
Bismuth single-atom alloying of palladium nanosheets promotes selective electrochemical valorization of glycerol to C3 products†
Zhenghao
Mao‡
ab,
Lin
Jia‡
ab,
Xinnan
Mao‡
ab,
Xue
Ding
ab,
Binbin
Pan
ab,
Tianran
Yan
ab,
Jie
Xu
 d,
Liang
Zhang
ab,
Lu
Wang
*ab,
Na
Han
*ab and
Yanguang
Li
*abc
d,
Liang
Zhang
ab,
Lu
Wang
*ab,
Na
Han
*ab and
Yanguang
Li
*abc
aInstitute of Functional Nano & Soft Materials (FUNSOM), Soochow University, Suzhou, 215123, China. E-mail: yanguang@suda.edu.cn; hanna@suda.edu.cn; lwang22@suda.edu.cn
bJiangsu Key Laboratory for Advanced Negative Carbon Technologies, Soochow University, Suzhou 215123, China
cMacao Institute of Materials Science and Engineering (MIMSE), MUST–SUDA Joint Research Center for Advanced Functional Materials, Macau University of Science and Technology, Taipa 999078, Macau SAR, China
dCollege of Chemistry and Materials Engineering, Wenzhou University, Wenzhou, 325035, China
First published on 7th August 2024
Abstract
The electrochemical oxidation of glycerol into value-added products (in particular C3 products) offers an appealing approach for biomass valorization. Precious-metal-based catalysts are so far the only candidates that can deliver appreciable C3 selectivity. Unfortunately, they generally have strong adsorption towards key reaction intermediates, leading to product over-oxidation and catalyst poisoning. To this end, we report that single atom alloying represents a promising solution, and demonstrate that bismuth atoms dispersed on palladium nanosheets significantly promote electrochemical glycerol oxidation to C3 products. Under the optimal conditions, our catalyst exhibits an extraordinary C3 faradaic efficiency of >90%, partial current density of >150 mA cm−2 and operational stability over 30 h. All these metrics greatly surpass those of other precious-metal-based competitors reported earlier. Theoretical simulations unveil that the single atom alloying with Bi modulates the electronic structure of Pd and lowers the adsorption energies of reaction intermediates. Finally, we use glycerol oxidation as the anodic half reaction to pair with the CO2 reduction reaction as the cathodic half reaction, and achieve the simultaneous valorization of glycerol and CO2 with improved high energy efficiency and economic viability.
 Na Han | Dr Na Han received her PhD degree in chemistry from Soochow University in 2018. After a three-year (2018–2021) stint as a postdoctoral fellow at Soochow University, she joined the Institute of Functional Nano & Soft Materials (FUNSOM) as an associate professor at Soochow University. Her current research is focused on energy electrocatalysis, including carbon dioxide reduction, water electrolysis and small molecule oxidation. In addition, she was awarded for the National Postdoctoral Program for Innovative Talents. |
1 Introduction
Glycerol is a major by-product of biodiesel manufacturing, and has been listed as one of the top twelve biobased building block chemicals by the U.S. Department of Energy.1 As the biodiesel industry is rapidly expanding, an excessive amount of crude glycerol is being produced every year, which is of relatively low economic value owing to its poor purity and sometimes discarded as waste.2 There are growing calls for the development of efficient approaches to convert crude glycerol into value-added products, such as 1,3-dihydroxyacetone (DHA), glyceric acid (GLA), tartronic acid (TA), lactic acid (LA), oxalic acid (OA) and formic acid (FA).3,4 In particular, all C3 products (DHA, GLA, TA and LA) have substantially higher economic value than glycerol (for example, DHA is over 100 times more expensive than the latter), and are also more desirable than C1 and C2 products.5,6 They are important chemical precursors with versatile applications in the cosmetic, pharmaceutical, fine chemical, and food industries.6,7 Among the various approaches currently under development to valorize glycerol, the electrochemical glycerol oxidation reaction (GOR) is gaining increasing attention by virtue of its mild reaction conditions and potential to couple with renewable electricity for rendering the whole process fully sustainable.8–11 However, it remains a largely underexplored field.The electrochemical GOR is a complex process involving 2–14 electron transfers depending on the oxidation products, and requires the assistance of suitable electrocatalysts in order to expedite the reaction and steer the selectivity.8,12,13 Among a variety of materials investigated, precious-metal-based catalysts (Pt, Pd and Au) are so far the only candidates that can achieve appreciable C3 product quantities from the GOR.14–16 Unfortunately, these catalysts generally have high surface affinities towards C3 intermediates, favoring their further oxidation and thereby causing unsatisfactory C3 product selectivity.14 The strong adsorption of reaction intermediates may also poison the catalyst surface, shut off the reaction cycle and result in poor catalyst stability.17–19 At present, the majority of precious-metal-based GOR electrocatalysts suffer from small current density (<10 mA cm−2), low C3 selectivity (<60%), and/or short operational stability (<5 h).11,14
Alloying represents a promising strategy in the rational design of metallic catalysts for a range of important reactions.20,21 The introduction of secondary metal components may effectively tune the electronic structure, modulate the surface adsorption properties, and thereby greatly impact the catalytic performances of the resultant alloys. Here, we reason that alloying precious metals with low-work-function metals may downshift their d-band centers, and therefore alleviate the strong adsorption of reaction intermediates according to the d-band theory.22 The presence of oxophilic metal components may also promote glycerol oxidation through a bifunctional mechanism and prolong the operational stability of alloy catalysts.23–26 As a proof-of-concept, we demonstrate Bi atoms dispersed on Pd nanosheets as an efficient GOR electrocatalyst. Through the synergistic alloying effect, the optimal sample (Bi1Pd NS) exhibits industry-level current density (>150 mA cm−2), high selectivity (>90%), and long-term stability (>30 h) for the GOR to C3 products. The Bi1Pd-catalyzed GOR can be further coupled with the CO2 reduction reaction (CO2RR) to realize the simultaneous valorization of glycerol at the anode and CO2 at the cathode.
2 Results and discussion
2.1 Synthesis and structural characterization of Bi1Pd NS
Fig. 1a illustrates the two-step synthetic procedure for Bi1Pd (see the ESI† for more details). Pd nanosheets (denoted as Pd NS) were first prepared using a solvothermal method under a CO atmosphere.27 During the reaction, CO specifically adsorbs on Pd (111), thereby inhibiting the growth of this particular facet, and directs the formation of ultrathin Pd nanosheets. Transmission electron microscope (TEM) imaging shows that the as-prepared Pd NS consists of polygonal nanosheets with an average lateral size of 100 nm (Fig. S1†), and their spectroscopic characterization is presented in Fig. S2.† Subsequently, Pd NS was loaded with dilute Bi atoms by reducing a calculated amount of bismuth chloride in ethylene glycol to yield the target product (Bi1Pd NS) under mild conditions. The X-ray diffraction (XRD) pattern of Bi1Pd NS is consistent with that of face-centered cubic Pd (JCPDS no. 05-0681) without any Bi signal owing to its dispersed state in the final product (Fig. 1b). After Bi atom deposition, the overall polygonal nanosheet morphology is preserved, and no additional nanoparticles or clusters are discernable under TEM (Fig. 1c). Energy-dispersive X-ray spectroscopy (EDS) elemental mapping under scanning transmission electron microscopy (STEM) reveals the uniform distribution of both Pd and Bi elements within Bi1Pd NS (Fig. 1d). Moreover, aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was employed to image the sample at the atomic scale (Fig. 1e). The bright spots on the nanosheet basal plane correspond to Bi atoms embedded in the Pd lattice due to the different Z-contrast of these two elements. They are presumably formed by trapping reduced Bi atoms at the defect sites of Pd nanosheets during the second step of the reaction. By controlling the Bi content to a low level (<8 at%), the majority of Bi atoms are observed to be singly dispersed on Pd nanosheets free of aggregation. | ||
| Fig. 1 Preparation and structural characterization of Bi1Pd NS. (a) Schematic synthetic procedure for Pd NS and Bi1Pd NS. (b) XRD pattern, (c) TEM image and (d) EDS elemental mapping of Bi1Pd NS. (e) Aberration-corrected HAADF-STEM image of Bi1Pd NS; the dashed blue circles indicate dispersed Bi atoms. | ||
The chemical environment and electronic state of Bi1Pd NS were investigated by X-ray based spectroscopy. Its Pd 3d X-ray photoelectron spectroscopy (XPS) spectrum indicates that Pd is predominantly in the metallic state (Fig. S3a†).28 The Bi 4f XPS spectrum can be deconvoluted into two sets of doublets with a major contribution from Bi0 and a minor one from Bi3+ (Fig. S3b†).23 The slight oxidation of both species is expected upon air exposure. X-ray absorption near edge structure (XANES) measurements were also conducted. At the Pd K-edge, the XANES spectrum of Bi1Pd closely resembles that of the Pd foil, corroborating the metallic nature of Pd in Bi1Pd (Fig. S4a†). At the Bi L3-edge, the adsorption edge of Bi1Pd lies between those of the Bi foil and Bi2O3 references, evidencing the mixed contributions of Bi metal and oxide (Fig. S4b†). Furthermore, the local coordination environment was probed by extended X-ray absorption fine structure (EXAFS) analysis. As shown in Fig. S4c,† a peak at 1.74 Å is observed likely attributed to the Bi–O bonding owing to its surface oxidation. The absence of direct Bi–Bi bonding at 2.96 Å attests to the single dispersion of Bi atoms in Bi1Pd.
2.2 Electrocatalytic GOR performance of Bi1Pd NS
Next, the electrocatalytic GOR performance of Bi1Pd NS was evaluated in a customized H-cell. Fig. S5† compares the cyclic voltammogram (CV) curves of Bi1Pd NS and Bi1Pd NS in 1 M KOH containing 0.5 M glycerol. The former exhibits much greater oxidation peak current density than the latter, indicative of a higher electrocatalytic activity. Fig. 2a, S6a and S6c† illustrate the polarization curves of Bi1Pd NS, Pd NS and commercial 20 wt% Pd/C. Among them, Bi1Pd NS exhibits the highest activity featuring a low onset potential of 0.6 V (versus the reversible hydrogen electrode or RHE, the same hereinafter), and delivers an anodic current density of 52 mA cm−2 at 0.75 V, which is substantially higher than those of Pd NS and 20 wt% Pd/C at the same precious metal loading. To quantitatively analyze the oxidation products, chronoamperometric (i–t) electrolysis was conducted at selected working potentials from 0.6 V to 0.79 V. The anolyte was collected at the end of electrolysis and analyzed by high-performance liquid chromatography (HPLC). Seven different oxidation products were detected for all the catalysts examined, including C1 (FA), C2 (OA and GA), and C3 (LA, GLA, TA and DHA), and they have distinct potential dependences. It is worth noting that the total faradaic efficiency (FE) for C3 products on Bi1Pd NS is measured to be 98% at 0.6 V (Fig. 2b). The C3 selectivity gradually decreases with the increasing working potential owing to product over-oxidation. Remarkably, Bi1Pd NS remains a total C3 FE of 70% even at 0.79 V. By comparison, both Pd NS and 20 wt% Pd/C exhibit inferior C3 selectivity, for example, their total C3 FEs are only 62% and 68% respectively at 0.6 V (Fig. 2c, S6d and S6b†). Moreover, the partial current density for C3 products (jC3) was derived for the three samples. It reaches a maximum value of 49 mA cm−2 at 0.79 V on Bi1Pd NS, 1.8–2.1 times higher than those of 20 wt% Pd/C and Pd NS (Fig. 2c, S6d and S6b†). These results attest to the great promoting effect of dispersed Bi atoms in Bi1Pd NS. | ||
| Fig. 2 Electrocatalytic GOR performance of Bi1Pd NS. (a) Polarization curves of Bi1Pd NS and Pd NS in an H-cell with 1 M KOH + 0.5 M glycerol. (b) Potential-dependent FEs for different oxidation products on Bi1Pd NS. (c) Potential-dependent C3 FE and partial current density on Bi1Pd NS and Pd NS. (d) Polarization curves of Bi1Pd NS and Pd NS in a flow cell with 4 M KOH + 2 M glycerol. (e) Potential-dependent FEs and partial current densities for C3 products on Bi1Pd NS at the flow rate of 12.5 mL min−1. (f) Comparison of the total C3 FE and partial current density on Bi1Pd NS and other reported GOR electrocatalysts from the literature. (g) Chronoamperometry curve of Bi1Pd NS at 0.71 V; the dotted lines indicate electrolyte refreshing every 5 h. | ||
So far, the majority of GOR studies have reported current densities less than 10 mA cm−2, which are far below the expected threshold value (>100 mA cm−2) for commercial applications.11,29 In order to show the practical viability of our catalyst and produce desirable C3 products at industrially relevant current densities, we constructed a flow cell electrolyzer using Bi1Pd-loaded carbon fiber paper (CFP) as the working electrode. Initial screening identifies the best electrolyte composition to be 4 M KOH solution with 2 M glycerol (Fig. S7†). The polarization current density of Bi1Pd NS similarly takes off at approximately 0.6 V and reaches an extraordinary value of 200 mA cm−2 at 0.75 V (Fig. 2d). The significantly larger current density observed here is partly due to the higher solution alkalinity and glycerol concentration, but more importantly the forced anolyte flow that enhances the glycerol mass transport and accelerates the removal of its oxidation products. As a result, we investigated the effect of the anolyte flow rate. A higher flow rate alleviates product over-oxidation and enhances the C3 selectivity (Fig. S8†). At the highest flow rate of 12.5 mL min−1 (attainable with our peristaltic pump), the total C3 FE on Bi1Pd NS stays above 75% over the entire potential range examined (Fig. 2e). In particular, the selectivity for DHA – one of the most desirable oxidation products from the GOR – is more than doubled compared to what is attainable in the H-cell under similar working potentials. In addition, an impressive C3 partial current density of 150 mA cm−2 is measured on Bi1Pd NS at 0.77 V. Fig. 2f summarizes and compares the C3 FEs and partial current densities of Bi1Pd NS and other state-of-the-art GOR electrocatalysts. Our sample stands out by demonstrating great C3 selectivity under 4–15 times larger current density, the combination of which has not been achieved in previous reports to our best knowledge.30–42
In addition to C3 selectivity, the deactivation of precious-metal-based catalysts is a daunting challenge for the electrochemical oxidation of alcohols including glycerol.43–45 Most Pd-based candidates are subjected to rapid activity declines within the first hour of electrolysis as a result of the surface poisoning by strongly adsorbed intermediates that block the catalyst surface.18,46,47 To assess the catalyst stability here, we conducted a chronoamperometric test of Bi1Pd NS at 0.71 V in the flow cell electrolyzer by continuously circulating 400 mL of 4 M KOH solution with 2 M glycerol as the anolyte. Starting at 110 mA cm−2, its anodic current density gradually decays over time before leveling off at 70 mA cm−2 after 10 h (Fig. S9a†). Product analysis indicates that C3 FE gradually lowers from 89% to 58% as the reaction proceeds owing to product over-oxidation (Fig. S9b†). Even though the stability is substantially improved compared to that of pristine Pd (Fig. S9c–f†), the activity loss reflects glycerol consumption and more importantly inevitable surface poisoning during the stability test.14,48,49 Interestingly, we find that the poisoning can be reversed. By periodically interrupting the reaction and refreshing the electrolyte every 5 h, the anodic current density of Bi1Pd NS can be recovered to close to its initial value, indicating no permanent degradation of our electrocatalyst (Fig. 2g). The average C3 FE within each reaction cycle is measured to be >60%.
2.3 Computational simulations of the GOR on Bi1Pd
To build deep understanding about the improved performance of Bi1Pd and reveal its atomic origin, density functional theory (DFT) calculations were employed to simulate the GOR to C3 products. On the Bi1Pd surface, the adsorption of glycerol molecules preferentially occurs at the Pd sites adjacent to Bi atoms, which are identified as the active sites for the GOR (Fig. S10†). The adsorption energy is calculated to be −1.23 eV, slightly higher than that on pristine Pd (111) (−1.21 eV). To ensure selective C3 production over C1–C2 production, an ideal catalyst should favor the rapid desorption of C3 products once formed, and suppress further C–C cleavage and over-oxidation. With this in mind, we consequently evaluated the adsorption energies of DHA, GLA and TA on Bi1Pd (Fig. 3a). Compared to pristine Pd, significantly weaker surface affinities are observed for all of them upon the introduction of Bi atoms in their proximity. For example, the adsorption energy of GLA is −1.12 eV on pristine Pd, whereas it reduces to −0.82 eV on Bi1Pd. Analysis of its optimized adsorption configuration indicates a relatively longer adsorption distance of 3.35 Å on Bi1Pd than that on Pd (3.31 Å).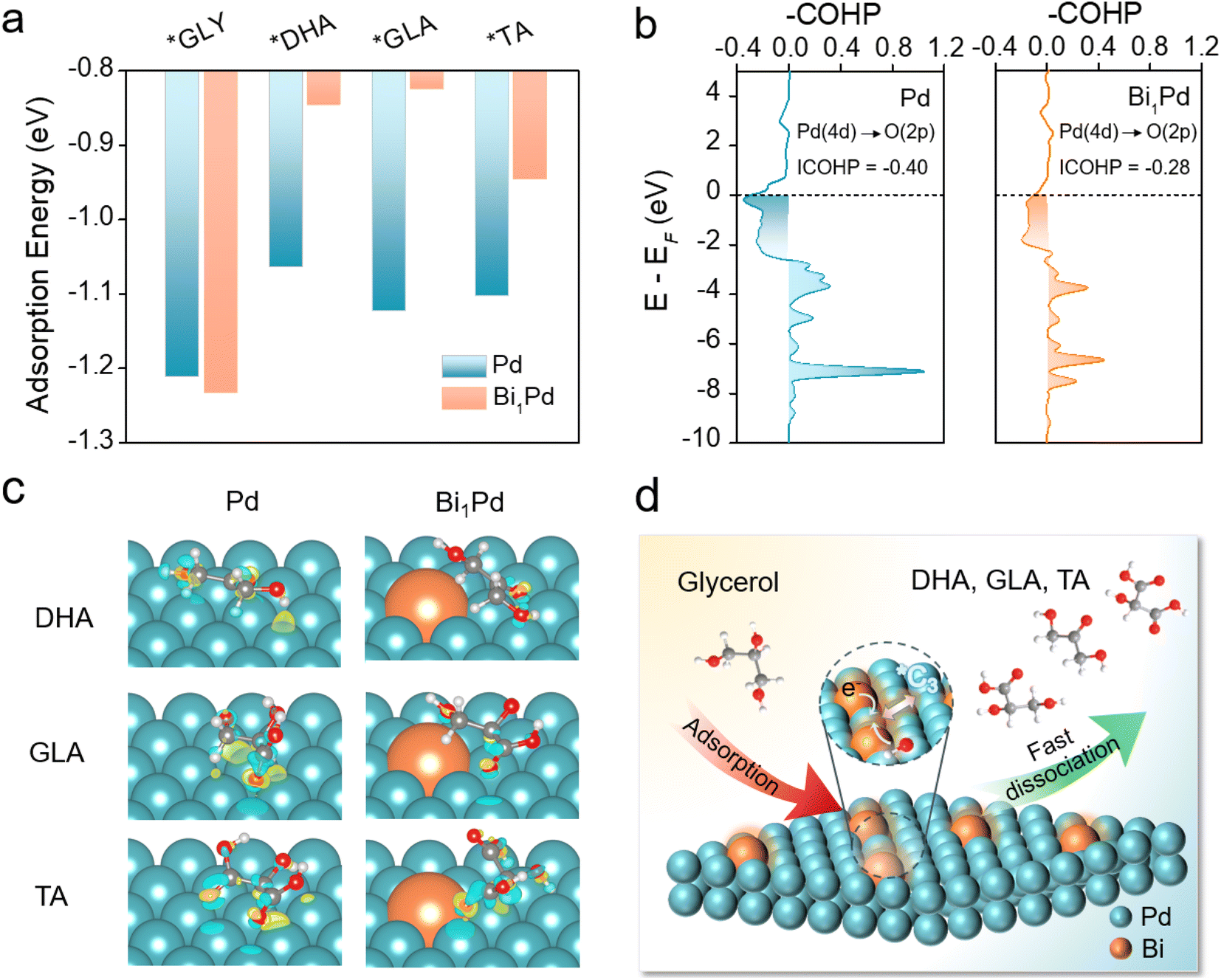 | ||
| Fig. 3 Computational simulations of the GOR on Bi1Pd and pristine Pd. (a) Adsorption energies of glycerol, DHA, GLA and TA on Bi1Pd or Pd. (b) Integrated COHP values between the O atom of adsorbed GLA and the Pd atom in Bi1Pd or pristine Pd. (c) Optimized geometric structures of C3 adsorbates on Bi1Pd or Pd, where yellow, cyan, grey, red and light-grey spheres represent Bi, Pd, C, O, and H atoms, respectively. (d) Schematic illustration showing the selective GOR to C3 products on Bi1Pd. | ||
We further conducted projected crystal orbital Hamilton population (COHP) analysis as illustrated in Fig. 3b. The integrated COHP value between the Pd atom and the O atom of adsorbed GLA is calculated to be −0.40 on pristine Pd and −0.28 on the Bi1Pd surface, evidencing the increasing occupation of the antibonding orbital of the Pd–O bond under the latter scenario. To corroborate this finding, charge density difference and Bader charge analysis are also presented in Fig. 3c. Compared to pristine Pd, Bi1Pd exhibits less charge accumulation between C3 products and the catalyst surface. The average charge per atom on the top layer of pristine Pd is determined to be −0.03|e|. On the Bi1Pd surface, Bi donates additional electrons to surrounding Pd, resulting in a more negative average charge on Pd (−0.09|e|). Consequently, relatively stronger electrostatic repulsion occurs between Bi1Pd and the adsorbates. These theoretical results strongly support that dispersed Bi atoms facilitate the desorption of GLA and other C3 products, thereby increasing the C3 selectivity and alleviating their over-oxidation (Fig. 3d). The decreased surface adsorption energies of intermediates are also responsible for the improved catalyst stability observed experimentally. Based on results from earlier literature and our theoretical study here, a possible reaction pathway for the GOR on Bi1Pd is proposed in Fig. S11.†
2.4 Coupling the GOR and CO2RR in a full cell
Lastly, we show that Bi1Pd-catalyzed GOR can be paired with cathodic reactions in a full cell. Earlier studies suggested the GOR as a promising substitute of the oxygen evolution reaction (OER) to couple with the CO2RR.50–52 This would not only lower the working voltage and hence improve the energy efficiency, but also increase the overall economic viability by valorizing low-cost feedstocks at both electrodes. The effective coupling between the GOR and CO2RR requires advanced anode and cathode catalysts able to deliver comparable current density (100–1000 mA cm−2) and high selectivity. The great electrocatalytic performance of our Bi1Pd reflects its unique potential for coupling with the CO2RR. To achieve this, we paired a Bi1Pd-loaded CFP as the anode with a Cu nanoparticle-sprayed gas-diffusion electrode as the cathode in a flow cell (Fig. 4a). Cu is well known for its capability to electrochemically reduce CO2 to C2+ products including ethylene, ethanol, acetate and propanol with appreciable FEs.53–55Fig. 4b illustrates the respective polarization curves of Cu-catalyzed CO2RR, Bi1Pd-catalyzed GOR and IrO2-catalyzed OER that are independently collected in the three-electrode configuration. It is worth highlighting that compared to the OER, the polarization curve of the GOR is negatively displaced by more than 780 mV at 100 mA cm−2. When integrated together in the two-electrode configuration, the GOR–CO2RR couple sustains a current density of 100 mA cm−2 at 1.75 V (free of iR compensation), about 710 mV lower than that of the OER–CO2RR couple (Fig. S12†), translating to a significant enhancement in energy efficiency. Furthermore, the products from both electrodes were analyzed at a few selected working voltages. At the anode side, the total C3 (DHA, GLA and TA) FE is measured to slightly decrease from 80% at 1.8 V to 66% at 2.3 V; at the cathode side, the total C2+ (ethylene, ethanol, acetate and propanol) FE correspondingly increases from 67% to 76%, in line with the typical performances of Cu-based electrocatalysts (Fig. 4c).53–55 Such paired co-electrolysis enables the simultaneous valorization of glycerol at the anode and CO2 at the cathode, and dramatically raises the economic viability of the whole system. Bi1Pd-catalyzed GOR can also be potentially coupled with other cathodic processes such as hydrogen evolution or nitrate/nitrite reduction. | ||
| Fig. 4 Coupled GOR–CO2RR in a full flow cell. (a) Schematic illustration of the full flow cell by coupling the Bi1Pd NS anode with a Cu-PTFE cathode in a two-electrode configuration. (b) Polarization curves of Bi1Pd NS-catalyzed GOR and OER and Cu-catalyzed CO2RR in a three-electrode configuration. (c) FEs of C3 products at the anode and C2+ products at the cathode at the indicated working voltage of the full flow cell. | ||
3 Conclusions
In summary, we reported that Bi atoms dispersed on Pd nanosheets promoted electrochemical GOR to C3 products. The catalyst material was prepared via a two-step solvothermal method, in which polygonal Pd nanosheets were first formed in the presence of CO and then decorated with dilute Bi atoms by reducing BiCl3 in ethylene glycol. Spectroscopic and microscopic characterizations corroborated the single dispersion of Bi atoms in the final product. Importantly, the introduction of Bi atoms was believed to modulate the electronic structure of Pd and lower the adsorption energy of key C3 intermediates, thereby alleviating product over-oxidation and catalyst poisoning. The electrocatalytic GOR performance of Bi1Pd NS was assessed in both an H-cell and flow cell. Under the optimal experimental conditions, the catalyst exhibited an outstanding C3 selectivity (>90%) and partial current density (>150 mA cm−2) in alkaline solution, the combination of which had been rarely attained in previous reports and far exceeded other precious-metal-based competitors. The surface decoration with Bi atoms also significantly retarded catalyst poisoning, and stability over 30 h was achieved by periodically refreshing the electrolyte. More interestingly, Bi1Pd-catalyzed GOR could substitute the OER to couple with Cu-catalyzed CO2RR, resulting in a substantially lower working voltage (by >780 mV at 100 mA cm−2) and improved energy efficiency. Our work here presents a new solution to valorize glycerol with improved C3 selectivity and stability, and might accelerate the technical maturity of room-temperature electrochemical GOR.Data availability
Data supporting the findings of this study are available within the article and its ESI,† or from the corresponding authors upon reasonable request.Author contributions
Y. G. L. conceived the project and designed the experiments, N. H. supervised the project, Z. H. M and L. J. synthesized the catalysts and conducted the structure analysis and electrocatalytic studies, L. W. and X. N. M. performed the DFT calculations, X. D and B. B. P. conducted the flow cell measurements, L. Z. and T. R. Y. carried out the X-ray absorption measurements and analysis, and J. X. performed TEM characterization. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (52161160331, 22279084, 52202373 and 22309126), the Science and Technology Development Fund Macau SAR (0077/2021/A2), the Natural Science Foundation of Jiangsu Province of China (BK20220027 and BK20210729), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (20KJA430002), the Postdoctoral Fellowship Program of CPSF (GZC20231878), Jiangsu Funding Program for Excellent Postdoctoral Talent (2022ZB543), and the Collaborative Innovation Center of Suzhou Nano Science and Technology. We are also thankful for the beamline support from SSRF (13SSW) for the XAS experiments.References
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC
.
- C. A. G. Quispe, C. J. R. Coronado and J. A. Carvalho Jr, Renewable Sustainable Energy Rev., 2013, 27, 475–493 CrossRef CAS
- G. Dodekatos, S. Schünemann and H. Tüysüz, ACS Catal., 2018, 8, 6301–6333 CrossRef CAS
- S. Bagheri, N. M. Julkapli and W. A. Yehye, Renewable Sustainable Energy Rev., 2015, 41, 113–127 CrossRef CAS
- M. Anitha, S. K. Kamarudin and N. T. Kofli, Chem. Eng. J., 2016, 295, 119–130 CrossRef CAS
- B. Katryniok, H. Kimura, E. Skrzynska, J. S. Girardon, P. Fongarland, M. Capron, R. Ducoulombier, N. Mimura, S. Paul and F. Dumeignil, Green Chem., 2011, 13, 1960–1979 RSC
- M. Guschakowski and U. Schröder, ChemSusChem, 2021, 14, 5216–5225 CrossRef CAS PubMed
- L. Fan, B. Liu, X. Liu, N. Senthilkumar, G. Wang and Z. Wen, Energy Technol., 2021, 9, 2000804 CrossRef CAS
- C. Coutanceau, S. Baranton and R. S. B. Kouamé, Front. Chem., 2019, 7, 100 CrossRef CAS PubMed
- M. Simões, S. Baranton and C. Coutanceau, ChemSusChem, 2012, 5, 2106–2124 CrossRef
- J. X. Wu, X. J. Yang and M. Gong, Chin. J. Catal., 2022, 43, 2966–2986 CrossRef CAS
- M. K. Goetz, M. T. Bender and K. S. Choi, Nat. Commun., 2022, 13, 5848–5858 CrossRef CAS PubMed
- M. S. Houache, K. Hughes and E. A. Baranova, Sustainable Energy Fuels, 2019, 3, 1892–1915 RSC
- T. Y. Li and D. A. Harrington, ChemSusChem, 2021, 14, 1472–1495 CrossRef CAS
- C. Dai, L. Sun, H. Liao, B. Khezri, R. D. Webster, A. C. Fisher and Z. J. Xu, J. Catal., 2017, 356, 14–21 CrossRef CAS
- Y. Liu, W. Yu, D. Raciti, D. H. Gracias and C. Wang, J. Phys. Chem. C, 2018, 123, 426–432 CrossRef
- H. B. Wang, L. Thia, N. Li, X. M. Ge, Z. L. Liu and X. Wang, ACS Catal., 2015, 5, 3174–3180 CrossRef CAS
- S. P. Li, J. P. Lai, R. Luque and G. B. Xu, Energy Environ. Sci., 2016, 9, 3097–3102 RSC
- H. J. Kim, S. M. Choi, S. Green, G. A. Tompsett, S. H. Lee, G. W. Huber and W. B. Kim, Appl. Catal., B, 2011, 101, 366–375 CrossRef CAS
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC
- X. H. Wu, Y. Wang and Z. S. Wu, Chem, 2022, 8, 2594–2629 CAS
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. B. Chorkendorff, J. K. Norskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed
- A. Zalineeva, A. Serov, M. Padilla, U. Martinez, K. Artyushkova, S. Baranton, C. Coutanceau and P. B. Atanassov, J. Am. Chem. Soc., 2014, 136, 3937–3945 CrossRef CAS PubMed
- S. Feng, J. Yi, H. Miura, N. Nakatani, M. Hada and T. Shishido, ACS Catal., 2020, 10, 6071–6083 CrossRef CAS
- W. J. Huang, X. L. Kang, C. Xu, J. H. Zhou, J. Deng, Y. G. Li and S. Cheng, Adv. Mater., 2018, 30, 1706962 CrossRef PubMed
- W. J. Huang, X. Y. Ma, H. Wang, R. F. Feng, J. G. Zhou, P. N. Duchesne, P. Zhang, F. J. Chen, N. Han, F. P. Zhao, J. H. Zhou, W. B. Cai and Y. G. Li, Adv. Mater., 2017, 29, 1703057 CrossRef PubMed
- X. Huang, S. Tang, X. Mu, Y. Dai, G. Chen, Z. Zhou, F. Ruan, Z. Yang and N. Zheng, Nat. Nanotechnol., 2011, 6, 28–32 CrossRef CAS PubMed
- M. Luo, Z. Zhao, Y. Zhang, Y. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. Qin, J. Y. Ma, F. Lin, D. Su, G. Lu and S. Guo, Nature, 2019, 574, 81–85 CrossRef CAS PubMed
- M. Yang, Z. R. Yuan, R. X. Peng, S. Y. Wang and Y. Q. Zou, Energy Environ. Sci., 2022, 5, 1117–1138 CAS
- L. Huang, J.-Y. Sun, S.-H. Cao, M. Zhan, Z.-R. Ni, H.-J. Sun, Z. Chen, Z.-Y. Zhou, E. G. Sorte, Y. J. Tong and S.-G. Sun, ACS Catal., 2016, 6, 7686–7695 CrossRef CAS
- S. Lee, H. J. Kim, E. J. Lim, Y. Kim, Y. Noh, G. W. Huber and W. B. Kim, Green Chem., 2016, 18, 2877–2887 RSC
- Y. Zhou, Y. Shen and J. Xi, Appl. Catal., B, 2019, 245, 604–612 CrossRef CAS
- Y. Kim, H. W. Kim, S. Lee, J. Han, D. Lee, J. R. Kim, T. W. Kim, C. U. Kim, S. Y. Jeong, H. J. Chae, B. S. Kim, H. Chang, W. B. Kim, S. M. Choi and H. J. Kim, ChemCatChem, 2017, 9, 1683–1690 CrossRef CAS
- Y. Zhou, Y. Shen, J. Xi and X. Luo, ACS Appl. Mater. Interfaces, 2019, 11, 28953–28959 CrossRef CAS PubMed
- X. Yu, R. B. Araujo, Z. Qiu, E. Campos dos Santos, A. Anil, A. Cornell, L. G. M. Pettersson and M. Johnsson, Adv. Energy Mater., 2022, 12, 2103750 CrossRef CAS
- J. González-Cobos, S. Baranton and C. Coutanceau, ChemElectroChem, 2016, 3, 1694–1704 CrossRef
- Y. Zhou, Y. Shen and J. Piao, ChemElectroChem, 2018, 5, 1636–1643 CrossRef CAS
- J. Li, Z. Li, Z. Zheng, X. Zhang, H. Zhang, H. Wei and H. Chu, ChemCatChem, 2022, 14, e202200509 CrossRef CAS
- T. G. Vo, G. S. Tran, C. L. Chiang, Y. G. Lin, H. E. Chang, H. H. Kuo, C. Y. Chiang and Y. J. Hsu, Adv. Funct. Mater., 2022, 33, 2209386 CrossRef
- H. Wan, C. Dai, L. Jin, S. Luo, F. Meng, G. Chen, Y. Duan, C. Liu, Q. Xu, J. Lu and Z. J. Xu, ACS Appl. Mater. Interfaces, 2022, 14, 14293–14301 CrossRef CAS
- L. Thia, M. S. Xie, Z. L. Liu, X. M. Ge, Y. Z. Lu, W. E. Fong and X. Wang, ChemCatChem, 2016, 8, 3272–3278 CrossRef CAS
- Z. X. Chen, C. B. Liu, X. X. Zhao, H. Yan, J. Li, P. Lyu, Y. H. Du, S. B. Xi, K. Chi, X. Chi, H. S. Xu, X. Li, W. Fu, K. Leng, S. J. Pennycook, S. Wang and K. P. Loh, Adv. Mater., 2019, 31, 1804763 CrossRef PubMed
- W. J. Huang, H. T. Wang, J. G. Zhou, J. Wang, P. N. Duchesne, D. Muir, P. Zhang, N. Han, F. P. Zhao, M. Zeng, J. Zhong, C. H. Jin, Y. G. Li, S. T. Lee and H. J. Dai, Nat. Commun., 2015, 6, 10035–10043 CrossRef CAS PubMed
- J. S. Spendelow and A. Wieckowski, Phys. Chem. Chem. Phys., 2007, 9, 2654–2675 RSC
- E. Antolini, Catalysts, 2019, 9, 980 CrossRef CAS
- X. Mo, X. Gao, A. V. Gillado, H. Y. Chen, Y. Chen, Z. Guo, H. L. Wu and E. C. M. Tse, ACS Nano, 2022, 16, 12202–12213 CrossRef CAS PubMed
- F. Yang, J. Y. Ye, Q. Yuan, X. T. Yang, Z. X. Xie, F. L. Zhao, Z. Y. Zhou, L. Gu and X. Wang, Adv. Funct. Mater., 2020, 30, 1908235 CrossRef CAS
- M. Simoes, S. Baranton and C. Coutanceau, Appl. Catal., B, 2010, 93, 354–362 CrossRef CAS
- J. Qi, N. Benipal, C. H. Liang and W. Z. Li, Appl. Catal., B, 2016, 199, 494–503 CrossRef CAS
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS
- R. Li, K. Xiang, Z. Peng, Y. Zou and S. Wang, Adv. Energy Mater., 2021, 11, 2102292 CrossRef CAS
- Á. Vass, A. Kormányos, Z. Kószó, B. Endrődi and C. Janáky, ACS Catal., 2022, 12, 1037–1051 CrossRef PubMed
- J. Yu, J. Wang, Y. Ma, J. Zhou, Y. Wang, P. Lu, J. Yin, R. Ye, Z. Zhu and Z. Fan, Adv. Funct. Mater., 2021, 31, 2102151 CrossRef CAS
- Y. Luo, K. Zhang, Y. Li and Y. Wang, InfoMat, 2021, 3, 1313–1332 CrossRef CAS
- J. Zhang, X. Mao, B. Pan, J. Xu, X. Ding, N. Han, L. Wang, Y. Wang and Y. Li, Nano Res., 2023, 16, 4685–4690 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03892d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |