
Assessing the effects of dealumination and bifunctionalization on 8-membered ring zeolite/zeo-type materials in the methanol-to-olefin catalytic process†
Shican
Jiang
,
Hexun
Zhou
,
Xin
Zhang
,
Xue
Zhou
and
Abhishek
Dutta Chowdhury
 *
*
College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, Hubei, P. R. China. E-mail: abhishek@whu.edu.cn
First published on 6th May 2024
Abstract
C2–C4 shorter olefins, particularly ethylene and propylene, are crucial building blocks in modern petrochemical, polymer, and chemical industries. However, their predominant sourcing from fossil resources raises concerns due to increased awareness of carbon emissions and diminishing petroleum reserves. Therefore, a necessary shift towards sustainable resources is underway. The zeolite-catalyzed methanol-to-olefin (MTO) process, particularly over 8-MR zeolite/zeo-type materials, has gained industrial prominence in this context. If methanol strictly originated from renewable sources, then the MTO process would actively promote the “methanol economy”. Despite the advantages of zeolite/zeo-type materials, they encounter deactivation due to the accumulation of coke precursors, limiting their lifetime. While achieving high olefin selectivity in the MTO process is not challenging, improving the catalytic lifetime without compromising preferential olefin selectivity is crucial. To achieve this objective, various surface modification approaches, such as dealumination through acid etching, steaming, and constructing bifunctional catalytic systems, are applied to numerous 8-MR zeolite/zeo-type materials, including industrially operational MTO catalysts. Combining catalytic studies with advanced characterization methods, including under operando conditions, has enhanced MTO process efficiency by mitigating the formation of coke precursors. Ultimately, this study contributes to a deeper understanding of zeolite-catalyzed MTO processes, paving the way for more efficient and sustainable production of low-carbon olefins.
1. Introduction
Low-carbon olefins, namely ethylene and propylene, are crucial foundational chemical raw materials extensively utilized in the petrochemical industry, playing a pivotal role in the realms of modern petroleum, polymer, and chemical production.1–3 Currently, the primary method for generating low-carbon olefins involves the petrochemical process of cracking light hydrocarbons, such as naphtha and light diesel oil.4,5 Given the escalating scrutiny on carbon emissions and the diminishing petroleum, there is a pressing need to shift towards sustainable resources to meet future demands for lower olefins. In this context, methanol derived from carbon dioxide (CO2) is poised to become the feedstock for numerous high-volume chemicals, including ethylene and propylene, in the coming decades.6 Transitioning from coal, crude oil, and natural gas to methanol, particularly when sourced rigorously from renewable sources like CO2, biomass, and waste, holds the promise of establishing a much-desired “methanol economy”.7 To achieve this objective, the zeolite-catalyzed methanol-to-olefin (MTO) process has emerged as a widely embraced catalytic technology.Although accidentally discovered in 1977 over the 10-membered ring (MR) zeolite ZSM-5, the current industrial prominence of the MTO process is attributed to the utilization of 8-MR zeo-type SAPO-34 materials.2,8–13 Presently, over 100 operational MTO plants in China employ SAPO-34-based materials.14–20 Beyond the conventional advantages associated with zeolite catalysts, such as a high specific surface area, uniform pore structure, inherent acidity, and remarkable hydrothermal stability, 8-MR zeolite/zeo-type materials exhibit significantly enhanced selectivity for low-carbon olefins.21–28 This heightened selectivity is attributed to their relatively small pore diameter, a characteristic known as “specific shape selectivity”.29–31 For the same reason, SAPO-34 or analogous 8-MR zeolite/zeo-type materials are prone to deactivation resulting from coke precursor accumulation, leading to a shorter lifetime.21,24,26–28,32–42 Although achieving higher selectivity for olefins is not a significant challenge in the MTO process over 8-MR zeolite/zeo-type materials,21–27 enhancing the catalytic performance with an extended catalyst lifespan is crucial for strengthening overall process efficiency. Within the space of 8-MR zeolite/zeo-type materials utilized in the MTO process, extensive literature has investigated SSZ-39,27,42 SAPO-18,43–47 SSZ-13,24,26,37,38,40 and SAPO-34 (ref. 21, 25, 28 and 35) (Fig. 1). Among these, SSZ-39 and SAPO-18 are with the AEI topology, whereas SSZ-13 and SAPO-34 belong to the CHA topology. However, SSZ-13 and SSZ-39 are aluminosilicate materials, whereas SAPO-18 and SAPO-34 are based on a zeo-type aluminophosphate (ALPO) framework. The AEI topology is characterized by a spatial network structure comprising eight-membered ring pore openings formed by different arrangements of dual six-membered rings. In contrast, CHA topology consists of a three-dimensional network of 8-MR channels with an ellipsoidal cage formed by stacking double six-membered rings and a three-dimensional intersecting channel structure.21,24–28,34–42,48,49 Regardless of their structural and topological distinctions, these materials share a common fate in the MTO process – exhibiting high olefin selectivity but with a shorter catalyst lifespan.
 | ||
| Fig. 1 The project design: (a) five zeolite/zeo-type materials were used in this study, namely SSZ-39, SAPO-18, SSZ-13, SAPO-34, and the industrial CCG-MTO material. The industrial MTO catalyst is also based on SAPO-34 (cf. XRD in the inset). (b) Illustration of the topological differences (AEI vs. CHA). (c) The representation of their SEM images indicates that only industrial CCG-MTO is a shaped catalytic body. (d) Three physical/chemical treatments were applied to these zeolites to compare their impact on MTO catalysis (i.e., dealumination by steaming and acid etching and constructing a bifunctional system upon combining with metallic Y2O3). | ||
In zeolite catalysis, three common approaches are typically employed to modify the surface and physicochemical properties, potentially extending the catalyst's lifetime. These techniques include dealumination through acid etching treatment,34,37,38,50 steaming treatment27,28,39 and constructing bifunctional catalytic systems that promote Brønsted–Lewis acid synergy in MTO catalysis.33,51,52 To facilitate a meaningful comparison among various 8-MR zeolite/zeo-type materials and gain insights into technical aspects, we employed these three distinct approaches on commercially available materials, namely SSZ-39, SAPO-18, SSZ-13, and SAPO-34 (Fig. 1). Additionally, we included the industrial MTO catalyst, CCG-MTO, utilized in China (procured from its producer, China Catalyst Group, CCG), which is also based on SAPO-34 (Fig. 1).53 Beyond evaluating catalytic performance, we utilized traditional zeolite characterization tools and operando characterization techniques to enhance our understanding of MTO catalysis over 8-MR zeolites. Remarkable catalytic performance was achieved, including extended catalyst lifetime and preferential C2–C4 olefin selectivity. Moreover, valuable insights were obtained, contributing to improving the MTO process efficiency. Notably, the dealumination technique exhibited distinct benefits depending on the material—steaming proved advantageous for the zeolite SSZ-39 and SAPO-18, while etching benefited SSZ-13, SAPO-34, and CCG-MTO materials. Dealumination treatment, whether steaming or chemical etching, enhanced both the catalyst lifetime and propylene selectivity by facilitating the olefin cycle of the dual-cycle mechanism. Operando studies unveiled that dealumination treatment positively influences the reaction by slowing the formation of poly-aromatics-based coke precursors. The most significant impact on catalyst lifetime resulted from bifunctional catalytic systems involving zeolite/zeo-type materials with Y2O3 in an inter- and intra-pellet mixing manner,52 although it did not significantly alter or promote specific product selectivity. Consequently, this study elevates the fundamental understanding of the zeolite-catalyzed MTO process, which might be beneficial to improving the process efficiency.
2. Results and discussion
2.1. Zeolite/zeo-type materials used and their characterization
As indicated before, this study focuses on five primary 8-MR zeolites: SSZ-39, SAPO-18, SSZ-13, SAPO-34, and CCG-MTO. SSZ-39 and SAPO-18 follow the AEI topology, while the rest belong to the CHA topology. The China Catalyst Group's (CCG) industrial MTO catalyst, CCG-MTO, employed in the DMTO process in China,14–16 is also based on SAPO-34 and independently acquired from its commercial producer, China Catalyst Group. All five zeolites underwent dealumination through chemical etching and hydrothermal steaming, denoted as “zeolite-etching” and “zeolite-ST750” (i.e., steamed at 750 °C), respectively. Both stand-alone and dealuminated zeolites were characterized by X-ray diffraction (XRD), scanning electron microscopy (SEM), X-ray fluorescence (XRF), thermal gravimetric analysis (TGA),27 Al magic angle spinning (MAS) solid-state NMR, ammonia temperature-programmed desorption (NH3-TPD), and nitrogen physisorption. The relevant experimental and characterization details are in the ESI† (see Tables S1–S3 and Fig. S1–S10). In addition, bifunctional zeolite systems were constructed by combining stand-alone and dealuminated zeolites with Y2O3 for catalytic performance evaluation. Given the extensive range of mono- and bifunctional catalytic systems, Table S1† provides a comprehensive list of zeolite-based systems used in this study.Fig. 2 displays the XRD patterns of untreated commercial 8-MR zeolite/zeo-type materials and their counterparts after dealumination treatments via both chemical etching and streaming. All zeolites, except for SSZ-39 and SAPO-18, exhibited the characteristic diffraction peaks of the CHA topology.21,24,26,36,42,43,54,55 SSZ-39 is an aluminosilicate material and SAPO-18 is an aluminophosphate material with an AEI topology demonstrating its characteristic XRD patterns.27,42,43,54,55 After the dealumination treatment, the zeolite framework structures of the corresponding materials were completely preserved for all five materials, as also revealed by their XRD. No impurity crystal or amorphous phase was observed either, implying that the crystalline structure of the commercial 8-MR zeolite/zeo-type materials was unaffected after both types of dealumination treatments. However, a decrease in intensity for certain diffraction peaks was observed after the dealumination treatment (except the zeolite SSZ-13, see Fig. S1 in the ESI†), suggesting a relatively lower crystallinity than that of the untreated stand-alone zeolite materials.27,39,41 Herein, the reduction in intensity of diffraction peaks was more pronounced in steamed zeolites than in untreated zeolites or their chemically etched counterparts. In contrast, SSZ-13 displayed minimal or no alteration in crystallinity following both types of dealumination treatments, suggesting the preservation of its higher crystalline nature.41 After creating a bifunctional system by incorporating zeolites with Y2O3, the characteristic diffraction peaks of metallic components (JCPD#65-3178) became apparent, alongside the typical CHA/AEI patterns associated with the utilized zeolite/zeo-type materials (Fig. S2 in the ESI†). It is important to highlight that there are no observable alterations in the XRD profiles of both the commercial SAPO-34 and the proprietary CCG-MTO materials.
 | ||
| Fig. 2 X-ray diffraction patterns of stand-alone untreated zeolite/zeo-type materials and their dealuminated forms after the steaming and chemical etching treatments: (a) SSZ-39, (b) SSZ-13, (c) SAPO-34, (d) CCG-MTO, and (e) SAPO-18. | ||
While the XRD profiles of dealuminated zeolites closely resembled those of their parent counterparts, with variations mainly observed in the intensity of certain diffraction peaks, the physicochemical properties were notably influenced by both steaming and chemical etching treatments. This impact was more pronounced in the case of zeo-type material sets (i.e., SAPO-34 and CCG-MTO), as evidenced by SEM images depicting the morphological changes in commercial 8-MR zeolites before and after treatment (see Fig. 3 and S4–S6 in the ESI†). The SSZ-39 zeolite exhibited uniform pseudo-cubic crystal sizes ranging from 0.5 to 2 μm,27,42 while its etched sample revealed smaller particle sizes (∼0.5–1 μm) with a distinct porous structure on its surface compared to the parent after etching (see Fig. 3a and b). The corresponding steamed SSZ-39 zeolite displayed some degree of crystal fragmentation, leading to zeolite crystals without noticeable pores (0.5–1.0 μm particle size, see Fig. 3c).27 SAPO-18 zeolite exhibited a uniform cubic crystal size from 0.5 to 1 μm, while its etched samples showed some particle fragmentation with no significant change in particle size (see Fig. 3d and e). The corresponding steamed SAPO-18 zeolite showed some degree of crystal fragmentation, resulting in no significant porosity of the zeolite crystals (particle size 0.5–1.0 μm, see Fig. 3f). The gradual decay of crystal size has been reflected in XRD profiles of both SAPO-18 and SSZ-39 materials (Fig. S1 in the ESI†). However, the cubic-shaped crystal sizes of both untreated and dealuminated SSZ-13 zeolites remained consistent (0.5–1.0 μm particle size, see Fig. 3g–i), aligning with their XRD profiles that indicate the preservation of high crystallinity.24,26,37–41 The zeo-type parent SAPO-34 material also exhibited typical cubic crystals with particle sizes of 2–7 μm (Fig. 3j). The chemically etched SAPO-34 crystals displayed butterfly-shaped porous surfaces (2–7 μm, Fig. 3k), distinct from the smoother surfaces of the parent crystals, covering half of a single crystal surface. In contrast, steamed SAPO-34 crystals exhibited smoother surfaces with a degree of grain fragmentation, lacking noticeable pores on the surface (2–7 μm particle size, Fig. 3l).21,28,34,35,48,49 For SAPO-34-based CCG-MTO materials, etching induces partial grain breakage, resulting in the formation of internal pores, and steaming further generates additional pores within the interior of the grains. However, their particle size remained within a range of 0.5–1.0 μm (Fig. 3m–o). As the severity of the treatment increases, SEM analysis reveals an increasingly evident fragmentation. Notably, the CCG-MTO material is a shaped catalytic body with dimensions of 35–45 μm, featuring a distinctive hollow shape in the middle of its spherical body (see Fig. S3 in the ESI†). This characteristic could potentially enhance diffusion dynamics during catalysis.56 Additionally, the industrial CCG-MTO material exhibited a significantly smaller average particle size (∼0.5–1.0 μm, components of shaped catalytic body) compared to the commercial SAPO-34 material (∼2.0–7.0 μm) employed in this study. As the severity of the treatment increases, SEM analysis also shows an increasing fragmentation for CCG-MTO. This is consistent with XRD, as the peak intensity decreases with increasing treatment severity. The unique structure of CCG-MTO undergoes significant damage after etching and steam treatment, with steam treatment causing the most severe damage (Fig. S5e–S6e in the ESI†). In contrast, SSZ-13 exhibits minimal fragmentation, consistent with the XRD testing results.
 | ||
| Fig. 3 SEM images of stand-alone untreated zeolite/zeo-type materials and their dealuminated forms after the steaming and chemical etching treatments: (a) SSZ-39, (b) SSZ-39-etching, (c) SSZ-39-ST750, (d) SAPO-18, (e) SAPO-18-etching, (f) SAPO-18-ST750, (g) SSZ-13, (h) SSZ-13-etching, (i) SSZ-13-ST750, (j) SAPO-34, (k) SAPO-34-etching, (l) SAPO-34-ST750, (m) CCG-MTO, (n) CCG-MTO-etching, and (o) CCG-MTO-ST750 materials. | ||
Next, we performed nitrogen adsorption–desorption experiments over both untreated and dealuminated zeolites (see Fig. S7 in the ESI† and Table 1). These isotherms exhibit type I characteristics, indicating the presence of micropores in all materials. Following dealumination, the microporous surface area (Smicro, refer to Table 1) of SSZ-39 zeolite sets was diminished, despite an increase in the external surface area content compared to the parent zeolite. As for the SAPO-18 material, the microporous surface area (Smicro, see Table 1) of SAPO-18 zeolite decreases after etching, although the external surface area content increases compared to the parent zeolite. After being steamed, the adsorption capacity of SAPO-18 decreases, and the surface area and pore volume also decrease (Table 1 and Fig. S7e†). Similar to their XRD and SEM characteristics, both untreated and dealuminated SSZ-13 zeolites exhibited comparable adsorption/desorption profiles (Fig. S7b in the ESI†). However, the total surface area (SBET, see Table 1) showed a slight increase in dealuminated versions, attributed to the expanded external surface area. In addition to the characteristic type I isotherm for microporous features, SSZ-13 materials also exhibited a relatively higher uptake at low relative pressures. The isotherm terminates with an H4 hysteresis loop at 0.45 < P/P0 < 0.99, indicative of narrow slit-like pores.37,38,57,58 After the dealumination via steaming, the SAPO-34 materials exhibited a reduced adsorption capacity and lower surface area and pore volume (Table 1). Notably, the etching treatments led to an increase in adsorption near saturation pressure (0.99 < P/P0 < 1.0), suggesting the presence of mesopores in the dealuminated materials, which is consistent with the surface pore structure observed in SEM (Fig. 3k). Herein, the steamed SAPO-34 material exhibited a relatively larger decrease in specific surface area (394 m2 g−1vs. 545 m2 g−1) and micropore volume (0.19 cm3 g−1vs. 0.27 cm3 g−1) compared to the untreated samples (Table 1). Those findings are consistent with previous research results.28 For the industrial CCG-MTO material, the etching treatment resulted in a substantial reduction in surface area (390 m2 g−1vs. 537 m2 g−1), aligning with SEM findings (Fig. 3m and n). Also, the difference between the steamed and the parent CCG-MTO materials was significant, suggesting the structural disruptions of its unique hollow-shaped spherical catalytic body, as supported by their SEM images (see Fig. 3o and S6e in the ESI†). To comprehensively assess the impact of different chemical treatments on the zeolites' elemental composition, we also conducted XRF elemental analyses on both pristine and dealuminated samples (see Table S2†).
| Samples | S BET (m2 g−1) | S micro (m2 g−1) | S ext (m2 g−1) | V micro (cm3 g−1) | V meso (cm3 g−1) | V total (cm3 g−1) |
|---|---|---|---|---|---|---|
| a S BET was calculated by the BET method. b S micro and Vmicro were calculated by the t-plot method. c S ext = SBET − Smicro. d V meso = Vtotal − Vmicro. e V total was calculated from the adsorption branch at P/P0 = 0.99. | ||||||
| SSZ-39 | 594 | 576 | 17 | 0.28 | 0.03 | 0.31 |
| SSZ-39-etching | 589 | 558 | 31 | 0.27 | 0.06 | 0.33 |
| SSZ-39-ST750 | 578 | 557 | 21 | 0.27 | 0.05 | 0.32 |
| SAPO-18 | 453 | 378 | 75 | 0.21 | 0.07 | 0.28 |
| SAPO-etching | 404 | 290 | 114 | 0.20 | 0.06 | 0.26 |
| SAPO-ST750 | 358 | 272 | 86 | 0.14 | 0.07 | 0.21 |
| SSZ-13 | 599 | 576 | 23 | 0.30 | 0.03 | 0.33 |
| SSZ-13-etching | 636 | 589 | 47 | 0.29 | 0.07 | 0.36 |
| SSZ-13-ST750 | 610 | 569 | 41 | 0.28 | 0.06 | 0.34 |
| SAPO-34 | 545 | 536 | 9 | 0.27 | 0.01 | 0.28 |
| SAPO-34-etching | 611 | 602 | 9 | 0.25 | 0.01 | 0.26 |
| SAPO-34-ST750 | 394 | 387 | 7 | 0.19 | 0.01 | 0.20 |
| CCG-MTO | 537 | 500 | 37 | 0.26 | 0.10 | 0.36 |
| CCG-MTO-etching | 390 | 346 | 43 | 0.17 | 0.15 | 0.32 |
| CCG-MTO-ST750 | 365 | 337 | 28 | 0.16 | 0.11 | 0.27 |
Afterward, temperature-programmed desorption of ammonia (NH3-TPD) was conducted to probe the acidity characteristics (Fig. S8 and S9 and Table S3 in the ESI†). Typically, these materials exhibited three NH3 desorption peaks at ≤200 °C, 200–300 °C, and ≥300 °C, classified as weak, medium, and strong acid sites, respectively.59–63 Generally, the dealumination treatment reduced the total acidity of each zeolite/zeo-type material set. Among these, steaming had a more pronounced effect on reducing material acidity compared to the chemical etching method (see Table S3 in the ESI†). As the severity of the treatment increases, both the total and the strong acid sites showed a decreasing trend (Fig. S9 and Table S3†), consistent with previous studies.23,27,28,32,34,37,39,41 In the case of SSZ-39 zeolite, steam treatment significantly reduces its stronger acidic content (presumably, Brønsted acid sites) while increasing the medium acidic (or presumably Lewis type) content.27,42 This trend is not very pronounced over etched SSZ-39 zeolite. After steaming, the stronger acid sites in SAPO-18 significantly decreased, while its etched version led to a less pronounced reduction than the steamed material. As for the SSZ-13 zeolite set, the steam treatment led to a substantial decrease in stronger acid sites than the chemically etched sample. However, both dealumination treatments increased the content of medium acid sites to the same extent (Table S3†), indicating that elevated Lewis acid-type content might promote the catalysts' durability.28,32,34 In the case of SAPO-34 material, both the total and the stronger acid sites showed a significant decrease upon dealumination, while the weak acid site content was enhanced only upon steaming (Table S3†). In the case of industrial CCG-MTO materials, the dealumination-led decrease of stronger acid sites appeared at the expense of increasing weak/medium acid sites. Interestingly, the parent SAPO-34 and CCG-MTO materials pose a similar amount of stronger acid sites, albeit the weaker acid site content was much lower for the industrial MTO catalyst.
To supplement these acidity assessments, additional structural insights into treated and dealuminated zeolite/zeo-type materials were provided through 27Al MAS solid-state NMR spectroscopy (Fig. S10 in the ESI†). As Brønsted acid sites typically stem from the bridging hydroxyl groups between Al and Si atoms in the zeolite, probing Al can be correlated to its acidic property.64 In the tetrahedral Al region, a singular highly intense peak around the 50 ppm region corresponds to tetrahedral aluminum associated with the Brønsted acid sites.65 Additionally, the much weaker peaks around the 15 ppm and −10 ppm regions were attributed to pentahedral and octahedral extra-framework Al species (EFAl), representing Lewis acid sites.65 The strikingly similar characteristics observed in the 27Al MAS solid-state NMR spectroscopy profiles of aluminosilicate zeolite sets, encompassing both untreated and dealuminated SSZ-39 and SSZ-13 zeolites, suggest that their aluminum distribution may not vary significantly, notwithstanding differences in their topology and dealumination treatments. However, the solid-state 27Al NMR spectra of steamed SSZ-39 zeolite indicated a sharp decrease in the tetrahedral-Al region peak at the expense of a relative increase of penta-/octa-coordinated-Al (Fig. S10a in the ESI†). Such a steaming-induced reduction of Brønsted acidic framework-Al and increasing EFAl content is consistent with their NH3-TPD profile (see Table S3†). However, in other cases, there is no evidence of a substantial rise in Lewis acidic EFAl species following dealumination treatment in both aluminosilicate zeolites (Fig. S10a and b in the ESI†), suggesting an internal rearrangement of acidity as indicated by NH3-TPD studies (Table S3 in the ESI†). This phenomenon is not observed in ALPO-based SAPO-34 and CCG-MTO materials (Fig. S10c and d in the ESI†). While untreated zeo-type SAPO-34 and CCG-MTO materials exhibited only a tetrahedral Al peak, the dealumination treatment resulted in the emergence of additional peaks attributed to penta- and octahedral EFAl species (Fig. S10c and d in the ESI†). For SAPO-18, a phenomenon similar to that of SAPO-34 and CCG-MTO was also observed. Untreated SAPO-18 exhibited only tetrahedral Al peaks, but dealumination treatment resulted in additional peaks attributed to pentahedral and octahedral EFAl species (Fig. S10e in the ESI†). Among the dealumination methods, steaming notably increased the octahedral EFAl species in ALPO-based zeo-type materials, particularly in the case of CCG-MTO materials. These changes indicate a significant alteration in the physicochemical environment of aluminum, where etching and steam treatment cause structural damage to the zeolite framework, resulting in a decrease in Brønsted acidic and an increase in Lewis acidic sites. This observation is in accordance with the NH3-TPD results, verifying the transformation of framework aluminum into EFAl species.
2.2. MTO catalytic performance over the monofunctional untreated and dealuminated zeolite/zeo-type materials
At the first stage of the catalytic performance evaluation, we focused on MTO reactions conducted exclusively over monofunctional zeolites, encompassing both untreated and dealuminated zeolites (see Fig. 4–6 and S11–S17, and Table S1 in the ESI†). All catalytic tests were conducted under atmospheric pressure at a temperature of 400 °C, with a weight hourly space velocity (WHSV) of 1 h−1. Catalyst lifetime was conventionally defined as the duration needed for methanol conversion to reach 80%. The catalytic performance of the untreated commercial zeolites is depicted in Fig. 4, serving as a benchmark in this study. When illustrating hydrocarbon selectivity, we incorporated the first GC injection point and GC data just before catalyst deactivation at 100% methanol conversion in this manuscript. This presentation of catalyst data aims to highlight the influence of zeolite physicochemical properties throughout the catalyst lifetime, with the first GC injection point also serving as a reference point for operando studies (vide infra). We refer to Fig. S11† for a detailed overview of the catalytic performance evaluation across all zeolites used in this study. | ||
| Fig. 4 Catalytic performance evaluation of untreated and stand-alone 8-MR zeolite and zeo-type materials: (a) methanol conversion and (b) hydrocarbon selectivity, which was assessed at the initial GC injection point and just before deactivation at 100% methanol conversion, offering insights into the catalytic behavior during the early and steady states of the MTO reaction (reaction conditions: 400 °C, WHSV = 1 h−1; also see Fig. S11† for their detailed catalytic profiles). | ||
 | ||
| Fig. 5 Comparison of the catalytic performance evaluation of untreated and dealuminated 8-MR aluminosilicate zeolites (a and b) SSZ-39 and (c and d) SAPO-18 materials: (a and c) methanol conversion and (b and d) hydrocarbon selectivity, which was assessed at the initial GC injection point and just before deactivation at 100% methanol conversion, offering insights into the catalytic behavior during the early and steady states of the MTO reaction (reaction conditions: 400 °C, WHSV = 1 h−1; also see Fig. S12–S14† for their detailed catalytic profiles). | ||
 | ||
| Fig. 6 Comparison of the catalytic performance evaluation of untreated and dealuminated 8-MR ALPO-based zeo-type (a and b) SSZ-13, (c and d) SAPO-34 and (e and f) CCG-MTO materials: (a, c and e) methanol conversion and (b, d and f) hydrocarbon selectivity, which was assessed at the initial GC injection point and just before deactivation at 100% methanol conversion, offering insights into the catalytic behavior during the early and steady states of the MTO reaction (reaction conditions: 400 °C, WHSV = 1 h−1; also see Fig. S12, S16 and S17† for their detailed catalytic profiles). | ||
Fig. 4 illustrates a comparable catalytic lifetime for aluminosilicate SSZ-39 and SSZ-13 zeolites, approximately 180 min, with SAPO-34 and SAPO-18 exhibiting around 150 min and 600 min, respectively. Notably, the industrial CCG-MTO material, also based on SAPO-34, demonstrated a lifetime of approximately 630 min. We must highlight that CCG-MTO is a shaped-catalytic material, and a direct lifetime comparison with other powdered materials should be taken cautiously. Despite limited differences in other physicochemical properties between SAPO-34 and CCG-MTO, the distinctive hollow-structured spherical catalytic body in CCG-MTO contributed to its superior performance. The relatively smaller particle size likely facilitated molecular diffusion, contributing to the prolonged catalyst lifetime. Initially, aluminosilicate SSZ-39 and SSZ-13 zeolites displayed a significant preference for short C2–C3 alkane selectivity (82% and 57%, respectively), gradually transitioning towards shorter C2–C4 olefins as the reaction progressed (reaching up to 86% and 88% over SSZ-39 and SSZ-13 zeolites, respectively; see Fig. 4 and S11a and b in the ESI†). In contrast, ALPO-based SAPO-34, SAPO-18, and CCG-MTO materials initiated the reaction directly with a preference for C2–C4 shorter olefins (i.e., around 57%, 80%, and 72%, respectively; Fig. 4 and S11c and e in the ESI†). At the steady state, their C2–C4 shorter olefin selectivity reached up to around 86%, 93%, and 94% over SAPO-34, SAPO-18, and CCG-MTO materials, respectively (Fig. 4 and S11c and e in the ESI†). Concerning preferential olefin selectivity, SSZ-39, SSZ-13, SAPO-18, and SAPO-34 materials demonstrated comparable ranges for ethylene (18–31%), propylene (41–47%), and butylene (16–24%). In contrast, the CCG-MTO material exhibited very high selectivity for ethylene and propylene in equal proportions (∼83% collectively). The exceptional performance of the CCG-MTO material in achieving higher ethylene and propylene selectivity is surprisingly noteworthy, justifying its application in the industrial DMTO process in China.11,14–20
Subsequently, the influence of dealumination on aluminosilicate zeolites was explored, with a particular emphasis on understanding the similarities and differences in the effects of acid-based chemical etching and steaming on catalytic performance (see Fig. 5 and 6 and S12–S17 in the ESI†). Usually, the chemical etching of both SSZ-39 and SAPO-18 zeolites did not cause a substantial change in the catalyst lifetime and hydrocarbon product selectivity (as compared to their untreated counterpart, see Fig. 5). Their preferential product selectivity remained C2–C3 alkanes and C2–C4 olefins at the beginning and before deactivation, respectively. In contrast, steaming these zeolites had a pronounced effect on treated SSZ-39 zeolites, leading to a significantly higher catalytic lifetime (∼870 min, i.e., ∼4.8 times increase compared to the parent SSZ-39 zeolite, see Fig. 5a). Remarkably, steaming induced a shift in product selectivity for SSZ-39 zeolites, particularly at the initial stages of the reaction (Fig. 5a and S12a in the ESI†). This observation is not obvious for the SAPO-18 material, given that our characterization indicated no significant physicochemical changes following the dealumination treatment, except for the alteration of acid sites (Table S1 in the ESI†). Herein, ∼87% and ∼80% selectivity of C2–C4 olefins were obtained at the beginning of the reaction over steamed SSZ-39 and SAPO-18, respectively (Fig. 5b and d). As the reaction progressed, these corresponding C2–C4 olefin selectivity reached up to ∼98% and ∼91% over steamed SSZ-39 and SAPO-18, respectively (Fig. 5b and d and S12a and e in the ESI†).
Given that chemical etching did not result in significant changes in product selectivity, additional materials were prepared using higher concentrations of citric acid to treat both aluminosilicate zeolites (Fig. S13 and S14 in the ESI†). However, even with increased acid content, we did not observe substantial alterations in hydrocarbon product selectivity for both SSZ-39 and SAPO-18 materials. Moreover, the catalyst lifetime decreased with increasing acid content in the treatment of zeolites (Fig. S13 and S14 in the ESI†). The optimal olefin selectivity and catalyst lifetime were attained with an acid etching concentration of 0.2 M for SSZ-39 and 0.005 M for SAPO-18. Acid-treated zeolites demonstrated an increase of only ∼8% and ∼4% shorter olefin selectivity over SSZ-39 and SAPO-18 materials, respectively. Additionally, both steamed zeolites exhibited lower undesired alkane levels (<5%), particularly at the initial stages of the reaction, representing a significant fundamental difference from the acid-treated dealuminated zeolites. NH3-TPD analysis indicated higher weak acid content in etched zeolites (Table S3†), which might be contributing to their longer lifetime.26,27,37,38,42
Subsequently, the influence of dealumination on aluminosilicate zeolites was explored on SSZ-13 zeolites. Usually, the chemical etching of SSZ-13 zeolite did not cause a substantial change in the catalyst lifetime and hydrocarbon product selectivity (as compared to their untreated counterpart, see Fig. 6). The preferential product selectivity remained C2–C3 alkanes (57–73%) and C2–C4 olefins (88–93%) at the beginning and before deactivation, respectively. In contrast, steaming these zeolites had a pronounced effect on treated SSZ-13 materials, leading to a significantly shorter catalytic lifetime (∼150 min, i.e., ∼180 min for the parent SSZ-13 zeolite, see Fig. 6a). Remarkably, steaming induced a shift in product selectivity for SSZ-13 zeolites, particularly at the initial stages of the reaction (Fig. 6a and S12b in the ESI†). This observation is noteworthy for zeolite SSZ-13, given that our characterization indicated no significant physicochemical changes following the dealumination treatment, except for the alteration of acid sites (Table S1 in the ESI†). Herein, ∼83% selectivity of C2–C4 olefins was obtained at the beginning of the reaction over steamed SSZ-13 zeolites (Fig. 6b). As the reaction progressed, this corresponding C2–C4 olefin selectivity reached up to ∼92% over steamed SSZ-13 zeolites (Fig. 6b and S12b in the ESI†). Analogous catalytic experiments were also performed over SAPO-34 and CCG-MTO materials (see Fig. 6c and e and S12, S16 and S17 in the ESI†). Intriguingly, etching-led dealumination treatment resulted in enhanced catalyst lifetime and preferential C2–C4 olefin selectivity than steaming, which is in stark contrast to what was observed with aluminosilicate zeolites (see Fig. 5vs.6). Strangely, the steaming led to inferior catalytic performance over the CCG-MTO material (Fig. 6e). The chemical etching led to ∼2.0 times and ∼1.1 times improvement in the catalyst lifetime over SAPO-34 and CCG-MTO materials, respectively. However, when examining product selectivity, there were no substantial changes in the preferential C2–C4 olefin selectivity regardless of the dealumination treatment applied to the SAPO-34 material (Fig. 6d). In contrast, for the CCG-MTO material, both dealumination treatments resulted in slightly improved preferential C2–C4 olefin selectivity, reaching up to 97% and 95% over steamed and etched CCG-MTO material, respectively (with respect to ∼94% over the parent material, see Fig. 6e).
Similar experiments were conducted on acid-treated SSZ-13, SAPO-34, and CCG-MTO materials prepared using varying acid concentrations (refer to Fig. S15–S17 in the ESI†). Generally, acid treatment positively impacted the catalyst lifetime for the SAPO-34 material. However, excessive etching concentrations led to a reduced lifetime, emphasizing the importance of an appropriate acid concentration. Notably, there were no significant changes in hydrocarbon product selectivity regardless of the amount of acid used in this study. Based on the characterization, we could correlate the enhanced Lewis acidic content to the extended catalyst life, where applicable.32,34 Subsequently, the post-reacted deactivated zeolites were probed by TGA analysis to investigate the quantity of deposited organic species/coke (see Fig. S30†) and SEM images (Fig. S28 and S29†). SEM studies revealed that apart from the CCG-MTO material, where the distinctive pore structure was disrupted, the morphology of the other zeolites remained largely unchanged after the reaction. A non-identical coke residual pattern was typically noted between AEI and CHA topology materials (Fig. S30†).
2.3. Operando MTO characterization over the monofunctional untreated and dealuminated zeolite/zeo-type materials
To explore reactivity differences and selectivity preferences of untreated and dealuminated zeolite/zeo-type materials, especially in the early stages of the MTO reaction, operando UV-vis diffuse reflectance spectroscopy (DRS) coupled with online mass spectrometry (MS) was employed (Fig. S18†). This approach proves valuable in understanding the formation of MTO-relevant carbenium-based reaction intermediates on the solid catalyst and their correlation with effluent products. Due to the extensive operando data generated in this study involving a large number of catalysts, the data are divided over the manuscript (see Fig. 7 and 8) and ESI files (see Fig. S19 and S20 in the ESI†), providing operando data for untreated stand-alone/untreated zeolites and their dealuminated counterparts, respectively. Regardless of the materials utilized, characteristic absorption bands were consistently observed around <290 nm, 320–340 nm, 400–420 nm, and >550 nm, which were attributed to neutral aromatic hydrocarbons with methyl/alkyl substitutions, dienylic carbocations or methylbenzenonium ions with lower alkyl substitution, highly methylated areniums (especially hexamethylbenzenonium, HMB+), and coke precursors based on polycyclic aromatic species, respectively.8,66–69 Although their band positions exhibited similarities, their time-dependent behaviors and their influence on the formation of MTO products differed significantly (Fig. S19 and S20 in the ESI†).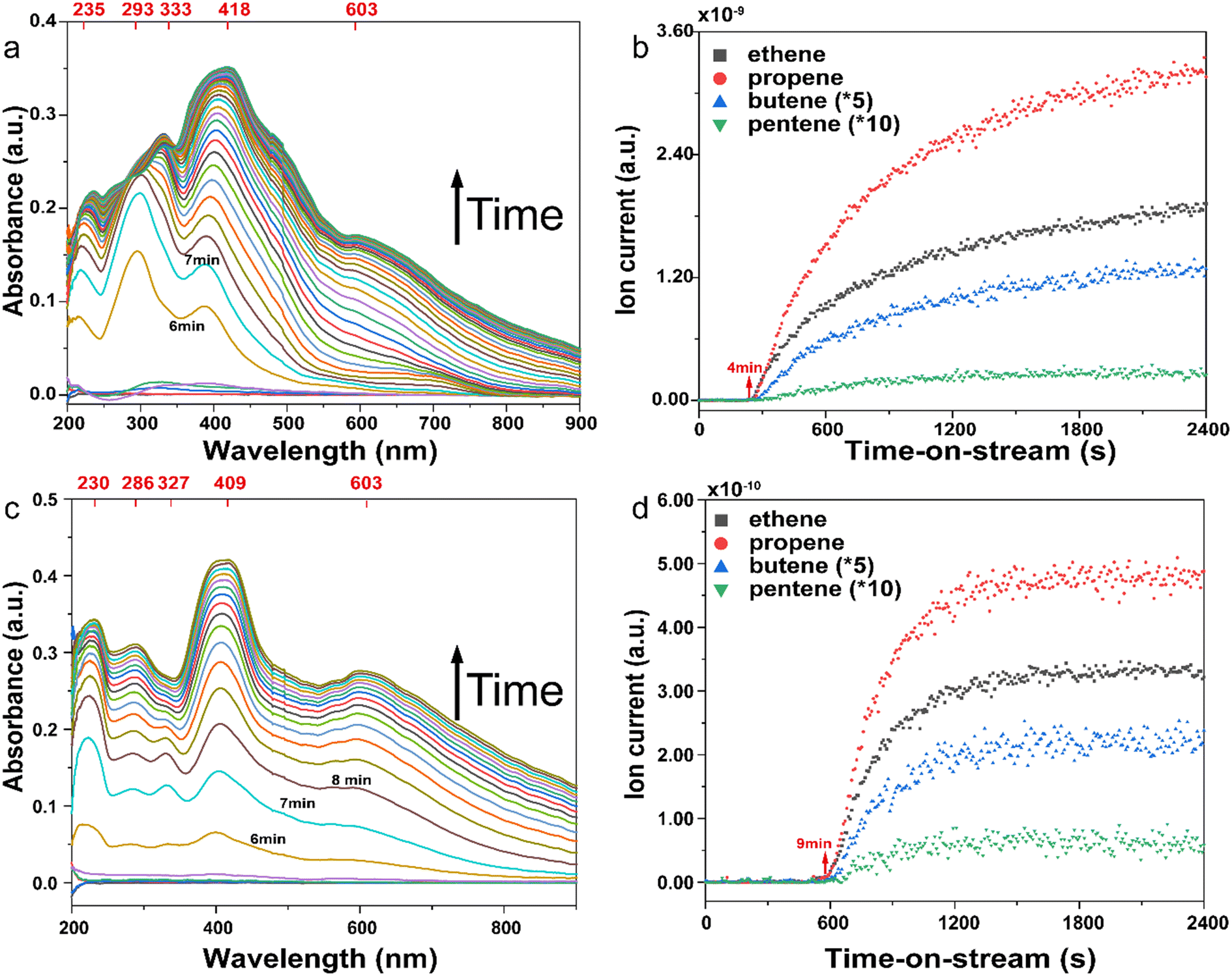 | ||
| Fig. 7 The operando investigation of the MTO reaction over stand-alone and untreated AEI-based (a and b) SSZ-39 and (c and d) SAPO-18 materials: the operando UV-vis profiles (a and c) and mass profiles (b and d) of methanol conversion over zeolites at 400 °C for 40 min (also see Fig. S19† for the analogous operando results over their dealuminated materials). | ||
 | ||
| Fig. 8 The operando investigation of the MTO reaction over stand-alone and untreated CHA-based (a and b) SSZ-13, (c and d) SAPO-34, and (e and f) CCG-MTO materials: the operando UV-vis profiles (a, c and e) and mass profiles (b, d and f) of methanol conversion over zeo-type materials at 400 °C for 40 min (also see Fig. S20† for the analogous operando results over their dealuminated materials). | ||
Fig. 7 presents an overview of the operando results for aluminosilicate zeolites SSZ-39 and SAPO-18. In both cases, there was a sudden increase in the intensity of UV-vis bands from ∼4 and ∼9 min onward, coinciding with the generation of gaseous MTO products, indicating a very brief induction period of a similar nature. Herein, SAPO-18 exhibited a relatively consistent rise in absorption band intensity compared to SSZ-39. However, SAPO-18 accumulated more coke precursors, as evidenced by the higher intensity of >550 nm bands related to poly-aromatic coke precursors.69 Dealuminated zeolites generally exhibited a slower increase in the intensity of the same absorption bands compared to the parent zeolites. Herein, both steamed SSZ-39 and SAPO-18 materials showed a relatively slower rise in relevant bands and effluent product formation compared to their etched counterparts, aligning with their superior lifetime (Fig. S12a in the ESI†). This operando study helps rationalize that superior performance is primarily governed by the slower accumulation of relevant HCP-based reaction intermediates.
The analogous operando characterization was conducted on SSZ-13 and ALPO-based zeo-type SAPO-34 and CCG-MTO materials (Fig. 8 and S20 in the ESI†). Comparatively, the intensity rise of all UV-vis bands was notably faster over SAPO-34 and coinciding with the main gaseous products. The operando UV–vis/MS profiles of untreated and dealuminated SAPO-34 materials showed minimal fundamental differences, except for the fact that etched SAPO-34 exhibited a slower change in absorption band intensity, aligning with its superior MTO performance in terms of catalyst lifetime (Fig. S16 in the ESI†). Conversely, steam-treated SAPO-34 exhibited a faster change in absorption band intensity, indicating its lower resistance to deactivation. For etched and steam-treated CCG-MTO (Fig. S20 in the ESI†), the steam-treated CCG-MTO material exhibited a faster change in absorption band intensity with relatively lower MTO product formation. In contrast, the etched CCG-MTO material displayed a slower change in absorption band intensity, reflecting its high resistance to deactivation and consistency with the reported catalytic performance (Fig. S17 in the ESI†). Interestingly, the UV-vis profile of etched and steamed versions of SSZ-13 seemed to be highly affected by the dealumination treatments (Fig. S19†). Although the slower formation of relevant HCP-based reaction intermediates, upon dealumination, is beneficial to catalysis, the large cages of CHA zeolites still could facilitate the accumulation and growth of aromatic species, ultimately promoting undesired catalyst deactivation.
2.4. MTO catalytic performance over the bifunctional zeolite/zeo-type materials
Our combined catalytic and operando studies have revealed that dealumination treatments, whether through steaming or chemical etching, primarily enhance catalyst lifetime by retarding the gradual formation of deactivation-related HCP-based reaction intermediates. However, the accumulation of poly-aromatics-based coke precursors cannot be entirely prevented; it can only be delayed, particularly in CHA-based materials. In existing literature, this deactivation phenomenon has often been associated with the detrimental role of formaldehyde (HCHO) or its derived oxymethylene species, which promote the generation of aromatics-based coke precursors and expedite catalyst deactivation.17,30,52,70,71 HCHO is a by-product in MTO catalysis, resulting from the disproportionation of methanol (2CH3OH → CH4 + HCHO).30 To mitigate the adverse impact of HCHO, metallic scavengers such as Y2O3 are typically combined with zeolites to form bifunctional catalytic systems, enhancing the reaction's durability.30,52,72 Hence, in the second phase of our catalytic performance evaluation, we assessed MTO performance over bifunctional catalytic systems, incorporating Y2O3 and untreated or dealuminated zeolites under identical reaction conditions (refer to Fig. 9 and 10 and S21–S25, and Table S1 in the ESI†). Fig. 4 is a benchmark for comparison in this study (i.e., MTO performance over monofunctional systems). For constructing these bifunctional systems, we specifically considered the superior dealuminated zeolite (either steamed or etched, based on the results in Fig. S12–S17 in the ESI†). Additionally, we employed two different mixing methods for zeolite/zeo-type materials and Y2O3: inter-pellet physical mixtures were prepared by separately sieving zeolite and Y2O3, while intra-pellet physical mixtures were prepared by pre-mixing zeolite and Y2O3 before sieving. As in previous analyses, we included both the first GC injection point and the GC data just before catalyst deactivation at 100% methanol conversion. For a detailed overview of the catalytic performance evaluation across all bifunctional zeolite systems used in this study, we refer to Fig. S21–S25.† | ||
| Fig. 9 Comparison of the catalytic performance evaluation over bifunctional AEI-zeolite-based systems consisting of Y2O3 and untreated and dealuminated (a and b) SSZ-39 and (c and d) SAPO-18 materials: (a and c) methanol conversion and (b and d) hydrocarbon selectivity, which was assessed at the initial GC injection point and just before deactivation at 100% methanol conversion, offering insights into the catalytic behavior during the early and steady states of the MTO reaction (reaction conditions: 400 °C, WHSV = 1 h−1; also see Fig. S21 and S22† for their detailed catalytic profiles). | ||
 | ||
| Fig. 10 Comparison of the catalytic performance evaluation over bifunctional CHA-based systems consisting of Y2O3 and untreated and dealuminated (a and b) SSZ-13, (c and d) SAPO-34 and (e and f) CCG-MTO materials: (a, c and e) methanol conversion and (b, d and f) hydrocarbon selectivity, which was assessed at the initial GC injection point and just before deactivation at 100% methanol conversion, offering insights into the catalytic behavior during the early and steady states of the MTO reaction (reaction conditions: 400 °C, WHSV = 1 h−1; also see Fig. S23–S25† for their detailed catalytic profiles). | ||
Fig. 9 represents the MTO catalytic outcomes for bifunctional systems utilizing AEI zeolites (SSZ-39 and SAPO-18). In the case of SSZ-39-based bifunctional catalytic systems, both inter- and intra-pellet mixtures of untreated SSZ-39 and Y2O3 did not exhibit an increase in their lifetimes, and their selectivity was comparable to that of the parent zeolite (Fig. 9a and b). However, bifunctional systems based on steamed SSZ-39 and Y2O3 enhanced catalyst lifetime (∼7.2 times) and C2–C4 shorter olefin selectivity (∼93%). For reference, the C2–C4 shorter olefin selectivity over the bifunctional system using untreated SSZ-39 and Y2O3 was ∼86% (also see Fig. S21 in the ESI†). In the bifunctional systems utilizing SAPO-18, both inter- and intra-pellet mixtures contribute to extended catalyst lifetimes. Specifically, the most durable performance was observed in the system incorporating steam-treated SAPO-18 and Y2O3, exhibiting a catalyst lifespan of approximately 1650 min (∼600 min longer) compared to untreated monofunctional SAPO-18 (Fig. 9c). Furthermore, irrespective of the specific SAPO-18-based catalytic system employed, consistent selectivity in the range of 96-97% towards C2–C4 short olefins is maintained (see Fig. 9c and S22 in the ESI†). Herein, the inter-pellet mixture method proves advantageous for SAPO-18-based systems.
Next, Fig. 10 illustrates the MTO catalytic outcomes for bifunctional systems employing CHA-based zeo-type materials (SSZ-13, SAPO-34 and CCG-MTO). Constructing bifunctional catalytic systems based on SSZ-13 zeolite did not significantly impact MTO performance (Fig. 10a and S23 in the ESI†). The only notable difference is that the intra-pellet bifunctional system using untreated and etched SSZ-13 zeolite exhibited a much slower rate of catalyst deactivation. It is important to highlight that the bifunctional catalytic systems demonstrated higher COx selectivity than their monofunctional counterparts, attributed to Y2O3's ability to decompose HCHO into CO and CO2 (see Fig. S26 in the ESI†). Herein, intra-pellet mixing was more advantageous. However, in the case of SAPO-34-based bifunctional catalytic systems, both inter- and intra-pellet mixing resulted in superior catalyst lifetime. Specifically, the most durable system was observed for the bifunctional catalytic system based on etched SAPO-34 and Y2O3, achieving ∼420 min of catalyst lifetime compared to monofunctional untreated SAPO-34 (∼150 min) (Fig. 10c). Apart from stability, there was no significant impact of bifunctional catalytic systems on the product distribution, with 92–95% of C2–C4 short olefin selectivity obtained regardless of the SAPO-34-based catalytic system employed (see Fig. 10c and S24 in the ESI†). The intra-pellet mixing was more advantageous for SAPO-34-based systems as well. Surprisingly, the bifunctional catalytic systems based on the CCG-MTO material did not follow the same trend as other materials (see Fig. 10e and S25 in the ESI†), although both inter- and intra-pellet mixing were beneficial for catalysis. Strangely, the bifunctional system based on dealuminated zeolite was not superior to its parent counterpart. The inter-pellet bifunctional system based on untreated CCG-MTO and Y2O3 led to ∼3.2 times enhancement of catalyst lifetime compared to its monofunctional counterpart. Although it is a remarkable MTO catalytic performance in terms of catalyst stability based on 8-MR zeolites, the bifunctional system did not impact preferential C2–C4 short olefin selectivity, which consistently remained within 95–97% during the steady state of MTO catalysis. It is worth noting again that the CCG-MTO material is a shaped catalytic body, where the inter-pellet mixing-based bifunctional system led to superior performance, in contrast to other 8-MR zeolite/zeo-type materials used that prefer intra-pellet mixing.52
3. Working protocol
The steady state of the zeolite-catalyzed MTO process is typically governed by a dual cycle mechanism consisting of arene- and alkene-based catalytic cycles.2,9–12,66,73 There are different postulations related to the efficacy of each component of the dual cycle and the preferential olefin selectivity. Based on the prior work, we could summarize that propylene and higher olefins were preferably sourced from the alkene cycle, while the arene cycle could lead to both ethylene and propylene.35,48,74–77 Thus, by examining the relative formation of C2–C4 short olefins, we can propose a hypothesis to explain the results obtained in this study (refer to Scheme 1). A simplified summary of the selectivity of C2–C4 short olefins from all catalytic experiments is provided in Table S1.† Combining results from both mono- and bifunctional zeolite systems, we draw the following conclusions. (i) Propylene and butylene selectivity consistently exceeded that of ethylene in all catalytic experiments, indicating the dominance of the alkene cycle over the arene cycle in 8-MR zeolites. (ii) SAPO-34-based materials, including CCG-MTO, exhibited relatively higher ethylene selectivity, suggesting the superior efficiency of the arene cycle in AlPO-based frameworks. The cage structure of ALPO-based CHA materials promotes the accumulation of aromatics-based HCP species, as confirmed by operando studies (Fig. S19 and S20 in the ESI†). (iii) SSZ-39 zeolite showed the lowest ethylene selectivity or highest propylene selectivity, suggesting a diminishing or dominating role of the arene or alkene cycles, respectively. (iv) Dealumination treatment, regardless of the zeolite or zeo-type material used, improved catalyst lifetime and propylene selectivity. This observation aligns with the idea that dealumination treatment reduces the quantity of Brønsted acid sites, promoting a more efficient alkene cycle, as confirmed by operando studies, indicating a slower rise of typical absorption bands over dealuminated materials (Fig. 7 and 8 and S19 and S20 in the ESI†). It is essential to note that zeolitic vicinal Brønsted acid sites are necessary for aromatics production, while the presence of isolated Brønsted acid sites is crucial for promoting the alkene cycle.27,78 (v) Constructing bifunctional catalytic systems, while enhancing system stability and durability, did not significantly impact hydrocarbon product selectivity. Bifunctional catalytic systems either led to similar C2–C4 short olefin selectivity or to slightly improved performance, as illustrated in Table S1.† Hence, the bifunctional catalytic system does not directly impact the respective dual-cycle mechanism. | ||
| Scheme 1 Simplified depiction of the role of arene- and alkene-based dual cycle mechanisms in MTO chemistry over untreated and dealuminated 8-MR zeolite/zeo-type materials. MTO catalysis over 8-MR zeolites is predominantly influenced by the alkene cycle, while a higher contribution from the arene cycle in CHA zeolite compared to AEI was also noted. Dealumination of zeolites enhances the efficiency of the alkene cycle, promoting propylene production. However, constructing a bifunctional system primarily enhances the stability and durability of the catalyst with minimal or no direct impact on the extent of the arene/alkene cycle. | ||
4. Conclusions
Intending to maximize MTO catalysis performance over 8-MR zeolite/zeo-type materials, including the industrially employed MTO catalyst, this study explores various strategies to achieve an extended catalyst lifetime, addressing their inherent deactivation issues without compromising preferential selectivity. The investigation focuses on five 8-MR zeolite/zeo-type materials: SSZ-39, SAPO-18, SSZ-13, SAPO-34, and CCG-MTO. Dealumination treatment is applied to these materials using both steaming and chemical etching approaches. Both untreated and dealuminated zeolite/zeo-type materials are combined with Y2O3 in an inter- and intra-pellet manner to construct corresponding bifunctional catalytic systems. The influence of dealumination on the original zeolite varied: while steaming proved advantageous for materials SSZ-39 and SAPO-18 (i.e., AEI-based materials), etching yielded benefits for SSZ-13, SAPO-34, and CCG-MTO materials (i.e., CHA-based materials). Additionally, it was observed that the effectiveness of the chemical etching method depended on the concentration of the acid used, with higher acidic amounts not necessarily leading to superior performance. The construction approach of bifunctional zeolites also influenced the MTO performance: inter-pellet mixing was advantageous for the industrial CCG-MTO and SAPO-18 catalysts, whereas intra-pellet mixing yielded benefits for SSZ-39, SSZ-13, and SAPO-34 materials. The steamed treatment of SSZ-39 and SAPO-18, AEI-structured materials, increased both C2–C4 short olefin selectivity and lifespan compared to the parent material. Combined with Y2O3 as a formaldehyde scavenger, a bifunctional catalytic system further enhances the catalyst lifespan without compromising the C2–C4 short olefin selectivity. The acid etching treatment of CHA-topology materials, including SSZ-13, SAPO-34, and CCG-MTO, also increases their lifespan while maintaining near-constant C2–C4 short olefin selectivity. Incorporation of Y2O3 intra-pellet as a mixture with zeolites results in a significant increase in lifespan for acid-etched SSZ-13 and SAPO-34 materials as compared to their monofunctional untreated counterparts, while the C2–C4 olefin selectivity again remains almost unchanged. The uniquely shaped catalytic body of CCG-MTO allowed a surprising increase in the catalyst lifetime (∼2000 min vs. ∼630 min), albeit with no change in C2–C4 olefin selectivity, by simply combining Y2O3 with the parent untreated material.To rationalize the MTO catalysis performance obtained in this work, a combination of traditional and advanced zeolite characterization tools was employed, including under operando conditions. Operando studies revealed that dealumination treatment positively impacted the reaction by slowing down the formation of poly-aromatics-based coke precursors. The most significant impact on catalyst lifetime resulted from bifunctional catalytic systems. Despite not significantly altering or promoting specific product selectivity, Y2O3, a well-known scavenger of formaldehyde (a methanol disproportionation product facilitating aromatic formation), played a crucial role in potentially mitigating aromatics' formation and slowing deactivation by preventing oxymethylene species formation.52,72 Moreover, the lifetime of catalysts in the MTO reaction over 8-MR zeolites can be extended by dealumination-driven changes in morphology (such as increased hierarchical porosity and smaller crystal size) or acidity (such as altered acid sites distribution or Brønsted–Lewis acid synergy), which improve product diffusion and reduce coke precursor deposition. In essence, this study not only maximizes the catalyst lifetime of 8-MR zeolite/zeo-type materials for the MTO process without affecting preferential product selectivity but also advances the understanding of zeolite-catalyzed methanol conversion chemistry, contributing to the promotion of the methanol economy initiative.
Author contributions
Shican Jiang: conceptualization, methodology, investigation, and writing – original draft. Hexun Zhou: formal analysis, investigation. Xin Zhang: formal analysis, investigation. Xue Zhou: investigation. Abhishek Dutta Chowdhury: conceptualization, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This project has been financially supported by the National Natural Science Foundation of China (NSFC) (Grant No. 22350610243, 22050410276), the Fundamental Research Funds for the Central Universities (Grant No.: 2042023kf0126) (China), and the start-up research grant from Wuhan University (China). The authors also thank the Core Research Facilities of CCMS (WHU) for the solid-state NMR characterization.References
- M. Wang, Z. Wang, S. Liu, R. Gao, K. Cheng, L. Zhang, G. Zhang, X. Min, J. Kang, Q. Zhang and Y. Wang, J. Catal., 2021, 394, 181–192 CrossRef CAS
.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS
- L. Zhong, F. Yu, Y. An, Y. Zhao, Y. Sun, Z. Li, T. Lin, Y. Lin, X. Qi, Y. Dai, L. Gu, J. Hu, S. Jin, Q. Shen and H. Wang, Nature, 2016, 538, 84–87 CrossRef CAS PubMed
- E. T. C. Vogt and B. M. Weckhuysen, Chem. Soc. Rev., 2015, 44, 7342–7370 RSC
-
E. T. C. Vogt, G. T. Whiting, A. Dutta Chowdhury and B. M. Weckhuysen, in Advances in Catalysis, Academic Press, 2015, vol. 58, pp. 143–314 Search PubMed
- A. Kätelhön, R. Meys, S. Deutz, S. Suh and A. Bardow, Proc. Natl. Acad. Sci. U. S. A., 2019, 166, 11187–11194 CrossRef PubMed
-
G. A. Olah, A. Goeppert and G. K. S. Prakash, in Beyond Oil and Gas: The Methanol Economy, 2nd edn, 2009, pp. 1–334 Search PubMed
- X. Gong, M. Çaǧlayan, Y. Ye, K. Liu, J. Gascon and A. Dutta Chowdhury, Chem. Rev., 2022, 122, 14275–14345 CrossRef CAS PubMed
- U. Olsbye, S. Svelle, M. Bjrgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed
- U. Olsbye, S. Svelle, K. P. Lillerud, Z. H. Wei, Y. Y. Chen, J. F. Li, J. G. Wang and W. B. Fan, Chem. Soc. Rev., 2015, 44, 7155–7176 RSC
- P. Tian, Y. Wei, M. Ye and Z. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS
- T. Li, T. Shoinkhorova, J. Gascon and J. Ruiz-Martinez, ACS Catal., 2021, 11, 7780–7819 CrossRef CAS
- S. Ilias and A. Bhan, ACS Catal., 2013, 3, 18–31 CrossRef CAS
-
S. Xu, Y. Zhi, J. Han, W. Zhang, X. Wu, T. Sun, Y. Wei and Z. Liu, in Advances in Catalysis, Academic Press Inc., 2017, vol. 61, pp. 37–122 Search PubMed
-
M. Ye, H. Li, Y. Zhao, T. Zhang and Z. Liu, in Advances in Chemical Engineering, Academic Press Inc., 2015, vol. 47, pp. 279–335 Search PubMed
- W. Zhang, S. Lin, Y. Wei, P. Tian, M. Ye and Z. Liu, Natl. Sci. Rev., 2023, 10, nwad120 CrossRef CAS PubMed
- S. Lin, Y. Zhi, W. Zhang, X. Yuan, C. Zhang, M. Ye, S. Xu, Y. Wei and Z. Liu, Chin. J. Catal., 2023, 46, 11–27 CrossRef CAS
-
W. Zhang, Y. Wei and Z. Liu, in The Chemical Transformations of C1 Compounds, 2022, pp. 71–126 Search PubMed
- M. Yang, D. Fan, Y. Wei, P. Tian and Z. Liu, Adv. Mater., 2019, 31, 1902181 CrossRef CAS PubMed
-
Z. Liu, G. Chen, J. Liang, Q. Wang and G. Cai, in Studies in Surface Science and Catalysis, ed. C. H. Bartholomew and J. B. Butt, Elsevier, 1991, vol. 68, pp. 815–818 Search PubMed
- L. Wu, Z. Liu, L. Xia, M. Qiu, X. Liu, H. Zhu and Y. Sun, Chin. J. Catal., 2013, 34, 1348–1356 CrossRef CAS
- D. Li, Y. Chen, J. Hu, B. Deng, X. Cheng and Y. Zhang, Appl. Catal., B, 2020, 270, 118881 CrossRef CAS
- Y. Ji, M. A. Deimund, Y. Bhawe and M. E. Davis, ACS Catal., 2015, 5, 4456–4465 CrossRef CAS
- Y. Mou, C. Xu, Z. Sun, S. Liu, F. Wang, D. Han, G. Wang and L. Bing, Inorg. Chem. Commun., 2023, 151, 110576 CrossRef CAS
- Q. Wang, W. Zhang, X. Ma, Y. Liu, L. Zhang, J. Zheng, Y. Wang, W. Li, B. Fan and R. Li, Fuel, 2023, 331, 125935 CrossRef CAS
- Q. Li, W. Cong, J. Zhang, C. Xu, F. Wang, D. Han, G. Wang and L. Bing, Microporous Mesoporous Mater., 2022, 331, 111649 CrossRef CAS
- M. Dusselier, M. A. Deimund, J. E. Schmidt and M. E. Davis, ACS Catal., 2015, 5, 6078–6085 CrossRef CAS
- Z. Li, J. Martínez-Triguero, J. Yu and A. Corma, J. Catal., 2015, 329, 379–388 CrossRef CAS
-
J. Hagen, in Industrial Catalysis, Wiley, 2015, pp. 239–260 Search PubMed
- X. Gong, Y. Ye and A. D. Chowdhury, ACS Catal., 2022, 12, 15463–15500 CrossRef CAS
- E. G. Derouane and Z. Gabelica, J. Catal., 1980, 65, 486–489 CrossRef CAS
- S. Ren, G. Liu, X. Wu, X. Chen, M. Wu, G. Zeng, Z. Liu and Y. Sun, Chin. J. Catal., 2017, 38, 123–130 CrossRef CAS
- Y. Wang, S. L. Chen, Y. L. Gao, Y. Q. Cao, Q. Zhang, W. K. Chang and J. B. Benziger, ACS Catal., 2017, 7, 5572–5584 CrossRef CAS
- W. Jin, B. Wang, P. Tuo, C. Li, L. Li, H. Zhao, X. Gao and B. Shen, Ind. Eng. Chem. Res., 2018, 57, 4231–4236 CrossRef CAS
- K. De Wispelaere, K. Hemelsoet, M. Waroquier and V. Van Speybroeck, J. Catal., 2013, 305, 76–80 CrossRef CAS
- L. Wu and E. J. M. Hensen, Catal. Today, 2014, 235, 160–168 CrossRef CAS
- V. Babić, S. Koneti, S. Moldovan, N. Nesterenko, J. P. Gilson and V. Valtchev, Microporous Mesoporous Mater., 2021, 314, 110863 CrossRef
- L. Sommer, D. Mores, S. Svelle, M. Stöcker, B. M. Weckhuysen and U. Olsbye, Microporous
Mesoporous Mater., 2010, 132, 384–394 CrossRef CAS
- M. A. Deimund, L. Harrison, J. D. Lunn, Y. Liu, A. Malek, R. Shayib and M. E. Davis, ACS Catal., 2016, 6, 542–550 CrossRef CAS
- X. Zhu, J. P. Hofmann, B. Mezari, N. Kosinov, L. Wu, Q. Qian, B. M. Weckhuysen, S. Asahina, J. Ruiz-Martínez and E. J. M. Hensen, ACS Catal., 2016, 6, 2163–2177 CrossRef CAS
- B. N. Bhadra, J. Y. Song, N. A. Khan, J. W. Jun, T. W. Kim, C. U. Kim and S. H. Jhung, J. Catal., 2018, 365, 94–104 CrossRef CAS
- N. Martín, Z. Li, J. Martínez-Triguero, J. Yu, M. Moliner and A. Corma, Chem. Commun., 2016, 52, 6072–6075 RSC
- M. Wen, L. Ren, J. Zhang, J. Jiang, H. Xu, Y. Guan and P. Wu, Chem. Commun., 2021, 57, 5682–5685 RSC
- C. Sun, Y. Wang, H. Chen, X. Wang, C. Wang and X. Zhang, Catal. Today, 2020, 355, 188–198 CrossRef CAS
- Z. Gao, Q. Wang, T. Yan, K. Zhao, W. Liu, Q. He, Q. Sun, Z. Li, J. Zhang and C. Liu, Ind. Eng. Chem. Res., 2023, 62, 8644–8653 CrossRef CAS
- Z. Gao, Q. Wang, T. Yan, K. Zhao, W. Liu, Q. He, Q. Sun, Z. Li, J. Zhang and C. Liu, Ind. Eng. Chem. Res., 2023, 62, 8644–8653 CrossRef CAS
- L. Zhang, B. Geng, S. Wang, P. Wang, H. Xiao and J. Jia, Microporous Mesoporous Mater., 2023, 361, 112744 CrossRef CAS
- A. Hwang, B. A. Johnson and A. Bhan, J. Catal., 2019, 369, 86–94 CrossRef CAS
- S. Wilson and P. Barger, Microporous Mesoporous Mater., 1999, 29, 117–126 CrossRef CAS
- L. Tan, N. Jiao, X. Bai, H. Wang, J. Wang, H. Wang and X. Zhang, Eur. J. Inorg. Chem., 2023, 26 Search PubMed
- Z. Xu, J. Li, Y. Huang, H. Ma, W. Qian, H. Zhang and W. Ying, Catal. Commun., 2020, 141, 106014 CrossRef CAS
- A. Hwang and A. Bhan, ACS Catal., 2017, 7, 4417–4422 CrossRef CAS
- https://www.china-catalyst.com/ .
- T. Lu, Y. Sun, Z. Wang, S. Zhang, Y. Yang, H. Zhang, P. Cheng and Z. Zhao, Ind. Eng. Chem. Res., 2023, 63, 5195–5205 CrossRef
- D. Fan, Y. Qiao, K. Cao, L. Sun, S. Xu, P. Tian and Z. Liu, Microporous Mesoporous Mater., 2020, 300, 110156 CrossRef CAS
- T. Shoinkhorova, A. Dikhtiarenko, A. Ramirez, A. Dutta Chowdhury, M. Caglayan, J. Vittenet, A. Bendjeriou-Sedjerari, O. S. Ali, I. Morales-Osorio, W. Xu and J. Gascon, ACS Appl. Mater. Interfaces, 2019, 11, 44133–44143 CrossRef CAS PubMed
- Y. Xing, G. Li, Z. Lin, Z. Xu, H. Huang, Y. Zhu, S. C. E. Tsang and M. M. J. Li, J. Mater. Chem. A, 2023, 11, 14058–14066 RSC
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS
-
J. M. Campelo, F. Lafont, J. M. Marinas and M. Ojeda, Studies of catalyst deactivation in methanol conversion with high, medium and small pore silicoaluminophosphates, 2000, vol. 192 Search PubMed
- Y. J. Lee, S. C. Baek and K. W. Jun, Appl. Catal., A, 2007, 329, 130–136 CrossRef CAS
- X. Wang, Z. Li, F. Gong, M. Ma and Y. Zhu, Mol. Catal., 2021, 499, 111312 CrossRef CAS
- F. Lónyi and J. Valyon, Microporous Mesoporous Mater., 2001, 47(2–3), 293–301 CrossRef
- Q. Zhu, J. N. Kondo, T. Tatsumi, S. Inagaki, R. Ohnuma, Y. Kubota, Y. Shimodaira, H. Kobayashi and K. Domen, J. Phys. Chem. C, 2007, 111, 5409–5415 CrossRef CAS
- L. Rodríguez-González, F. Hermes, M. Bertmer, E. Rodríguez-Castellón, A. Jiménez-López and U. Simon, Appl. Catal., A, 2007, 328, 174–182 CrossRef
- G. T. Whiting, S. H. Chung, D. Stosic, A. D. Chowdhury, L. I. Van Der Wal, D. Fu, J. Zecevic, A. Travert, K. Houben, M. Baldus and B. M. Weckhuysen, ACS Catal., 2019, 9, 4792–4803 CrossRef CAS
- H. Sharbini Kamaluddin, X. Gong, P. Ma, K. Narasimharao, A. Dutta Chowdhury and M. Mokhtar, Mater. Today Chem., 2022, 26, 101061 CrossRef CAS
- W. Chen, X. Yi, Z. Liu, X. Tang and A. Zheng, Chem. Soc. Rev., 2022, 51, 4337–4385 RSC
- A. D. Chowdhury, K. Houben, G. T. Whiting, S. H. Chung, M. Baldus and B. M. Weckhuysen, Nat. Catal., 2018, 1, 23–31 CrossRef CAS
- A. D. Chowdhury, K. Houben, G. T. Whiting, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 15840–15845 CrossRef CAS PubMed
- W. Wen, S. Yu, C. Zhou, H. Ma, Z. Zhou, C. Cao, J. Yang, M. Xu, F. Qi, G. Zhang and Y. Pan, Angew. Chem., 2020, 132, 4903–4908 CrossRef
- Y. Liu, F. M. Kirchberger, S. Müller, M. Eder, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2019, 10, 1462 CrossRef PubMed
- X. Zhang, H. Zhou, Y. Ye, X. You, X. Zhou, S. Jiang, K. Liu and A. Dutta Chowdhury, Inorg. Chem. Front., 2023, 10, 6632–6645 RSC
- S. Xu, A. Zheng, Y. Wei, J. Chen, J. Li, Y. Chu, M. Zhang, Q. Wang, Y. Zhou, J. Wang, F. Deng and Z. Liu, Angew. Chem., Int. Ed., 2013, 52, 11564–11568 CrossRef CAS PubMed
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K. P. Lillerud, S. Kolboe and M. Bjørgen, J. Am. Chem. Soc., 2006, 128, 14770–14771 CrossRef CAS PubMed
- M. Bjørgen, F. Joensen, K. P. Lillerud, U. Olsbye and S. Svelle, Catal. Today, 2009, 142, 90–97 CrossRef
- X. Sun, S. Mueller, Y. Liu, H. Shi, G. L. Haller, M. Sanchez-Sanchez, A. C. Van Veen and J. A. Lercher, J. Catal., 2014, 317, 185–197 CrossRef CAS
- X. Sun, S. Mueller, H. Shi, G. L. Haller, M. Sanchez-Sanchez, A. C. Van Veen and J. A. Lercher, J. Catal., 2014, 314, 21–31 CrossRef CAS
- S. Bailleul, I. Yarulina, A. E. J. Hoffman, A. Dokania, E. Abou-Hamad, A. D. Chowdhury, G. Pieters, J. Hajek, K. De Wispelaere, M. Waroquier, J. Gascon and V. Van Speybroeck, J. Am. Chem. Soc., 2019, 141, 14823–14842 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cy00110a |
| This journal is © The Royal Society of Chemistry 2024 |