
Reduction reactions at metal/non-aqueous interfaces can be sensed with the turn-on fluorophore resazurin†
Zachary
Gatland
a,
Daniel
Madrid
a,
Mark
Siegel
a and
Lydia
Kisley
 *ab
*ab
aDepartment of Physics, Case Western Reserve University, Cleveland, Ohio 44106-7079, USA. E-mail: lydia.kisley@case.edu
bDepartment of Chemistry, Case Western Reserve University, Cleveland, Ohio 44106-7079, USA
First published on 21st March 2023
Abstract
Interfacial oxidation–reduction reactions have important applications in corrosion and catalysis, but traditional electrochemical cell methods cannot be used to study these reactions in non-conducting environments such as non-aqueous solvents. We demonstrate that the molecule resazurin that can be reduced to highly-fluorescent resorufin is compatible for sensing in non-aqueous solvents. We characterize the spectral properties of the dyes in ethanol, dimethylformamide (DMF), acetone, and dimethyl sulfoxide (DMSO), showing a ∼10× increase in intensity for “turned-on” resorufin compared to resazurin in all four solvents. We then apply resazurin to sense corrosive reduction reactions at iron surfaces. Increases in fluorescence intensity due to resazurin reduction to resorufin are observed in ethanol, acetone, and DMSO, while DMF had no turn-on. Our work shows that fluorescent dyes have considerable potential to be used to understand redox reactions in non-aqueous solvents, but care must be taken to understand the interplay between the dye, the solvent, and the reactions occurring.
Introduction
Interfacial oxidation–reduction reactions include corrosion which degrades the quality of metal, leading to structural damage, costly repairs, and fatal incidents. The global cost of corrosion is estimated to be US$2.5 trillion annually.1 The oil and gas industry is one area where the threat of corrosion is keenly felt. The annual cost of corrosion in the oil and gas industry is roughly US$1.3 billion,2 and failure in oil and gas storage or transport infrastructure can pose danger to both human and environmental health. Additionally, metal ions originating from the corrosion process can accelerate the formation of harmful deposits that will lead to problems in fuel distribution systems.3 While many industries are only concerned with corrosion occurring in aqueous environments, the oil and gas industry must uniquely contend with corrosion occurring in non-aqueous environments. In a survey of reported natural gas pipeline incidents in Canada, failure statistics revealed that 28% of the pipeline incidents were due to internal corrosion, which occurs within the pipeline at the interface of the organic mixture and metal.4Corrosion is less predictable in organic or mixed-solvent environments compared to aqueous environments. The molecular structure and physical properties of an organic solvent influence the reactivity of metals.5 An organic solvent may act as the corrosive species that facilitates corrosion if it has an oxidizing group within the molecules structure, or it may act simply to transport corrosive reactants and products formed during the corrosion process, in which case oxidizing species exist as solutes and the solvent itself is non-reactive with the metal.5 The inclusion of a small amount of H2O in an otherwise non-aqueous solvent will increase the rate of corrosion, even if corrosion would otherwise not occur in that solvent. Generally, the rate of corrosion will continue to increase with increasing water content as more protons are made available by the water molecules, though the inclusion of water can also contribute to the formation of a passive film that forms at the corroded surface and inhibits further corrosion of the metal.6
Corrosion is difficult to study in situ in non-aqueous environments due to sensing limitations in electrochemical and temporal limitations in nanoscale methods. Macroscale electrochemical measurements quantify redox reaction rates at the ensemble level, but have low sensitivities and are unable to resolve nanoscale heterogeneities.6,7 Electrochemical methods also require the ability of electrons to reach electrodes through a conductive aqueous solution, and cannot be applied to non-aqueous fluids due to the lack of conductivity (Fig. 1a). Higher resolution methods can be used to study the effects and products of redox reactions at the micro- and nanoscales. For example, the morphological effects and products of corrosion of steel and iron has been probed at these scales with the use of atomic force microscopy (AFM),8,9 scanning electron microscopy (SEM),10,11 X-ray photoelectron spectroscopy (XPS),12 vertical scanning interferometry (VTI), and Raman spectroscopy.13 AFM, SEM, XPS, and VTI can all provide nanoscale spatial resolution, but these methods require the specimen to be removed from the environment of the reaction, so dynamic temporal information is lost. Optical methods can be performed in situ, but spatial resolution is restricted to ∼250 nm by the diffraction limit of visible light.
 | ||
| Fig. 1 Resorufin can detect reduction reactions in non-aqueous environments. (a) Illustration of experimental system showing that the turn-on of a dye at iron colloid/non-aqueous solvent interface can sense reduction as an increase in fluorescence over time in environments that would not work for traditional electrochemical methods due to lack of conductivity in the solvent. (b) The molecular structure of weakly fluorescent resazurin, highly fluorescent resorufin, and non-fluorescent dihydroresorufin. Resazurin is irreversibly reduced to resorufin. Resorufin can be reversibly reduced to dihydroresorufin. | ||
Fluorescence “turn-on” probes, such as the resazurin–resorufin fluorescent dye system have proven to be useful tools for studying interfacial redox reactions including corrosion in aqueous environments at the ensemble and single-molecule level. Fluorescence spectroscopy and microscopy can detect in situ chemical reactions, the latter with high spatial and temporal resolution.14 Resazurin turn-on has been used to monitor redox reactions including corrosion of iron,15 catalysis by gold nanoparticles,16 and aerobic processes of microorganisms.17 Weakly fluorescent resazurin is irreversibly reduced to highly fluorescent resorufin in the presence of these redox reactions (Fig. 1b), resulting in an increase in fluorescence intensity that can be detected at the single-molecule and ensemble level (Fig. 1a, inset). Resorufin can be further reduced to non-fluorescent dihydroresorufin, though this reaction is reversible (Fig. 1b). Other fluorophores can be used to detect the oxidation half of reactions, such as dyes which turn-on upon chelation to the oxidation product or undergo oxidation itself.14,18 The use of resazurin turn-on as a tool to monitor redox reactions has been useful, but largely limited to aqueous environments.
There has been some study of the photophysical properties of resazurin and resorufin in non-aqueous environments, but the system's effectiveness as a turn-on probe in non-aqueous media has not received much attention. Previous studies have measured some of the photophysical properties of resorufin in non-aqueous solvents, including relaxation time, effect of pH, and UV-vis absorption in various organic solvents,19–21 but there has been no comprehensive study comparing the fluorescence emission of resazurin to resorufin in non-aqueous environments. Further, the application and understanding of resazurin turn-on in non-aqueous environments is even more limited, though it has been shown that resazurin can be reduced to resorufin through photoreduction in methanolic environments at the ensemble level.19
Here, we investigate the fluorescence emission properties of the dyes resazurin and resorufin in several non-aqueous solvents and assess the effectiveness of resazurin as a turn-on probe in these solvents by monitoring the cathodic corrosion of iron. We show the measured fluorescence intensity of non-aqueous solutions containing resorufin is higher compared to solutions created from the same solvent containing resazurin, and therefore could have a turn-on readout based on the chemical change regardless of solvent. We report that the shape and location of the emission spectra differs from solvent to solvent which we ascribe to differences in the intermolecular interactions between dye and solvent molecules. Finally, to show resazurin can be used as a turn-on probe to monitor redox reactions in non-aqueous environments, we monitor the corrosion of iron in non-aqueous solutions by measuring the increase in fluorescence intensity over time as resazurin is reduced to resorufin in the presence of the cathodic reaction. Overall, our results suggest that resazurin is an effective sensor in three of the four non-aqueous solvents investigated.
Methods
Resazurin and resorufin solutions
Resazurin sodium salt (Tokyo Chemical Industry Co.) and resorufin (Tokyo Chemical Industry Co.) were each dissolved in DMSO (99.9%; Cambridge Isotope Laboratories, Inc.) to create 1 mM stock solutions. Dilutions were created from these stock solutions in ethanol (99.5%; Fisher bioreagents), DMSO, DMF (>99%; Sigma Aldrich), and acetone (99.5%; Fisher chemical) with a final dye concentration of 5 μM and 0.5% v/v DMSO. All dilutions were stored protected from light until spectral measurements were performed.Iron corrosion turn-on experiment solutions
Stock solutions of 50 mM LiCl (98.5%; Fisher Chemical) and 100 μM resazurin for each solvent (ethanol, DMSO, DMF, acetone) were used to make 10 mL sample solutions of 2 mM LiCl, 5 μM resazurin, 1–10% v/v H2O and 500 mg Fe powder (< 10 μm, >99.9%; Sigma–Aldrich). Samples measuring 1 mL of each solution were collected every hour after iron was added over the course of six hours. Samples were placed in an Eppendorf 5702 R centrifuge, and centrifuged at 2500 × g for 2.5 minutes to separate any remaining iron from the solution. A volume of 0.95 mL of iron-free supernatant was collected and protected from light until spectral measurements were performed.Fluorescence spectroscopy measurements
The fluorescence emission spectra of all samples were measured with a Jasco FP-8350 spectrofluorometer, with a PCT-118 temperature-controlled four-cuvette reader attachment, at ambient temperature (∼20–22 °C). Jasco quartz cuvettes (J/1103-1643F) were used for all iron corrosion measurements, and optical glass cuvettes (Labomed Inc; G-204) were used for resazurin and resorufin spectral measurements. Emission spectra were collected with 561 nm excitation wavelength produced by a Xe lamp, 2.5 nm excitation and emission slit bandwidth, 50 ms response time, 400 V power, 0.5 nm data interval, and 500 nm/min scanning speed.Results and discussion
The fluorescence emission spectra of resazurin and resorufin exhibit solvatochromatism (Fig. 2). We measure the location of peak emission for resazurin and resorufin in solvents with a report of their hydrogen-bond donor acidity (α), a measure of the ability of the solvent to donate a proton in a solvent-to-dye hydrogen bond,22 and relative polarity, a measure of the capabilities of a solvent to dissolve charged vs. neutral, apolar vs. dipolar, dyes.23 We observe the peak emission wavelength for the dye pairs in the protic solvent ethanol (α = 0.83;22 relative polarity = 0.65423) at 593 nm (Fig. 2a), and aprotic solvents solvents DMF (α = 0.00;22 relative polarity = 0.38623), acetone (α = 0.08;22 relative polarity = 0.35523) and DMSO (α = 0.00;22 relative polarity = 0.44423), at 597.5 nm (Fig. 2b), 592.5 nm (Fig. 2c) and 600.5 nm (Fig. 2d), respectively. For reference we measured the location of peak emission for resazurin and resorufin in H2O (α = 1.17;22 relative polarity = 1.0023) at 584 nm (Fig. S1, ESI†).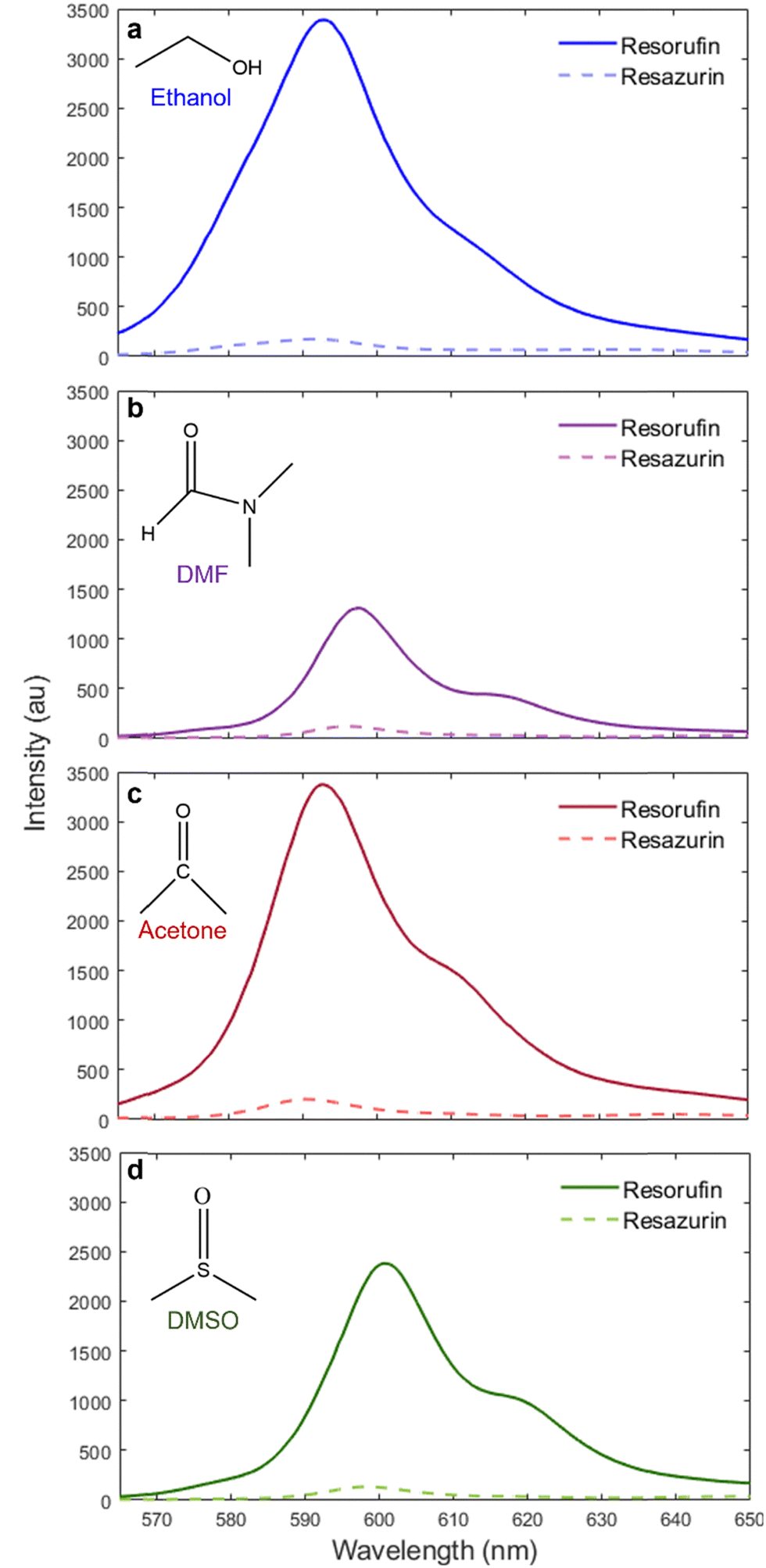 | ||
| Fig. 2 Resorufin has increased emission intensity compared to resazurin in non-aqueous environments. The fluorescence emission spectra of resazurin and resorufin in (a) ethanol, (b) DMF, (c) acetone, and (d) DMSO along with the molecular structure of each solvent. | ||
The change in the peak emission wavelength of resazurin and resorufin supports that protic solvents exhibit negative solvatochromism with respect to α, while in aprotic solvents the emission spectra exhibit positive solvatochromism with respect to solvent polarity. Generally, fluorescent molecules exhibit positive solvatochromism based on solvent polarity. As the fluorophore transitions from the ground state to the excited state, the molecule's dipole moment shifts. Before the molecule can transition back to the ground state a period of reorientation occurs between the fluorophore and the surrounding solvent molecules.24 The greater the energy expended during this period of reorientation, the lower the energy emitted by radiative processes. The reorientation of the fluorophore due to the shift in dipole moment is more pronounced in solvents with high polarity, resulting in a redder emission than observed in solvents with low polarity.24 However, previous reports have observed a blue shift of the emission spectra of resorufin with increasing solvent polarity.21,25–27 This effect is likely due to solvent molecules donating hydrogen bonds with the carboxyl groups of resazurin and resorufin. A solvent with a greater hydrogen-bond donor acidity (α) is able to create a more stable complex with the dye molecules, resulting in less energy lost during reorientation and a bluer emission.26 Our measurements support these observations that the interplay between solvent polarity and solvent-dye hydrogen bonding determine solvatochromism in resazurin and resorufin.
A significant increase in fluorescence emission intensity from resazurin to resorufin was observed in all solvents tested. For all solvents investigated the increase in total fluorescence emission intensity from resazurin to resorufin was between 14–15× with the exception of DMF, where the fluorescence emission intensity of resorufin is only 9× greater than that of resazurin (Fig. 2b). To understand why this is the case we turn to the fluorescence excitation spectra of resazurin and resorufin (Fig. S2, ESI†). Resorufin absorption in ethanol, acetone, and DMSO at 561 nm is comparable, while absorption in DMF is 0.4× less intense. Resazurin on the other hand is excited in all solvents at 561 nm comparably. The mechanism of this difference in absorption is beyond the scope of our work here that is focused on turn-on sensing. Overall, the increase in total fluorescence emission intensity from resazurin to resorufin is significant enough in all solvents for resazurin to be used as a turn-on probe in these environments if the dye can be reduced to resorufin.
With this characterization of the spectral properties of the dyes in non-aqueous solvents, we then turn to introducing resazurin to an environment where reduction can occur via the corrosion of iron colloid particles in the solvents. We vary the amount of water content from 1–10% as our potential corrosive agent within the non-aqueous solvents that can increase the rate of corrosion as more protons are made available by the water. Control measurements of resazurin in the non-aqueous solvents in the absence of iron are reported in Table S2 and Fig. S3 (ESI†).
We observe a significant increase in fluorescence emission intensity in ethanol-based solutions containing 5%, 7%, and 10% H2O after exposure to iron (Fig. 3a and d). We observe a 1.09 ± 0.04×, 1.35 ± 0.02×, and 1.47 ± 0.01× increase in fluorescence emission intensity respectively after six hours of exposure to iron (Table S1, ESI†). We observed a greater reduction of resazurin to resorufin in ethanol solutions with higher water content. We observe no significant change in the fluorescence of ethanol-based solutions containg 3% and 1% H2O (Table S1, ESI†). These results suggest that resazurin is an effective iron corrosion turn-on probe in ethanol-based environments with ≥5% v/v water for the concentrations and instrumentation used. Future work could pursue lower limit of detection methods such as single-molecule microscopy15,18 to determine if no changes were observed at 1% and 3% H2O either due to the detection limit of the fluorometer or absence of corrosion taking place (no dye turn on).
 | ||
| Fig. 3 Turn-on of resorufin senses the corrosive reduction of Fe in non-aqueous solutions. (a–c) The time-dependent fluorescence emission spectra of (a) ethanol, (b) DMF, and (c) acetone based corrosive solutions containing resazurin and 10% v/v H2O over time after exposure to iron. (d–f) The integrated total fluorescence intensity over time for (d) ethanol, (e) DMF, and (f) acetone based corrosive solutions containing resazurin and various concentrations of H2O after exposure to iron. Dashed lines in d–f are provided as a guide for the eye. | ||
We observe no significant change in fluorescence intensity in DMF-based solutions, regardless of H2O content (Fig. 3b and e). No significant increase in fluorescence emission intensity was observed after six hours of exposure to iron (Table S1, ESI†). Although we do not observe resazurin reducing to resorufin in DMF-based solutions after exposure to iron, we know the corrosion reaction is progressing. First, previous potentiodynamic measurements have shown that the corrosion reaction should progress in DMF-based solutions with as little as 1% H2O and comparable concentrations of electrolyte.6 Additionally, a qualitative experiment shows the effects of corrosion appear on a piece of A109 low-carbon steel shim after being exposed to DMF solution containing 10% H2O by volume (Fig. S4, ESI†). Despite these signs of corrosion, no significant change in fluorescence intensity is observed in this solution even after several days of exposure. The absence of any observed reduction of resazurin to resorufin despite the presence of the corrosion reaction indicates that resazurin is not an effective turn-on probe for detection corrosion in DMF-based environments.
DMF has been shown to inhibit inter-molecular proton transfer for other fluorescent turn-on probes.28 Previous studies have shown that weak intermolecular interactions between DMF and fluorescent probes have introduced an additional potential energy barrier that inhibits inter-molecular proton transfer necessary for the fluorescent probe to turn-on.27 Similar intermolecular interactions between DMF solvent molecules and resazurin solute molecules could increase the reduction potential of resazurin above the potential generated by the cathodic corrosion of iron. This increased reduction potential would explain why we do not observe the reduction of resazurin to resorufin in DMF-based environments, despite the progression of the corrosion reaction. Further study should be given to determine the reduction potential of resazurin in DMF-based environments, as it is possible that it could prove useful as a fluorescent turn-on probe for other oxidation–reduction reactions with higher oxidation potentials.
We observe significant reduction of resazurin to resorufin at all H2O concentrations in acetone-based solvents, with a higher rate of reduction observed in solutions with lower H2O concentration (Fig. 3c and f). In acetone-based solutions containing 1%, 3%, 5%, 7%, and 10% H2O by volume we observe a 0.8 ± 0.2×, 1.7 ± 0.8×, 10.0 ± 0.3×, 11 ± 3×, and 11 ± 1× change in total fluorescence intensity respectively after six hours of exposure to iron (Table S1, ESI†). These end point results are somewhat deceptive. If we focus instead on the fluorescence intensity after only one hour we observe a change of 0.7 ± 0.2×, 5 ± 4×, 6.2 ± 0.5×, 5 ± 1×, and 3.7 ± 0.3× respectively (Fig. 3f and Table S1, ESI†). Additionally, it should be noted that the change in fluorescence observed in the solution containing 1% H2O is due to the complete reduction of resazurin to resorufin, followed potentially by the further reduction from resorufin to non-fluorescent dihydroresorufin. This can be observed by the shift in solution color from pink to clear (Fig. S5, ESI†), and the fluorescence emission spectra for the solution containing 1% H2O (Fig. S6, ESI†). This further reduction indicates a more aggressive rate of corrosion induced reduction.
The observed results indicate a greater rate of corrosion occurs in acetone-based solutions containing lower H2O content which can be explained by the solvent properties of acetone. While the rate of corrosion of iron in acetone has been shown to be relatively comparable to that in H2O,29 acetone is seldom used as a solvent for corrosion experiments. Acetone is highly effective at removing the products of corrosion from the metal surface. Acetone is so effective in that capacity that it is often used in experiments to clean potential corrosive products prior to the exposure to the corrosive environment so the effects of corrosion on un-corroded metal surface can be observed.30–32 As the iron surface is corroded, the corrosive products can form a passive film, inhibiting the rate of corrosion at those locations.6 The constant removal of this film will prevent the inhibition of corrosion at previously corroded locations. Because H2O is a key component in these passive films,6 the solutions with higher H2O content are more capable of forming and maintaining passive films, while the solutions with lower H2O content (and greater acetone content) are more capable of removing these films.
Despite the unexpected trend, our results suggest resazurin could be used an effective turn-on probe for studying corrosion in acetone-based environments. However, care should be taken with the concentration of dyes, metal, and equilibrium properties to avoid subsequent turn off of resorufin as it could be further reduced to non-fluorescent dihydroresorufin. Additionally, further study should be given to determine the electrochemical potentials that these reductions occur in acetone-based environments.
Finally, we observed an increase in DMSO-based solutions (Fig. S7, ESI†). Similar to ethanol, an increase in fluorescence is observed in DMSO-based solutions containing 10%, 7%, and 5% v/v H2O when exposed to iron. No significant change in fluorescence is observed in DMSO-based solutions containing 3% and 1% H2O. Measurements have been difficult to conduct in DMSO given the solvent degrades in the presence of oxygen, reducing apparent solvent pH.33 Resorufin is not an effective turn-on probe at the wavelenghts we are concerned with in solutions with pH ≤ 6.17 We intend to continue our investigation of resazurin as an iron corrosion fluorescence turn-on probe in DMSO-based solutions, though our preliminary observations indicate that resazurin can be effective in solutions with ≥ 5% v/v water.
Conclusion
Overall, we show reduction reactions at metal/non-aqueous interfaces can be sensed with the resazurin turn-on to resorufin. We measured the solvatochromatic fluorescence emission spectra of resazurin and resorufin in ethanol, DMF, acetone, and DMSO, showing solvent polarity and solvent-dye hydrogen bonding change dye peak emission wavelength properties. In all four solvents we observed a 9–15× increase in the fluorescence emission intensity of resorufin compared to resazurin. Therefore, if reduction can take place, resazurin/resorufin should be able to be used as a turn-on probe. When we applied our turn-on dye to sense reduction in the presence of iron, resazurin/resorufin successfully sensed increases in corrosion with increasing water content in ethanol and DMSO solvents, while aggressive reduction in acetone led to further reduction of resorufin to the turned-off product dihydroresorufin at long timescales of observation (≥3 h). No dye turn-on was observed in DMF despite corrosion taking place due to a possible increase in reduction potential that has been observed for DMF with other turn-on fluorophores. Our work highlights that turn-on fluorophores can be used to sense reduction reactions at metal/non-aqueous interfaces for some, but not universally all, organic solvents. Care must be taken to understand the intricacies between dye, solvent, and reaction of interest. Careful use of fluorescence turn-on sensing has much potential for understanding a broad range of heterogeneous interfacial reduction reactions in non-aqueous solvents that would be relevant to corrosion and catalysis where electrochemical methods cannot be used due to lack of conductivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Acknowledgment is made to the donors of The American Chemical Society Petroleum Research Fund for partial support of this research, along with the National Science Foundation Award #2142821 made through the CHE CMSD-A program. The authors thank the Case Western Reserve University College of Arts and Sciences for further financial support. We would also like to thank Kisley Lab members for providing helpful discussion.References
-
G. Koch, in Trends in Oil and Gas Corrosion Research and Technologies, ed. A. M. El-Sherik, Woodhead Publishing, Boston, 2017, pp. 3–30 Search PubMed
.
- Oil and Gas Production - AMPP, https://www.ampp.org/resources/what-is-corrosion/corrosion-reference-library/oil-gas, (accessed 16 December 2022).
- Corrosiveness of Fuels During Storage Processes | IntechOpen, https://www.intechopen.com/chapters/47919, (accessed 16 December 2022).
- M. Askari, M. Aliofkhazraei and S. Afroukhteh, J. Nat. Gas Sci. Eng., 2019, 71, 102971 CrossRef
-
H. S. Ahluwalia and R. Thodla, in ASM Handbook, Vol. 13C: Corrosion: Environments and Industries, ed. S. M. Cramer and B. S. Covino Jr., ASM International, 2006, vol. 13C Search PubMed
- G. Capobianco, P. Venti and F. Bellucci, Br. Corros. J., 1990, 25, 133–135 CrossRef CAS
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS
- K. H. Anantha, C. Örnek, S. Ejnermark, A. Medvedeva, J. Sjöström and J. Pan, J. Electrochem. Soc., 2017, 164, C85 CrossRef CAS
- D. Sebastian, C.-W. Yao and I. Lian, MRS Commun., 2020, 10, 305–311 CrossRef CAS
- D. Sebastian and C.-W. Yao, MRS Commun., 2021, 11, 70–77 CrossRef CAS
- H. Masuda, Corrosion, 1996, 52, 435–439 CrossRef CAS
- Y. Wan, Y. Sun, D. Cai, L. Yin, N. Dai, L. Lei, Y. Jiang and J. Li, Front. Chem., 2020, 8 DOI:10.3389/fchem.2020.00529
- J. Heuer and A. Luttge, Npj Mater. Degrad., 2018, 2, 1–8 CrossRef
-
L. Kisley, in Spectroscopy and Dynamics of Single Molecules, ed. C. K. Johnson, Elsevier, Amsterdam, 2019, pp. 117–161 Search PubMed
- A. Saini, H. Messenger and L. Kisley, ACS Appl. Mater. Interfaces, 2021, 13, 2000–2006 CrossRef CAS PubMed
- P. Chen, X. Zhou, H. Shen, N. M. Andoy, E. Choudhary, K.-S. Han, G. Liu and W. Meng, Chem. Soc. Rev., 2010, 39, 4560 RSC
- J. L. A. Knapp, R. González-Pinzón and R. Haggerty, Water Resour. Res., 2018, 54, 6877–6889 CrossRef
- A. Saini, Z. Gatland, J. Begley and L. Kisley, MRS Commun., 2021, 11, 804–810 CrossRef CAS
- C. Bueno, M. L. Villegas, S. G. Bertolotti, C. M. Previtali, M. G. Neumann and M. V. Encinas, Photochem. Photobiol., 2002, 76, 385–390 CrossRef CAS PubMed
- M. Krishnamurthy, K. K. Khan and S. Doraiswamy, J. Chem. Phys., 1993, 98, 8640–8646 CrossRef CAS
- L. Flamigni, E. Venuti, N. Camaioni and F. Barigelletti, J. Chem. Soc., Faraday Trans. 2, 1989, 85, 1935–1943 RSC
- M. J. Kamlet, J.-L. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS
-
T. Welton and C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, John Wiley & Sons, Weinheim, 4th edn, 2011 Search PubMed
-
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 2006 Search PubMed
- J. P. Rasimas, K. A. Berglund and G. J. Blanchard, J. Phys. Chem., 1996, 100, 17034–17040 CrossRef CAS
- B. R. Gayathri, J. R. Mannekutla and S. R. Inamdar, J. Mol. Struct., 2008, 889, 383–393 CrossRef CAS
- E. F. G. Templeton, E. L. Quitevis and G. A. Kenney-Wallace, J. Phys. Chem., 1985, 89, 3238–3243 CrossRef CAS
- C. Li, B. Hu and Y. Liu, Spectrochim. Acta, Part A, 2020, 225, 117487 CrossRef PubMed
- P. Hronsky, Corrosion, 1981, 37, 161–170 CrossRef CAS
- A. Prithiraj, I. O. Otunniyi, P. Osifo and J. van der Merwe, Eng. Failure Anal., 2019, 104, 977–986 CrossRef CAS
- G. Herting, I. Odnevall Wallinder and C. Leygraf, J. Food Eng., 2008, 87, 291–300 CrossRef CAS
- R. A. King, C. K. Dittmer and J. D. A. Miller, Br. Corros. J., 1976, 11, 105–107 CrossRef CAS
- Y. Deguchi, M. Kono, Y. Koizumi, Y. Izato and A. Miyake, Org. Process Res. Dev., 2020, 24, 1614–1620 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qm01309f |
| This journal is © the Partner Organisations 2023 |