
Electrochemical hydrotrifluoromethylation of alkynes†
Jihoon Jang and
Eun Jin Cho *
*
Department of Chemistry, Chung-Ang University, 84, Heukseok-ro, Dongjak-gu, Seoul 06974, Republic of Korea. E-mail: ejcho@cau.ac.kr
First published on 1st July 2025
Abstract
We report the electrochemical hydrotrifluoromethylation of alkynes using the Langlois reagent as a CF3 source. The mild conditions enable broad functional group tolerance and allow late-stage functionalization. Mechanistic studies reveal that DMSO serves a dual role as both solvent and hydrogen atom donor.
The trifluoromethyl (CF3) group is regarded as one of the most privileged functional groups for modulating the physicochemical properties of bioactive molecules, including metabolic stability, lipophilicity, bioavailability, and binding selectivity.1 In particular, CF3-substituted alkenes have emerged as valuable motifs in medicinal chemistry2 and as versatile intermediates for further functionalization.3 Among various synthetic approaches, the hydrotrifluoromethylation of alkynes has gained attention as a highly atom-economical strategy for accessing CF3-alkenes.4
Recent advancements in environmentally friendly radical chemistry, particularly through photochemical5 and electrochemical6 methods, have enabled the sustainable generation of radical intermediates using photons or electric current as green energy sources. Photochemical protocols developed by Scaiano,7 Yang,8 and our own group9 have utilized electrophilic CF3 radical precursors, such as Togni's reagent and CF3I, in combination with photocatalysts to yield CF3-alkenes (Fig. 1a). Despite substantial progress in photochemical radical methodologies, the hydrotrifluoromethylation of alkynes had yet to be realized in the realm of electrochemistry.
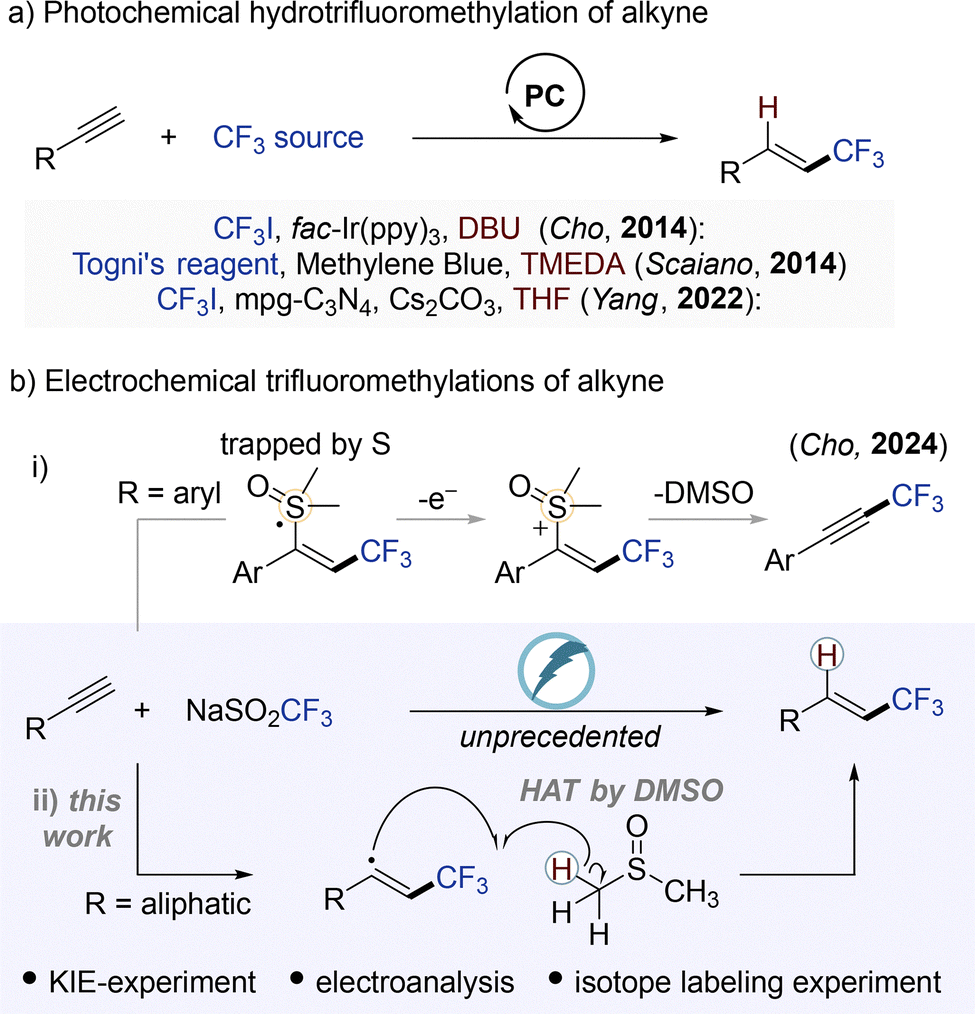 | ||
| Fig. 1 Synthesis of CF3-alkenes via radical processes. | ||
In this work, we report the first electrochemical hydrotrifluoromethylation of alkynes using a nucleophilic CF3 source, the Langlois reagent (NaSO2CF3), under mild and sustainable conditions (Fig. 1b). This transformation proceeds through a distinct mechanistic pathway that contrasts with our previous study on aromatic alkynes, where the vinyl radical intermediates underwent further electrochemical oxidation to furnish CF3-substituted alkynes (Fig. 1b-i).10 In contrast, we envisioned that aliphatic alkynes could engage in a selective hydrotrifluoromethylation pathway, wherein the resulting vinyl radical intermediate undergoes rapid hydrogen atom abstraction from DMSO, which acts as an effective hydrogen donor (Fig. 1b-ii). The proposed mechanism is supported by comprehensive mechanistic investigations, including kinetic isotope effect (KIE) measurements and isotopic labeling experiments.
The investigation began with but-3-yn-1-ylbenzene (1a) as a model substrate and NaSO2CF3 (2) as the CF3 radical source in DMSO (Table 1). The reaction was performed under constant-voltage electrolysis in an undivided cell, employing tetrabutylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte, graphite [C(+)] as the anode (working electrode), and nickel foam [Ni foam (−)] as the cathode (counter electrode).
|
||
|---|---|---|
| Entry | Variation | Yieldb (%) |
| a 0.2 mmol scale.b Yields were determined by 19F NMR spectroscopy using 2,2,2-trifluoroethanol as an internal standard. | ||
| 1 | None | 58 |
| 2 | Stainless steel(+) instead of C(+) | 0 |
| 3 | C(−) instead of Nifoam(−) | 54 |
| 4 | MeCN instead of DMSO | 4 |
| 5 | DMF instead of DMSO | 21 |
| 6 | DCE instead of DMSO | 0 |
| 7 | TBANO3 instead of TBAPF6 | 32 |
| 8 | No electricity | 0 |
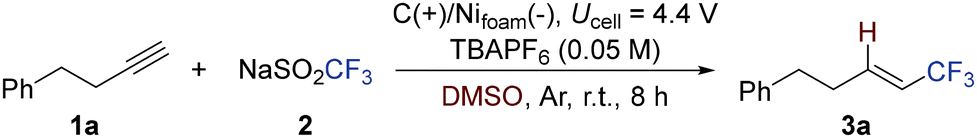
At a cell potential of 4.4 V, the desired product 3a was obtained in 58% yield (entry 1). Replacing the graphite working electrode with stainless steel completely suppressed the reaction (entry 2), while using graphite as both electrodes led to a slightly decreased yield (entry 3). Despite potential challenges, DMSO proved uniquely effective, likely due to its role as a hydrogen atom donor in this transformation. In contrast, alternative solvents such as MeCN, DMF, and DCE gave poor or no conversion (entries 4–6). When TBAPF6 was replaced with TBANO3, the reaction still proceeded but with reduced efficiency (entry 7). Notably, no product was formed in the absence of electrical input, confirming the essential role of electrochemical activation (entry 8).
Next, a series of mechanistic studies was conducted to validate the involvement of key intermediates and to elucidate the role of each reaction component—particularly DMSO as a hydrogen atom donor (Fig. 2). Initially, cyclic voltammetry experiments were carried out (Fig. 2a). Initially, cyclic voltammetry experiments were carried out (Fig. 2a). In MeCN with TBAPF6 as the supporting electrolyte, but-3-yn-1-ylbenzene (1a, black), NaSO2CF3 (2, red), and DMSO (blue) each exhibited irreversible oxidation peaks at 2.23 V, 0.97 V, and 1.98 V vs. Ag/AgCl, respectively. Notably, when 1a and 2 were combined (green), the oxidation peak of 1a at 2.23 V disappeared, and a new peak emerged at 2.34 V, which was attributed to the oxidation of the alkenyl–CF3 radical intermediate. Upon the addition of DMSO (light brown), this new peak vanished, and the oxidation peak of 1a reappeared, suggesting the involvement of DMSO in quenching the radical intermediate.
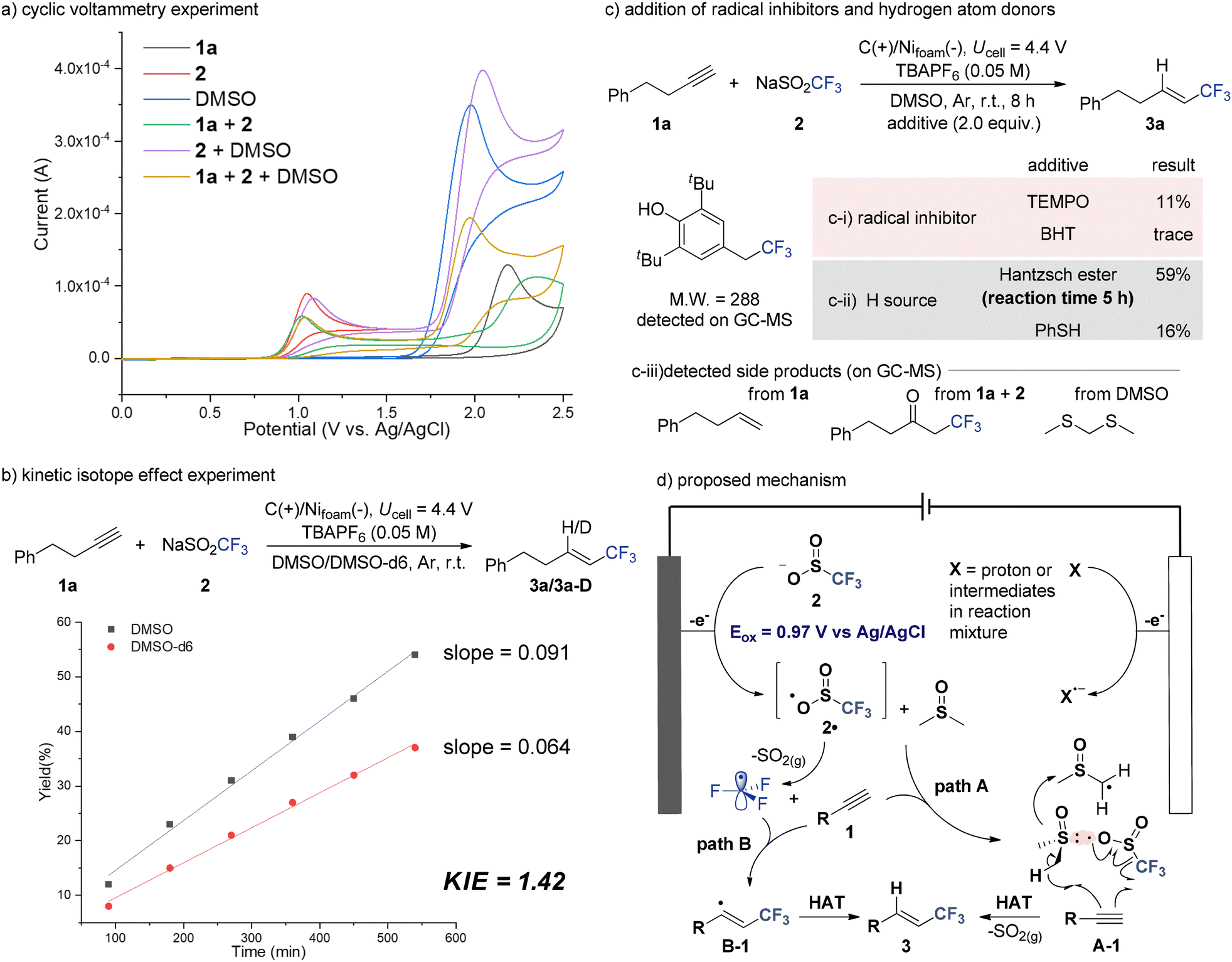 | ||
| Fig. 2 Mechanistic investigations and proposed mechanism. | ||
To further probe the role of DMSO, kinetic isotope effect (KIE) and isotopic labeling experiments were performed (Fig. 2b). The use of deuterated DMSO (DMSO-d6) resulted in decreased reactivity, yielding a KIE value of 1.42, which supports the involvement of DMSO in the reaction pathway. Furthermore, the formation of deuterium-labeled product (3a-D, 73%) was confirmed by GC-MS (Fig. S2 and S3, ESI†) and NMR analyses (Fig. S4, ESI†), clearly demonstrating DMSO's function as the main H source in the final HAT step.
In support of this conclusion, additional experiments were conducted (Fig. 2c). When a Hantzsch ester was added as an additional H-donor, the reaction rate increased, shortening the reaction time from 8 h to 5 h (Fig. 2c-i). In contrast, the use of thiophenol (PhSH) led to a decrease in efficiency, suggesting that competitive HAT pathway could negatively affect the reaction outcome. The radical nature of the reaction was confirmed by radical-trapping experiments (Fig. 2c-ii). The addition of radical quenchers such as butylated hydroxytoluene (BHT) and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) significantly suppressed product formation. In particular, GC-MS analysis detected a BHT–CF3 adduct (Fig. S5, ESI†), further supporting the involvement of CF3 radicals. Finally, GC-MS analysis of the crude reaction mixture revealed the formation of several side products, such as reduced alkenes11 and α-CF3 ketones12 (Fig. 2c-iii and Fig. S6, ESI†). Notably, bis(methylthio)methane (Fig. S6, ESI†)—a derivative of DMSO—was also identified, suggesting that DMSO may undergo further transformation after the HAT process.
Based on the above results, a plausible mechanism for the hydrotrifluoromethylation of alkynes is proposed (Fig. 2d). The reaction is initiated by the anodic oxidation of NaSO2CF3 (2), generating a trifluoromethyl sulfinyl radical intermediate (2˙). Subsequent extrusion of SO2 furnishes the CF3 radical, which adds to the alkyne substrate (1) to form a vinyl radical intermediate (B-1). A hydrogen-atom transfer from DMSO then delivers the desired product (3) (path A). Alternatively, path B may involve the initial coordination of the sulfinyl radical 2˙ with DMSO. Within this complex (A-1), the CF3 radical is generated and adds to the alkyne to form a vinyl radical, which subsequently undergoes an intra-complex HAT with nearby DMSO molecule to afford product 3.
To evaluate the utility of this transformation, the substrate scope of the hydrotrifluoromethylation was examined under the optimized conditions using a variety of alkynes (Table 2). Notably, the protocol tolerated a range of redox-sensitive functional groups, including phthalimide (3e), free alcohols (3f, 3l), malonate (3g), and esters (3h–3k). Protected alcohol-containing substrates furnished the desired CF3-alkenes without deprotection (3m, 3n).
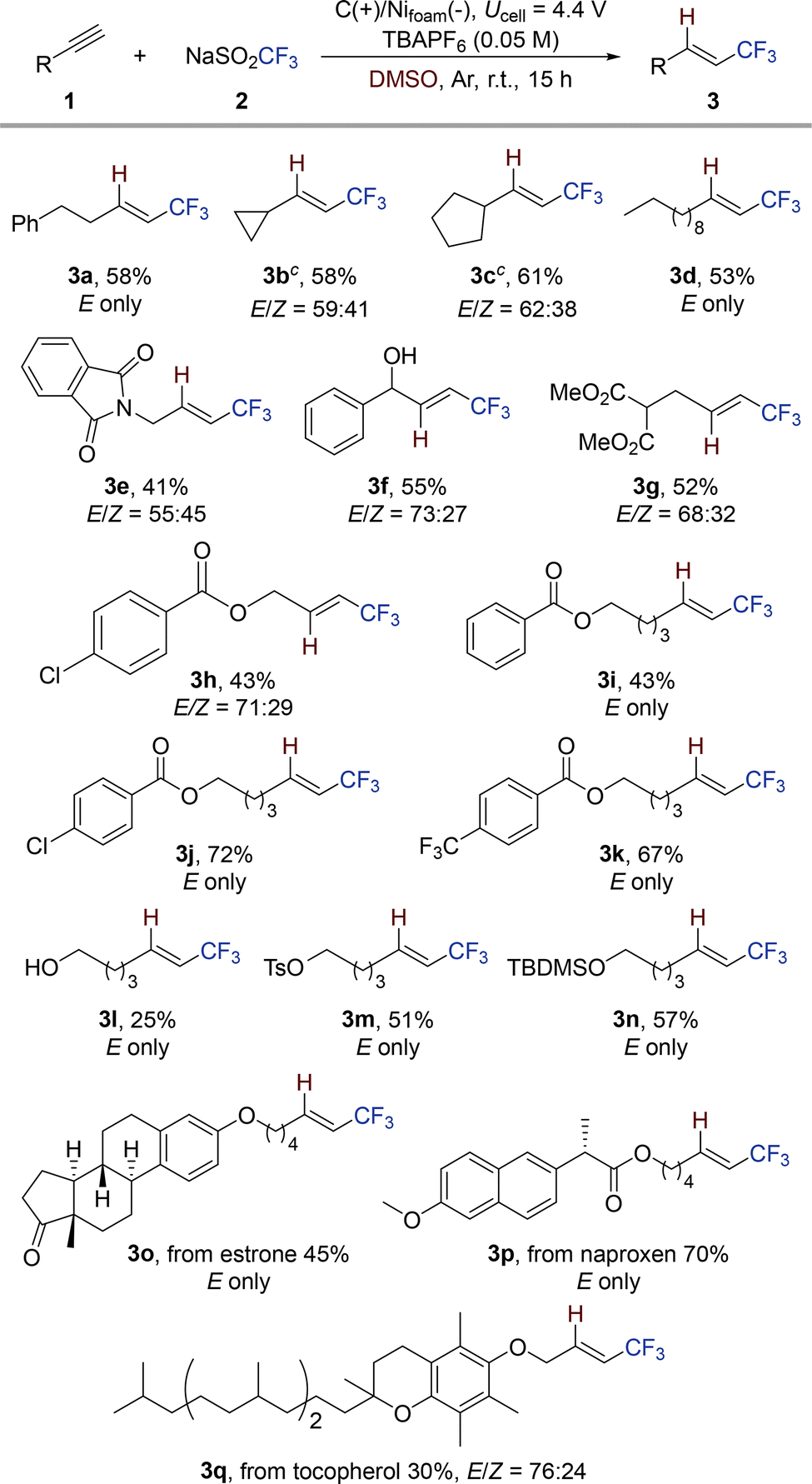
To further demonstrate the broad applicability of this methodology, the reaction was extended to the late-stage functionalization of complex bioactive molecules. Structurally complex derivatives based on estrone (3o) and the anti-inflammatory agent naproxen (3p), as well as the antioxidant natural product α-tocopherol (3q), underwent efficient hydrotrifluoromethylation under the standard conditions. The exclusive formation of the E-isomer was observed for substrates with extended aliphatic chains, suggesting that chain length plays a significant role in governing stereoselectivity. This observation is consistent with previously reported chain length-dependent stereocontrol in a related system.13 As mentioned earlier (Fig. 2c), side products such as reduced alkenes (lacking CF3) and α-CF3 ketones were observed. In the case of a strained cyclopropyl-containing alkyne (3b), formation of a minor ring-opened byproduct (∼5%) was observed, further supporting the radical-mediated nature of this process. However, internal alkynes were not suitable substrates for the transformation. This limitation is likely due to the steric hindrance encountered by the DMSO-stabilized SO2CF3 radical intermediate, which impedes efficient radical addition to internal alkynes.
In conclusion, we have developed the first electrochemical hydrotrifluoromethylation of alkynes using the Langlois reagent as the CF3 source. The mild reaction conditions tolerated a variety of functional groups and demonstrated applicability in the late-stage functionalization of complex molecules. Mechanistic studies revealed that DMSO plays a dual role as both solvent and hydrogen atom donor. This work highlights the potential of electrochemical strategies for accessing valuable CF3-functionalized alkenes with high efficiency and selectivity.
We gratefully acknowledge the National Research Foundation of Korea (RS-2025-00559692 and RS-2024-00409659) and the Ministry of Trade, Industry and Energy, Korea (Technology Innovation Program, RS-2023-00266039).
Conflicts of interest
There are no conflicts to declare.Data availability
The data underlying this study are available in the published article and its ESI.†Notes and references
-
(a) A. Abula, Z. Xu, Z. Zhu, C. Peng, Z. Chen, W. Zhuan and H. A. Aisa, J. Chem. Inf. Model., 2020, 60, 6242–6250 CrossRef CAS PubMed
; (b) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed
-
(a) G. Németh, R. Kapiller-Dezsöfi, G. Lax and G. Simig, Tetrahedron, 1996, 52, 12821–12830 CrossRef
-
(a) C. Pritanka, R. Adepu and N. Punna, Eur. J. Org. Chem., 2025, e202401168 CrossRef
-
(a) A. A. Kazi, N. Manjuladevi, S. S. Kumar, A. Sharma and L. R. Singh, Chem. Commun., 2024, 60, 9376–9379 RSC
-
(a) R. Shaw, N. Sihag, H. Bhartiya and M. R. Yadav, Org. Chem. Front., 2024, 11, 954–1014 RSC
-
(a) S. Kim and H. Kim, ACS Catal., 2025, 15, 6826–6851 CrossRef CAS
- S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, ACS Catal., 2014, 4, 2530–2535 CrossRef CAS
- X. Shi, T. Song, Q. Li, X. Guo and Y. Yang, Org. Lett., 2022, 24, 8724–8728 CrossRef CAS PubMed
- N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., Int. Ed., 2014, 53, 539–542 CrossRef CAS PubMed
- J. Jang, H. S. Hwang, H. Jeong and E. J. Cho, Chem. Sci., 2024, 15, 19739–19744 RSC
-
(a) Y. Zhang, X. Zhao and G. Qing, Chem. Synth., 2024, 4, 16 CAS
- W. Jud, C. O. Kappe and D. Cantillo, Org. Biomol. Chem., 2019, 17, 3529–3537 RSC
- M. K. Amstrong, M. B. Goodstein and G. Lalic, J. Am. Chem. Soc., 2018, 140, 10233–10241 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc02370j |
| This journal is © The Royal Society of Chemistry 2025 |