944–5945 CrossRef CAS PubMed
- Q. Li, S. Zhang, L. Dai and L. S. Li, J. Am. Chem. Soc., 2012, 134, 18932–18935 CrossRef CAS PubMed
- X. Yan, B. Li, X. Cui, Q. Wei, K. Tajima and L. S. Li, J. Phys. Chem. Lett., 2011, 2, 1119–1124 CrossRef CAS PubMed
- G. D. Nguyen, H. Z. Tsai, A. A. Omrani, T. Marangoni, M. Wu, D. J. Rizzo, G. F. Rodgers, R. R. Cloke, R. A. Durr, Y. Sakai, F. Liou, A. S. Aikawa, J. R. Chelikowsky, S. G. Louie, F. R. Fischer and M. F. Crommie, Nat. Nanotechnol., 2017, 12, 1077–1082 CrossRef CAS PubMed
- J. R. Sanchez-Valencia, T. Dienel, O. Groning, I. Shorubalko, A. Mueller, M. Jansen, K. Amsharov, P. Ruffieux and R. Fasel, Nature, 2014, 512, 61–64 CrossRef CAS PubMed
- R. S. K. Houtsma, J. de la Rie and M. Stohr, Chem. Soc. Rev., 2021, 50, 6541–6568 RSC
- A. P. Johnson, H. V. Gangadharappa and K. Pramod, J. Controlled Release, 2020, 325, 141–162 CrossRef CAS PubMed
- L. Chen, Y. Hernandez, X. Feng and K. Mullen, Angew. Chem., Int. Ed., 2012, 51, 7640–7654 CrossRef CAS PubMed
- J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Mullen and R. Fasel, Nature, 2010, 466, 470–473 CrossRef CAS PubMed
- Y. Choi, S. S. Kim, J. H. Kim, J. Kang, E. Choi, S. E. Choi, J. P. Kim, O. Kwon and D. W. Kim, ACS Nano, 2020, 14, 12195–12202 CrossRef CAS PubMed
- S. Ligima, Nature, 1991, 354, 56–58 CrossRef
- A. Yahyazadeh and B. Khoshandam, Results Phys., 2017, 7, 3826–3837 CrossRef
- F. Yuan, T. Yuan, L. Sui, Z. Wang, Z. Xi, Y. Li, X. Li, L. Fan, Z. Tan, A. Chen, M. Jin and S. Yang, Nat. Commun., 2018, 9, 2249 CrossRef PubMed
- Z. Wang, F. Yuan, X. Li, Y. Li, H. Zhong, L. Fan and S. Yang, Adv. Mater., 2017, 29, 1702910 CrossRef PubMed
- A. Hussain, Y. Liao, Q. Zhang, E. X. Ding, P. Laiho, S. Ahmad, N. Wei, Y. Tian, H. Jiang and E. I. Kauppinen, Nanoscale, 2018, 10, 9752–9759 RSC
- P. X. Hou, F. Zhang, L. Zhang, C. Liu and H. M. Cheng, Adv. Funct. Mater., 2021, 32, 2108541 CrossRef
- Y. M. Manawi Ihsanullah, A. Samara, T. Al-Ansari and M. A. Atieh, Materials, 2018, 11, 822 CrossRef PubMed
- H. Omachi, Y. Segawa and K. Itami, Acc. Chem. Res., 2012, 45, 1378–1389 CrossRef CAS PubMed
- H. Omachi, T. Nakayama, E. Takahashi, Y. Segawa and K. Itami, Nat. Chem., 2013, 5, 572–576 CrossRef CAS PubMed
- H. Omachi, Y. Segawa and K. Itami, Org. Lett., 2011, 13, 2480–2483 CrossRef CAS PubMed
- Z. Zhu, Y. Zhai, Z. Li, P. Zhu, S. Mao, C. Zhu, D. Du, L. A. Belfiore, J. Tang and Y. Lin, Mater. Today, 2019, 30, 52–79 CrossRef CAS
- H. Ding, J. S. Wei, P. Zhang, Z. Y. Zhou, Q. Y. Gao and H. M. Xiong, Small, 2018, 14, e1800612 CrossRef PubMed
- C. Xia, W. Wu, T. Yu, X. Xie, C. van Oversteeg, H. C. Gerritsen and C. de Mello Donega, ACS Nano, 2018, 12, 8350–8361 CrossRef CAS PubMed
- R. Czarnomysy, A. Bielawska and K. Bielawski, Int. J. Nanomed., 2019, 14, 7123–7139 CrossRef CAS PubMed
- A. J. Perise-Barrios, R. Gomez, A. L. Corbi, J. de la Mata, A. Dominguez-Soto and M. A. Munoz-Fernandez, Nanoscale, 2015, 7, 3857–3866 RSC
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |