
Atomic strain and catalytic properties of formate oxidation and dehydrogenation in AgPd nanoalloys†
Tao
Jin
ab,
Longfei
Guo
ab,
Quan
Tang
ab,
Junpeng
Wang
ab,
Bowei
Pan
ab,
Zhen
Li
ab,
Chongyang
Wang
ab,
Shuang
Shan
ab and
Fuyi
Chen
 *ab
*ab
aState Key Laboratory of Solidification Processing, Northwestern Polytechnical University, Xi'an, 710072, China. E-mail: fuyichen@nwpu.edu.cn
bSchool of Materials Science and Engineering, Northwestern Polytechnical University, Xi'an, 710072, China
First published on 29th May 2023
Abstract
Formate is a promising hydrogen carrier for safe storage and transport and a fuel for direct formate fuel cells. However, the sluggish kinetics of catalysts for formate dehydrogenation (FDH) and oxidation reactions (FORs) significantly limit the potential applications of formate. Strain effects can effectively modulate catalytic properties by altering the electronic structure. Nevertheless, the lack of theoretical principles to quantify atomic strain and its effects on FDH and FOR catalytic activity has made experimental efforts laborious. In this work, we establish a database of atomic strain distributions for AgPd nanoalloys, reveals that the presence of compressive strain at the edges and corners and compressive strain exerted on the surface of Ag@Pd nanoalloys, particularly the one with an icosahedral shape, boost the FDH and FOR catalytic activity by shifting down the d-band center, thus weakening the adsorption of key intermediate Had. This study provides a theoretical perspective on the development and use of formate as a hydrogen carrier and fuel.
1. Introduction
In recent years, proton-exchange membrane fuel cells (PEMFCs) have gained considerable attention from researchers due to the pressing issues of the energy crisis and environmental pollution.1,2 However, the storage of hydrogen (H2) remains a significant challenge due to its low density and flammability. Formate, on the other hand, serves as a promising hydrogen carrier, which can produce hydrogen through the formate dehydrogenation reaction (FDH) under ambient conditions.3 Furthermore, formate can be utilized as a fuel for a direct formate fuel cell (DFFC), which offers a theoretical voltage of 1.45 V, surpassing methanol and ethanol fuel cells4 by 0.24 V and 0.31 V, respectively. Additionally, formate can be easily obtained from renewable sources through photocatalytic5 or electrocatalytic6 reduction of CO2, rendering it sustainable and eco-friendly.Formate has several advantages as a hydrogen carrier, including high energy density, non-toxicity, and safe handling in aqueous solutions.7 For instance, potassium formate (KHCOO) can provide a theoretical hydrogen capacity of 29 g H2 per L at room temperature.8 The dehydrogenation of a formate solution can produce high-purity hydrogen under milder operating conditions and has the advantage of avoiding CO products.9 Therefore, it is a promising candidate for the safe storage and transportation of hydrogen at ambient temperatures and pressures. In recent decades, researchers have found that Pd-based catalysts are considered the best candidates for FDH, including Pd single-atom catalysts,10 Pd/C catalysts with O-functional groups,9 Ag–Pd core–shell catalysts,7 and Au3Pd1 catalysts.11 However, the primary issues for FDH catalysts remain poor catalyst stability and sluggish kinetics.
The commercial application of DFFCs is hindered by the sluggish kinetics and poor stability of the formate oxidation reaction (FOR) catalysts.12–17 Pd-based catalysts have emerged as a promising alternative for FOR catalysts without CO poisoning.17 To enhance the FOR catalytic activity of Pd-based catalysts, several strategies have been employed. Firstly, the alloying effect has been utilized, including AgPd,13 AgPdCu,12 AgPdCo,18 and AgPdAu,19 which not only improves the activity and stability of catalysts but also reduces the amount of Pd used in catalysts and lowers the cost. Secondly, catalysts with different microstructures have been synthesized, such as AgPdIr nanoflowers,17 AgPdRh nanosheets,16 and Janus-shaped AgPdNi.20 Lastly, catalysts with high-valence oxides21 and fluorides22 have been employed. However, varying the catalyst composition and micromorphology alone has its limits in controlling catalytic activity.23 The strain effect adds an additional dimension of tunability to traditional catalytic variables, including the types of metals, surfaces, and surface sites. Different responses to strain can be utilized to regulate catalytic reactions,24 and strain effects have received significant attention due to their correlation with the intrinsic activity of catalysts.25–27
At the atomistic scale, the strain effect can induce changes in lattice parameters, modify intrinsic interatomic distances and work function28 and alter the energy levels of bonding electrons.29 Furthermore, strain can change the electronic structure of the active site in the catalyst structure,30 thereby altering the adsorption strength of reactive intermediates and modulating the reaction performance. Theoretical studies have demonstrated that a surface strain of only 1% can shift the d-band center of Pt by approximately 0.1 eV, leading to significant changes in the adsorption energy of reactive intermediates.24,31–33 Moreover, recent research has found that lattice strain can enhance the activity and selectivity of CO2 reduction reactions (CO2RR) by breaking the linear scaling relationship.33 The authors propose that the binding-energy response to strain depends on the coupling of the adsorbate-induced eigenstress with the applied strain, indicating that tensile strain can either strengthen or weaken the binding energy of the adsorbate.
Various experimental approaches have been implemented to investigate the catalytic properties of nanomaterials by inducing strain. These methods involve both externally applied strain through mechanical methods and bombardment and internally or structurally induced strain through the construction of heterostructures and the synthesis of shape- and size-controlled nanoparticles.34 Certain polyhedral shapes such as highly symmetric polyhedra (e.g. octahedron and tetrahedron) and multiple twinned structures (e.g., decahedron and icosahedron) may self-induce surface strain.35 For instance, Wang et al.36 synthesized Au75Pd25 icosahedral bimetallic nanocatalysts that exhibited 0.51% tensile strain on their surface and −1.26% compressive strain at their corners. Huang et al.37 observed an average of 1.8% tensile strain on the Pd icosahedral surface and an average of −0.5% compressive strain on the octahedral surface. The tensile strain on the surface of icosahedra boosts the CO2 reduction reaction activity by shifting up the d-band center, thus strengthening the adsorption of key intermediate COOH*. The catalytic activity of Pd-based core–shell nanoalloys, such as H2O2 synthesis38 and other physical properties, such as heating-induced reshaping preference,39 have also been systematically investigated by theoretical calculations.
To promote the development and utilization of formate, it is crucial to design a catalyst with high catalytic activity for FDH and FOR, particularly considering the increasing price of precious metals.40,41 However, to our knowledge, limited research has been conducted on the impact of the strain effect on the properties of catalysts used for FDH and FOR. Therefore, this study aims to analyse the atomic strain distribution within AgPd nanoalloys and explore how atomic strain affects the catalytic activities of FDH and FOR.
To accomplish this goal, a comprehensive investigation of the aforementioned properties was conducted. Firstly, AgPd nanoalloys were constructed utilizing a genetic algorithm combined with the Gupta potential. Subsequently, molecular dynamics simulations were performed to analyse the atomic strain and stress of the nanoalloys. The simulations revealed the presence of compressive strain at the edges and corners of the nanoalloys and compressive strain exerted on the surface of Ag@Pd nanoalloys. Density functional theory (DFT) calculations were carried out to investigate the effect of compressive and tensile strain on the FDH and FOR activities on Pd(100), Pd(111), AgPd(100), and AgPd(111) surfaces. The results indicate that compressive strain can increase the activities of FDH and FOR on Pd(111), AgPd(100), and AgPd(111) surfaces. In contrast, tensile strain can increase the activities of FDH and FOR on the Pd(100) surface, as a result of changes in the preferred adsorption site of the intermediate Had. Therefore, the compressive strain exerted on the surface of Ag@Pd nanoalloys, particularly ones with an icosahedral shape, boosts the FDH and FOR catalytic activity by shifting down the d-band center and thus weakening the adsorption of the key intermediate Had. This study provides insight into the influence of the strain effect on the catalytic activity of FDH and FOR, shedding light on the design of highly active catalysts for these reactions. It offers a theoretical basis for the development of Pd-based catalysts for FDH and FOR with high intrinsic activity, which has significant implications for the commercialization of direct formate fuel cells and provides a promising approach for hydrogen storage and transportation.
2. Methods
2.1 Nanoalloy structure generation
In this study, the Birmingham cluster genetic algorithm (BCGA) software42,43 was utilized, in conjunction with the many-body Gupta potential,44 to produce AgPd nanoalloys in cuboctahedral (Oh), decahedral (Dh) and icosahedral (Ih) shapes, consisting of 55, 147, and 309 atoms. The Gupta potential can more accurately describe the surface energy of a metal45 than the embedded atom method (EAM) potential, and can be applied at the appropriate time corresponding to each structure, and the structure converges directly to its stable conformation regardless of the initial configuration.44 The Oh- and Dh-shaped nanoalloys are enclosed by both (100) and (111) surfaces, whereas the Ih-shaped nanoalloy is exclusively enclosed by (111) surfaces. We constructed two types of pure metal nanoalloys, donated as Ag and Pd, and two core–shell nanoalloys, donated as Ag@Pd and Pd@Ag.2.2 Molecular dynamics (MD) simulations
The stress distribution and strain fields of these nanoalloys were obtained through MD simulations using the LAMMPS46 software with the EAM interatomic potential47 for AgPd nanoalloys. To obtain the equilibrium configuration, we first applied the conjugate gradient (CG) minimization method to the entire nanoalloy, followed by performing MD simulations in the canonical ensemble (NVT) at 50 K with the Nose–Hoover thermostat48 for 100 picoseconds at a timestep of 1 femtosecond. The atomic strain was calculated by comparing the final configuration with the initial configuration, and then mapped atom-wise onto the final configuration. The OVITO49 software was used to calculate and visualize the atomic strain. Detailed instructions for calculating the atomic strain using the OVITO software are provided in the ESI.† This methodology is consistent with previously reported studies.37,502.3 Density functional theory (DFT) calculations
The DFT calculations were performed using the Vienna ab initio simulation package (VASP) code with the periodic plane-wave method.51–53 We adopted the projected augmented wave (PAW) scheme54 in conjugation with the generalized gradient approximation (GGA) of the exchange-correlation functional in the Perdew–Burke–Ernzerhof (PBE) form.55 Full geometry optimizations were performed for all model atomic configurations. A plane-wave cutoff of 450 eV was set for all calculations, which was determined by convergence tests, as shown in Fig. S2.† The self-consistent field and atomic force convergence criterion were set to be 1 × 10−7 eV and 0.02 eV Å−1, respectively, for geometry optimizations and other calculations. For the Brillouin zone integration, we used 7 × 7 × 1 and 13 × 13 × 13 Monkhorst–Pack k-point mesh samplings for slabs and metal unit cells, respectively. The slab model was simulated by a four-atomic-layer slab with the bottom two atomic layers fixed, and a vacuum space of 20 Å in the z-direction was included to minimize the interaction between periodic images. Furthermore, ab initio molecular dynamics simulations (AIMD) were performed in the canonical ensemble (NVT)56 with a Nose–Hoover thermostat57 at 300 K for a time period of 2 ps.2.4 Thermodynamic treatment
Lattice strain is defined as the deviation from the equilibrium spacing between atoms in the material lattice. The strain is calculated as the change in the lattice constant (astrain) normalized to the unstrained bulk (abulk). The change in crystal strain was simulated by expanding and contracting the lattice of the unstrained metal unit cell with the lattice constant (abulk) used in each DFT simulation, and the lattice strain was given by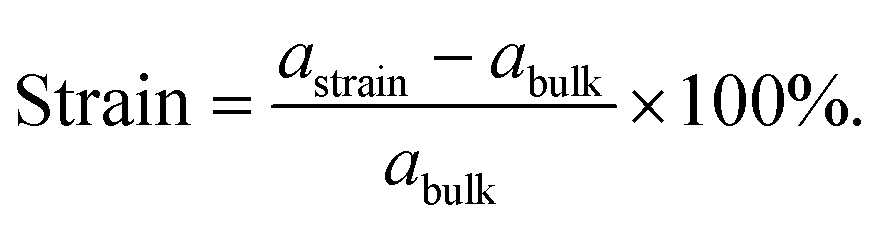 | (1) |
The adsorption energy is defined as follows:
| Ead = Eslab+x − Eslab − Ex, | (2) |
The thermodynamics of the formate oxidation and formate dehydrogenation reaction mechanism was evaluated by calculating the Gibbs free energy to construct a reaction diagram depicting the various reaction steps as follows:
| ΔG = ΔH − T × ΔS, | (3) |
3. Results and discussion
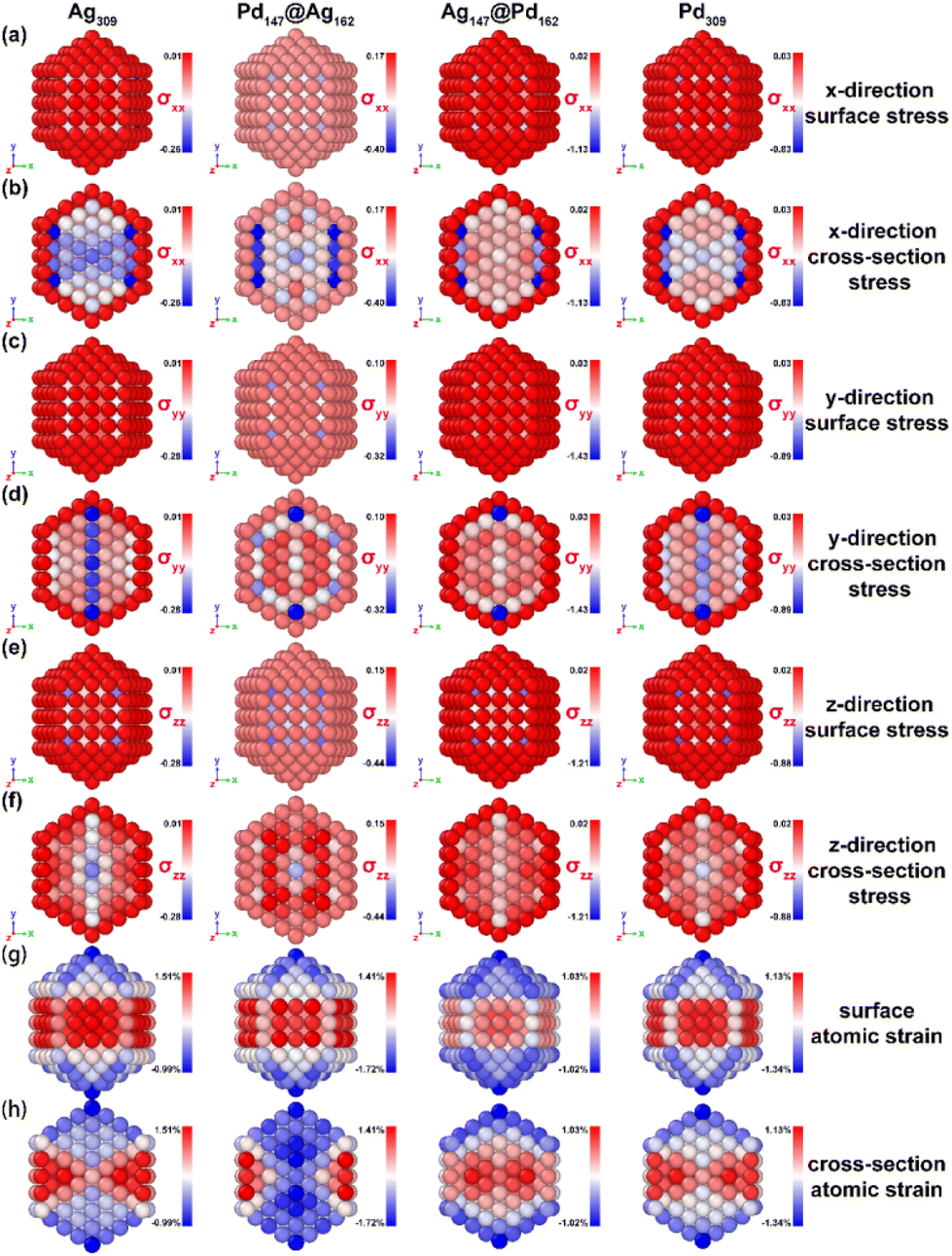
We conducted theoretical calculations to gain insight into the atomic strain related to formate dehydrogenation and oxidation activity. To examine the influence of the size effect on atomic strain, we compared three isomers of nanoalloys comprising 55, 147, and 309 atoms. Fig. S1† illustrates the atomic structures of the 309-atom cuboctahedral (Oh), decahedral (Dh) and icosahedral (Ih) nanoalloys, donated as Ag309, Pd147@Ag162, Ag147@Pd162, and Pd309. The Oh- and Dh-shaped nanoalloys are enclosed by both the (100) and (111) surfaces, whereas the Ih-shaped nanoalloys are enclosed by the (111) surface. Additionally, the common neighbor analysis (CNA) reveals twinning within the nanoalloys, with no twinning in Oh, fivefold twinning in Dh, and tenfold twinning in Ih nanoalloys. As depicted in Fig. S1,† the surfaces of Oh-, Dh-, and Ih-shaped nanoalloys have an abundance of edge and corner sites. During the minimization stage of the MD simulation, surface atoms tend to seek a lower energy state, resulting in deviations from the original model positions, which is termed atomic strain.45 The displacements of the AgPd nanoalloy in the Oh-, Dh-, and Ih-shaped structures after the MD simulations are shown in Fig. S3.†3.1 Stress and atomic strain distribution of Oh-shaped AgPd nanoalloys
In order to investigate the atomic strain of the nanoalloys in depth, we conducted calculations and analyzed the stress along the x-, y- and z-directions on their surfaces and cross-sections. Fig. 1 illustrates the stress distribution and atomic strain field for the Ag309, Pd147@Ag162, Ag147@Pd162, and Pd309 nanoalloys in an Oh shape. | ||
| Fig. 1 The stress distribution and atomic strain field maps of Ag309, Pd147@Ag162, Ag147@Pd162, and Pd309 nanoalloys in the cuboctahedral (Oh) shape. (a and b) The x-direction, (c and d) y-direction, and (e and f) z-direction for the surface and cross-sectional stress. (g and h) The atomic strain at the surface and in the cross-section. The unit of measurement used for stress in the diagram is bars. The nature and intensity of stress and atomic strain are represented by color maps, with blue indicating negative values corresponding to compressive stress and strain, red indicating positive values corresponding to tensile stress and strain, and white representing zero stress and strain. The surface strain is calculated based on the equilibrium bond length in bulk Ag and Pd. | ||
Fig. 1(a, c, and e) and (b, d, and f) show the stress distribution along the x-, y- and z-directions in the cross-section of the Oh-shaped nanoalloys, revealing that the subsurface of the nanoalloy experiences compressive stress, consistent with prior research.53 In addition, since the outer surface atoms lack coordinating atoms, they are always under tensile stress. Fig. 1(g) illustrates the common characteristics of Oh-shaped Ag309, Pd147@Ag162, Ag147@Pd162, and Pd309 nanoalloys: the nanoalloys tend to shrink slightly to achieve a lower surface energy state at optimal lattice parameters, inducing significant compression strain at the edges and corners.45 Specifically, the edges and corner sites exhibit compressive strain (averaging −0.93%), while the surfaces of Ag309, Pd147@Ag162, and Pd309 nanoalloys exhibit tensile strain (averaging 1.35%). In contrast, the surface of the Ag147@Pd162 nanoalloy shows negligible atomic strain. The average atomic strain on the surface of the Oh-shaped nanoalloys can be ranked in the following order: Pd@Ag (0.34%) > Pd (0.22%) > Ag (0.11%) > Ag@Pd (−0.32%).
Fig. 1(h) depicts the cross-sectional atomic strain field within Ag309, Ag147@Pd162, and Pd309 nanoalloys, highlighting the presence of tensile strain (averaging 1.13%) within their interiors. This contrasts with the compressive strain (−1.04%) observed within Pd147@Ag162 nanoalloys, which can be attributed to differences in atomic radii and atomic interactions between Ag and Pd.
3.2 Stress and atomic strain distribution of Dh-shaped AgPd nanoalloys
The stress distribution and atomic strain field observed in the Dh-shaped nanoalloys are comparable to those of the Oh-shaped nanoalloys, despite the differences in their shapes. In addition, both types of nanoalloys are enclosed by (100) and (111) surfaces.Fig. 2 summarizes the stress distribution and atomic strain field of Ag309, Pd147@Ag162, Ag147@Pd162, and Pd309 nanoalloys with Dh shapes. Fig. 2(a, c, and e) and (b, d, and f) display the surface and cross-section stress of Dh-shaped nanoalloys along the x-, y-, and z-directions, respectively. Similar to Oh-shaped nanoalloys, Dh-shaped nanoalloys exhibit tensile stress on their surfaces due to the lack of coordination atoms. Conversely, compressive stress is present in the interior of the nanoalloys along the x-, y-, and z-directions.
 | ||
| Fig. 2 The stress distribution and atomic strain field maps of Ag309, Pd147@Ag162, Ag147@Pd162, and Pd309 nanoalloys in the decahedral (Dh) shape. (a and b) The x-direction, (c and d) y-direction, and (e and f) z-direction for surface and cross-sectional stress. (g and h) The atomic strain at the surface and in the cross-section. The unit of measurement used for stress in the diagram is bars. The nature and intensity of stress and atomic strain are represented by color maps, with blue indicating negative values corresponding to compressive stress and strain, red indicating positive values corresponding to tensile stress and strain, and white representing zero stress and strain. The surface strain is calculated based on the equilibrium bond length in bulk Ag and Pd. | ||
Fig. 2(g) displays the atomic strain on the surface of Dh-shaped nanoalloys. The distribution of atomic strain on the surface exhibits a common feature, with compressive strain (averaging −1.27%) at the edges and corners of the (111) surface of nanoalloys and tensile strain (averaging 1.27%) on the (100) surface. Additionally, the (111) surface of the Ag147@Pd162 nanoalloy experiences a slight compressive strain (−0.51%) due to the smaller radius of Pd atoms compared to Ag, causing the inward contraction of Pd atoms to attain a lower energy state. The average atomic strain on the surface of the Dh-shaped nanoalloys can be ranked in the following order: Ag (0.13%) > Pd (0.01%) ≈ Pd@Ag (0.01%) > Ag@Pd (−0.25%).
Fig. 2(h) presents the atomic strain in the cross-section of the nanoalloy. The centers within the Ag309, Ag147@Pd162, and Pd309 nanoalloys have tensile strain, whereas the center inside the Pd147@Ag162 nanoalloy experiences compressive strain. This observation is consistent with the atomic strain distribution within the Oh-shaped nanoalloys since Pd has a smaller atomic radius than Ag.
3.3 Stress and atomic strain distribution of Ih-shaped AgPd nanoalloys
The stress distribution and strain fields of the Ih-shaped nanoalloys with higher symmetry exhibit slight differences when compared to Oh- and Dh-shaped nanoalloys. Fig. 3 illustrates the stress distribution along the x-, y-, and z-directions and the atomic strain fields of the Ih-shaped nanoalloys, all of which are enclosed by the (111) surface. | ||
| Fig. 3 The stress distribution and atomic strain field maps of Ag309, Pd147@Ag162, Ag147@Pd162, and Pd309 nanoalloys in the icosahedral (Ih) shape. (a and b) The x-direction, (c and d) y-direction, and (e and f) z-direction for surface and cross-sectional stress. (g and h) The atomic strain at the surface and in the cross-section. The unit of measurement used for stress in the diagram is bars. The nature and intensity of stress and atomic strain are represented by color maps, with blue indicating negative values corresponding to compressive stress and strain, red indicating positive values corresponding to tensile stress and strain, and white representing zero stress and strain. The surface strain is calculated based on the equilibrium bond length in bulk Ag and Pd. | ||
Fig. 3(a, c, and e) and (b, d, and f) depict the surface and cross-sectional stress along the x-, y-, and z-directions, respectively.
The surface of the Ih-shaped nanoalloys is under tensile stress in all x-, y- and z-directions, which agrees with the Oh and Dh-shaped nanoalloys. However, the Ih-shaped nanoalloys exhibit compressive stress at the center, as shown by the strain distribution diagram in the cross-section. This is in contrast to the Oh and Dh-shaped nanoalloys, which exhibit compressive stress only at the subsurface.
Fig. 3(g) displays the atomic strain distribution on the surface of the Ih-shaped nanoalloys, which exhibits some similarities across different materials. Specifically, the corners exhibit a compressive strain (averaging −1.08%), while the surfaces exhibit a tensile strain (averaging 1.41%). This observation agrees with the strain distribution previously reported for Ih-shaped Pt,35 Pd,30 and AuPd36 nanoalloys, where compressive strain is present at the edges and corners, and tensile strain is present on the surface. The average atomic strain on the surface of the Ih-shaped nanoalloys can be ranked in the following order: Ag (0.65%) > Pd@Ag (0.56%) > Pd (−0.01%) > Ag@Pd (−0.33%).
Fig. 3(h) illustrates the atomic strain fields in the cross-section of Ih-shaped nanoalloys, where Ag, Pd@Ag, and Pd nanoalloys exhibit compressive strain at their centers, while Ag@Pd nanoalloys exhibit tensile strain at their centers, which is consistent with the atomic strain distribution in the cross-section of Oh- and Dh-shaped nanoalloys. The surface and cross-section atomic strain fields of the 55 and 147-atomic nanoalloys are depicted in Fig. S4 and S5,† which are in good agreement with the 309-atomic nanoalloys.
To summarize, the analysis conducted indicates that there is a negative average atomic strain on the surface of Ag@Pd nanoalloys of Oh, Dh, or Ih shape, indicating a compressive strain.
Given that a large library of atomically precise metal NCs has been structurally resolved, it is important to probe the segregation or anti-segregation during the reactions from ab initio calculations to advance our fundamental understanding of catalytic stability.60 As shown in Fig. S6,† the vacuum Eseg of Oh- and Ih-shaped core–shell Ag@Pd nanoalloys is −0.254 and −0.512 eV, respectively, showing a strong Ag surface segregation trend; on the contrary, the segregation energy under the adsorbed of H and O is higher than zero, indicating a tendency for Pd atom surface segregation during the reaction.
Ag@Pd core–shell alloys exhibit a preference for Ag surface segregation under vacuum conditions; however, it is important to note that the thermodynamic feasibility of Ag surface segregation diminishes once H intermediates are formed during formate dehydrogenation and oxidation reactions.
The structural stability of core–shell Ag@Pd nanoalloys, both with and without Ag surface segregation, was examined through additional AIMD simulations, as depicted in Fig. S7 and S8.† These simulations provide further evidence that Ag@Pd nanoalloys in both Oh and Ih shapes under vacuum, H and O conditions maintain their structural integrity.
3.4 Effect of strain on electronic properties
The activity of a catalyst is determined by its electronic properties, including the d-band center and adsorption of reactants and intermediates. The position of the d-band center plays a crucial role in determining the strength and nature of the interaction between the metal surface and adsorbates. If the d-band center is close to the Fermi level, implying strong adsorption, it can lead to the occupation of active sites and result in catalyst poisoning. Conversely, if the d-band center is far from the Fermi level, indicating weak adsorption, it may have a negative impact on the reaction. To investigate the effect of strain on adsorption strength, we calculated the adsorption energy of H, OH, and HCOO−, projected density of states (PDOS), and d-band center of the Pd(100), Pd(111), AgPd(100), and AgPd(111) surfaces in the strain range of −5–5%.Fig. 4(a–c) depict the relationship between adsorption energy of HCOO−, H and OH and strain. The strain effect was found to have a significant impact on the adsorption energy of HCOO−, OH and H on various metallic surfaces. As shown in Fig. 4(a) and (c), compressive strain tends to decrease the adsorption energy of HCOO− and OH on Pd(100), Pd(111), AgPd(100), and AgPd(111) surfaces. This is because the compressive strain causes a negative shift of the d-band center, which in turn decreases the adsorption energy. However, the effect of compressive strain on intermediate H varies depending on the surface type. For instance, compressive strain reduces the adsorption energy on the Pd(111) and AgPd(111) surfaces, while it tends to increase the adsorption energy on the Pd(100) and AgPd(100) surfaces. Notably, strain effects can alter the preferred adsorption site of H on the Pd(100) surface. As shown in Fig. S9,† strain effects cause the H preference adsorption site to change from the hollow site to the bridge site at 5% tensile strain, and the OH preference adsorption site remains at the bridge site. This is due to the effect of adsorbate-induced eigenstress and applied strain. In the absence of applied strain, the adsorption of an H atom at the hollow (four-fold) or bridge (two-fold) site results in a pull or push of the neighboring atoms inward or outward, leading to tension or compression eigenstress. As a result, compressive or tensile strain should stabilize the adsorption, with the preferred H atom adsorption site being the hollow site at −5% compressive strain or the bridge site at 5% tensile strain. These findings are consistent with previous research indicating that the binding-energy response to strain depends on the coupling of the adsorbate-induced eigenstress with the applied strain, and the strain effect can break the scaling law of the adsorbent and change independently with strain variations.33
 | ||
| Fig. 4 The adsorption energies of (a) HCOO−, (b) H, and (c) OH on Pd(100), Pd(111) AgPd(100) and AgPd(111) surfaces, under varying strains. (d) The d-band center as a function of strain. | ||
Fig. 4(d) illustrates the change in the d-band center of the Pd(100), Pd(111), AgPd(100), and AgPd(111) surfaces with varying strains. It reveals that compressive strain can cause a negative shift in the d-band centers because of the decrease in interatomic spacing of the d-band metal surface atoms. This decrease results from the compressive strain, which in turn leads to an increase in the overlap of d orbitals and widening of the d-bandwidth. As the number of d electrons and fractional filling of the d-band remain constant, this widening results in a negative shift of the d-band centers, which is evidenced in the electron density plots as shown in Fig. S10.† As the d-band center shifts up, a distinctive antibonding state appears above the Fermi level.61 The antibonding states above the Fermi level are empty, and the bond becomes increasingly stronger as their numbers increase.62 Thus, strong bonding occurs if the antibonding states are shifted up through the Fermi level (and become empty), and weak bonding occurs if the antibonding states are shifted down through the Fermi level (and become filled).63 The projected state density (PDOS) is shown in Fig. S11.† It is crucial to note that a continuous increase in applied tensile strain does not lead to a consistent positive shift in the d-band center. Fig. S12† illustrates that when a 10% tensile strain is applied to the Pd(111) surface, the resulting d-band center is positioned farther away from the Fermi level compared to the case with a 5% tensile strain.
3.5 Effect of strain on catalytic properties of FDH and FOR
To investigate the catalytic activity of FDH and electrocatalytic activity of FOR, we utilized DFT calculations on Pd(100), Pd(111), AgPd(100), and AgPd(111) surfaces with strain levels ranging from −5% to 5%.Formate acts as an organic hydrogen carrier to facilitate hydrogen storage and transport. We investigated the catalytic activity of formate dehydrogenation reactions (FDH) on the Pd(100), Pd(111), AgPd(100), and AgPd(111) surfaces in the strain range of −5–5%. The elementary steps of FDH54 are as follows:
| HCOO− + * → HCOOad | (R1) |
| HCOOad → CO2 + Had | (R2) |
| Had → 1/2H2 + * | (R3) |
As illustrated in Fig. 5(a and b), the primary obstacle for FDH is the desorption of the H intermediate from the catalyst surface, which is the thermodynamic uphill step, and can be impeded by excessive hydrogen adsorption free energy. Therefore, the free energy change (ΔG3) of this uphill step is a descriptor for FDH. Fig. 5(c) displays the variation of ΔG3 with strain, revealing that the AgPd(111) surface exhibits the worst FDH activity, whereas the AgPd(100) surface with a strain of −5% exhibits the best FDH activity, with a ΔG3 value of 0.022 eV. Fig. 5(d) presents the volcano-type relationship between the H adsorption free energy and the ΔG3 of FDH. A moderate H adsorption free energy significantly reduces the ΔG3 and increases the catalytic activity.
 | ||
| Fig. 5 The free energy diagrams for the dehydrogenation of formate to CO2 and 1/2H2 on (a) Pd(100) and Pd(111) surfaces and (b) AgPd(100) and AgPd(111) surfaces under varying surface strains. (c) ΔG3 of FDH versus surface and strain. (d) Volcano plot of ΔG3 of the formate dehydrogenation reaction versus hydrogen adsorption free energy. | ||
The FOR plays a crucial role in determining the performance of DFFC. Based on early research,21,64–66 the electrochemical conversion of HCOO− oxidation to CO2 and H2O in an alkaline environment typically involves the following elementary steps:
| HCOO− + * → HCOOad− | (R4) |
| HCOOad− → CO2 + Had + e− | (R5) |
| OH− + Had → H2O + * + e− | (R6) |
Fig. 6(a and b) show that the desorption of H intermediates is hindered due to the strong hydrogen adsorption free energy on the Pd(100), Pd(111), and AgPd(111) surfaces, which significantly restricts the reaction. On the other hand, the weak H adsorption free energy on the AgPd(100) surface hinders the breaking of the C–H bond by the formate, which is the crucial step hindering the reaction. Fig. 6(c) presents the strain-dependent overpotential for the FOR. The FOR overpotentials on Pd(100), Pd(111), AgPd(100), and AgPd(111) surfaces without strain were 0.659, 0.607, 0.357, and 0.448 V, respectively, indicating that the FOR catalytic activity of AgPd is significantly superior to that of pure Pd in the absence of strain.
 | ||
| Fig. 6 The free energy diagrams for the oxidation of formate to CO2 and H2O on (a) Pd(100) and Pd(111) surfaces and (b) AgPd(100) and AgPd(111) surface under varying surface strains. (c) The overpotential of FOR versus surface and strain. (d) The volcano plot of the FOR overpotential versus hydrogen adsorption free energy. | ||
The AgPd(100) surface with −5% strain showed the lowest overpotential of 0.244 V, which was reduced by 31.65% compared to the unstrained surface. Fig. 6(d) shows the volcano-type relationship between the H adsorption free energy and the overpotential of the FOR. A moderate H adsorption free energy significantly reduces the overpotential and increases catalytic activity. The increased FOR activity observed on the Pd(100) surface at 5% strain may be attributed to the alteration of preferred H adsorption sites and the enhanced OH adsorption energy. Fig. S15† shows that compressive strain boosts FDH and FOR catalytic activity by shifting down the d-band center, thus weakening the adsorption of the key intermediate Had.
Based on the calculation results presented above, it can be inferred that Ag@Pd nanoalloys with Oh, Dh, and Ih shapes exhibit superior catalytic activities for FDH and FOR. This is attributed to the presence of compressive strain on the surface of these nanoalloys, which can cause the d-band center to shift negatively and weaken the adsorption of intermediate Had. The Ih-shaped Ag@Pd nanoalloy is the optimal catalyst candidate for FDH and FOR, as it has the maximum compressive strain on its surface.
4. Conclusions
In this study, we utilized the Birmingham cluster genetic algorithm software package in conjunction with the Gupta potential to generate Ag, Pd@Ag, Ag@Pd and Pd nanoalloys in cuboctahedral, decahedral and icosahedral shapes. We employed molecular dynamics simulations to observe the presence of twin structures and stress inside the nanoalloy structures. Notably, we found that the strain distribution in Pd@Ag and Ag@Pd nanoalloys is opposite due to the different atomic radii of Ag and Pd. Spontaneous atomic strain was observed on the surfaces and interiors of nanoalloys, with significant compressive strain observed at the edges and corners of nanoalloy surfaces and tensile strain at the face centers. Ag@Pd core–shell alloys exhibit a preference for Ag surface segregation under vacuum conditions; however, it is important to note that the thermodynamic feasibility of Ag surface segregation diminishes once H intermediates are formed during formate dehydrogenation and oxidation reactions. Furthermore, we investigated the impact of strain on dehydrogenation (FDH) and formate oxidation reactions (FOR) using density functional theory (DFT) calculations. Our findings suggest that compressive strain can make the d-band center shift negatively and regulate the adsorption free energy of the reaction intermediate H, leading to optimal activity for FDH and FOR. Therefore, the Ih-shaped Ag@Pd nanoalloys may have the best FDH and FOR activity due to the average compressive strain of the surface. Our studies have revealed that compressive strain enhances the activity of FDH and FOR on the Pd(111), AgPd(100), and AgPd(111) surfaces, while tensile strain enhances the activities of FDH and FOR on the Pd(100) surface due to the altered H preference adsorption site. This work offers valuable theoretical insights for the design and development of highly efficient formate dehydrogenation catalysts and anode catalysts for direct formate fuel cell.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 51874243, 51271148 and 50971100), the Research Fund of State Key Laboratory of Solidification Processing (NPU), China (grant no. 2020-TS-02), the Project of Transformation of Scientific and Technological Achievements of NWPU (grant no. 19-2017), the Funding for Innovation and Venture Capital of Student Work Department of Party Committee of NWPU (grant no. 2021-CXCY-018), and the Open Fund of State Key Laboratory of Advanced Technology for Material Synthesis and Processing (Wuhan University of Technology grant no. 2018-KF-18). We would like to thank the Analytical & Testing Center of Northwestern Polytechnical University for TEM characterizations.References
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed
.
- L. Zhang, Q. Chang, H. Chen and M. Shao, Nano Energy, 2016, 29, 198–219 CrossRef CAS
- J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed
- L. An, T. S. Zhao, S. Y. Shen, Q. X. Wu and R. Chen, Int. J. Hydrogen Energy, 2010, 35, 4329–4335 CrossRef CAS
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS PubMed
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Nat. Commun., 2018, 9, 1320 CrossRef PubMed
- K. Tedsree, T. Li, S. Jones, C. W. A. Chan, K. M. K. Yu, P. A. J. Bagot, E. A. Marquis, G. D. W. Smith and S. C. E. Tsang, Nat. Nanotechnol., 2011, 6, 302–307 CrossRef CAS PubMed
- K. Grubel, H. Jeong, C. W. Yoon and T. Autrey, J. Energy Chem., 2020, 41, 216–224 CrossRef
- Z. Dong, A. Mukhtar, T. Ludwig, S. A. Akhade, S. Kang, B. Wood, K. Grubel, M. Engelhard, T. Autrey and H. Lin, Appl. Catal., B, 2023, 321, 122015 CrossRef CAS
- C. Liu, Q. Bing and J. Liu, Appl. Surf. Sci., 2022, 604, 154510 CrossRef CAS
- X.-T. Guo, J. Zhang, J.-C. Chi, Z.-H. Li, Y.-C. Liu, X.-R. Liu and S.-Y. Zhang, RSC Adv., 2019, 9, 5995–6002 RSC
- Q. Wang, F. Chen, L. Guo, T. Jin, H. Liu, X. Wang, X. Gong and Y. Liu, J. Mater. Chem. A, 2019, 7, 16122–16135 RSC
- L. Guo, F. Chen, T. Jin, H. Liu, N. Zhang, Y. Jin, Q. Wang, Q. Tang and B. Pan, Nanoscale, 2020, 12, 3469–3481 RSC
- Q. Tang, F. Chen, T. Jin, L. Guo, Q. Wang and H. Liu, J. Mater. Chem. A, 2019, 7, 22996–23007 RSC
- Q. Tang, F. Chen, Q. Wang, T. Jin, L. Guo, Y. Wu, S. Yu and Z. Li, J. Mater. Chem. A, 2021, 9, 23072–23084 RSC
- Y. Jin, F. Chen, L. Guo, J. Wang, B. Kou, T. Jin and H. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 26694–26703 CrossRef CAS PubMed
- Y. Jin, F. Chen, T. Jin, L. Guo and J. Wang, J. Mater. Chem. A, 2020, 8, 25780–25790 RSC
- Q. Wang, F. Chen, Q. Tang, L. Guo, T. Jin, B. Pan, J. Wang, Z. Li, B. Kou and W. Bian, Nano Res., 2021, 14, 2268–2276 CrossRef CAS
- B. Pan, F. Chen, B. Kou, J. Wang, Q. Tang, L. Guo, Q. Wang, Z. Li, W. Bian and J. Wang, Nanoscale, 2020, 12, 11659–11671 RSC
- Q. Wang, F. Chen, Q. Tang, L. Guo, T. T. Gebremariam, T. Jin, H. Liu, B. Kou, Z. Li and W. Bian, Appl. Catal., B, 2020, 270, 118861 CrossRef CAS
- T. Jin, F. Chen, L. Guo, Q. Tang, J. Wang, B. Pan, Y. Wu and S. Yu, J. Phys. Chem. C, 2021, 125, 19497–19508 CrossRef CAS
- T. Jin, L. Guo, Q. Tang, J. Wang, B. Pan, C. Wang, Z. Li and F. Chen, J. Phys. Chem. C, 2022, 126(23), 9683–9695 CrossRef CAS
- R. P. Jansonius, L. M. Reid, C. N. Virca and C. P. Berlinguette, ACS Energy Lett., 2019, 4, 980–986 CrossRef CAS
- F. Liu, C. Wu, G. Yang and S. Yang, J. Phys. Chem. C, 2015, 119, 15500–15505 CrossRef CAS
- H. Li, K. Shin and G. Henkelman, J. Chem. Phys., 2018, 149, 174705 CrossRef PubMed
- J. Fan, H. Du, Y. Zhao, Q. Wang, Y. Liu, D. Li and J. Feng, ACS Catal., 2020, 10, 13560–13583 CrossRef CAS
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- S. Thomas, M. S. Manju, K. M. Ajith, S. U. Lee and M. Asle Zaeem, Phys. E, 2020, 123, 114180 CrossRef CAS
- H. Zhu, G. Gao, M. Du, J. Zhou, K. Wang, W. Wu, X. Chen, Y. Li, P. Ma, W. Dong, F. Duan, M. Chen, G. Wu, J. Wu, H. Yang and S. Guo, Adv. Mater., 2018, 30, 1707301 CrossRef PubMed
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Phys. Rev. Lett., 1998, 81, 2819–2822 CrossRef
- M. Escudero-Escribano, P. Malacrida, M. H. Hansen, U. G. Vej-Hansen, A. Velázquez-Palenzuela, V. Tripkovic, J. Schiøtz, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, Science, 2016, 352, 73–76 CrossRef CAS PubMed
- I. G. Shuttleworth, Appl. Surf. Sci., 2016, 378, 286–292 CrossRef CAS
- L. Wang, Z. Zeng, W. Gao, T. Maxson, D. Raciti, M. Giroux, X. Pan, C. Wang and J. Greeley, Science, 2019, 363, 870–874 CrossRef CAS PubMed
- S. Yang, F. Liu, C. Wu and S. Yang, Small, 2016, 12, 4028–4047 CrossRef CAS PubMed
- J. Wu, L. Qi, H. You, A. Gross, J. Li and H. Yang, J. Am. Chem. Soc., 2012, 134, 11880–11883 CrossRef CAS PubMed
- L. Wang, S. Zhao, C. Liu, C. Li, X. Li, H. Li, Y. Wang, C. Ma, Z. Li and J. Zeng, Nano Lett., 2015, 15, 2875–2880 CrossRef CAS
- H. Huang, H. Jia, Z. Liu, P. Gao, J. Zhao, Z. Luo, J. Yang and J. Zeng, Angew. Chem., 2017, 129, 3648–3652 CrossRef
- H. Xu, D. Cheng and Y. Gao, ACS Catal., 2017, 7, 2164–2170 CrossRef CAS
- Z. Zhao, H. Xu, Y. Gao and D. Cheng, Nanoscale, 2019, 11, 1386–1395 RSC
- L. An and R. Chen, J. Power Sources, 2016, 320, 127–139 CrossRef CAS
- M. E. Scofield, H. Liu and S. S. Wong, Chem. Soc. Rev., 2015, 44, 5836–5860 RSC
- R. L. Johnston, Dalton Trans., 2003, 4193–4207 RSC
- S. Núñez and R. L. Johnston, J. Phys. Chem. C, 2010, 114, 13255–13266 CrossRef
- R. P. Gupta, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 23, 6265–6270 CrossRef CAS
- M. J. Mehl and D. A. Papaconstantopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 4519–4530 CrossRef CAS PubMed
- A. P. Thompson, H. M. Aktulga, R. Berger, D. S. Bolintineanu, W. M. Brown, P. S. Crozier, P. J. in ‘t Veld, A. Kohlmeyer, S. G. Moore, T. D. Nguyen, R. Shan, M. J. Stevens, J. Tranchida, C. Trott and S. J. Plimpton, Comput. Phys. Commun., 2022, 271, 108171 CrossRef CAS
- S. M. Foiles, M. I. Baskes and M. S. Daw, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 7983–7991 CrossRef CAS PubMed
- D. J. Evans and B. L. Holian, J. Chem. Phys., 1985, 83, 4069–4074 CrossRef CAS
- A. Stukowski, Model. Simul. Mater. Sci. Eng., 2009, 18, 015012 CrossRef
- T. Wu, M. Sun and B. Huang, Angew. Chem., Int. Ed., 2021, 60, 22996–23001 CrossRef CAS PubMed
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed
- S. Nosé, J. Chem. Phys., 1984, 81, 511–519 CrossRef
- B. Raju Karimadom, D. Meyerstein and H. Kornweitz, Phys. Chem. Chem. Phys., 2021, 23, 25667–25678 RSC
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS
- X. Zhang, S. Han, B. Zhu, G. Zhang, X. Li, Y. Gao, Z. Wu, B. Yang, Y. Liu, W. Baaziz, O. Ersen, M. Gu, J. T. Miller and W. Liu, Nat. Catal., 2020, 3, 411–417 CrossRef CAS
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, J. Chem. Phys., 2004, 120, 10240–10246 CrossRef CAS PubMed
- B. Hammer and J. K. Nørskov, Surf. Sci., 1995, 343, 211–220 CrossRef CAS
-
B. Hammer and J. K. Nørskov, Advances in Catalysis, Academic Press, 2000, vol. 45, pp. 71–129 Search PubMed
- N. Zhang, F. Chen and L. Guo, Phys. Chem. Chem. Phys., 2019, 21, 22598–22610 RSC
- V. Grozovski, F. J. Vidal-Iglesias, E. Herrero and J. M. Feliu, ChemPhysChem, 2011, 12, 1641–1644 CrossRef CAS PubMed
- H. Ma, G. Wang, Y. Morikawa and J. Nakamura, Sci. China, Ser. B: Chem., 2009, 52, 1427–1433 CrossRef CAS
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef
- N. Zhang, F. Chen, X. Wu, Q. Wang, A. Qaseem and Z. Xia, J. Mater. Chem. A, 2017, 5, 7043–7054 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr01221b |
| This journal is © The Royal Society of Chemistry 2023 |