
Engineering twin Cu0.3Zn0.7S for CO2 photocatalytic reduction to ethylene†
Hang Zhaoab,
Hao Songab,
Zhefei Panab,
Xun Zhu ab,
Dingding Yeab,
Yang Yangab,
Hong Wangab,
Qiang Liaoab and
Rong Chen*ab
ab,
Dingding Yeab,
Yang Yangab,
Hong Wangab,
Qiang Liaoab and
Rong Chen*ab
aKey Laboratory of Low-grade Energy Utilization Technologies and Systems (Chongqing University), Ministry of Education, Chongqing 400030, China. E-mail: rchen@cqu.edu.cn
bInstitute of Engineering Thermophysics, School of Energy and Power Engineering, Chongqing University, Chongqing 400030, China
First published on 8th July 2025
Abstract
A twin Cu0.3Zn0.7S solid solution is developed for carbon dioxide photocatalytic reduction, in which ordered stacking faults induce homojunctions to enhance the separation and migration of photogenerated charge carriers. The high-coverage of the *CO intermediate is responsible for promoting C–C coupling. The developed photocatalyst exhibits efficient CO2-to-C2H4 conversion with a C2H4 production rate of 8.45 μmol g−1 h−1 and an electron selectivity of 53.6%.
Ethylene (C2H4), as a high-value multicarbon product widely used in the production of plastics and cosmetics, is highly prized for its energy density and commercial importance.1 However, conventional synthesis primarily relies on steam cracking of oil or shale gas at elevated temperatures (800–900 °C), which is energy-intensive and environmentally detrimental.2 Carbon dioxide photocatalytic reduction (CO2PR), mimicking natural photosynthesis, has been recognized as a promising environmentally-friendly strategy as it can reduce CO2 into fuels or industrial feedstocks utilizing solar energy under mild conditions.3–5 Despite significant progress in CO2PR, the challenges of poor activity and selectivity remain. In particular, the photocatalytic conversion of CO2 to multicarbon products is limited by slow C–C bond formation kinetics and inefficient multielectron transfer.3 Previous studies have revealed that efficient C–C coupling in CO2PR necessitates the following prerequisites: (i) precise control of multi-electron and proton transfer processes to favor the generation of C2 products;6 (ii) optimal stabilization of key intermediates (e.g. *CO and *CHO) to ensure sufficient surface residence time while preventing catalyst poisoning or premature desorption.7,8
Copper (Cu)-based catalysts have emerged as distinctive candidates for driving CO2 reduction over typical two-electron products (CO and formate) to enable the generation of high-order hydrocarbons in photocatalytic systems.4 The key reason lies in that the Cu active sites can stably adsorb crucial C1 intermediates.4 Additionally, when Cu forms bimetallic sites with other metals, the electron density on the Cu sites can be modulated, creating a locally asymmetric electronic structure. This facilitates the formation of the transition state for efficient C–C coupling.7,9–11 Hence, the design of Cu-based bimetallic photocatalysts is highly viable to advance CO2PR.
In addition, CO2PR also faces a grand challenge of the recombination of photogenerated charge-carriers due to slow charge carrier kinetics, limiting photocatalytic efficiency. A potential strategy to enhance charge transport is the construction of homojunctions via coherent twin boundaries, where mirror-symmetric atomic arrangements introduce planar defects that facilitate charge separation by generating internal electric fields.12–14 Zinc sulfide (ZnS), a typical semiconductor photocatalyst, readily forms such a twin structure due to its cubic close-packed crystal structure and intrinsically low twin boundary energy.14 The incorporation of heteroatoms can introduce internal strain and local lattice distortions, thereby promoting the formation of twin boundaries within the crystals, such as CdZnS15 and MnZnS.16 This distinctive surface structure effectively facilitates the separation of photogenerated carriers, demonstrating unique advantages in photocatalytic applications. As mentioned above, when the bimetallic sites are formed between Cu and other metals, the C–C coupling can be enhanced. However, the CuZnS with a twin structure has not yet been reported for CO2PR. Herein, we developed a twin Cu0.3Zn0.7S by introducing Cu into the ZnS lattice. This newly developed photocatalyst not only enhances surface charge carrier kinetics but also optimizes the adsorption of key intermediates (*CO), steering the reaction pathway toward CO2-to-C2H4 conversion. Benefiting from the structural and electronic modulations, the twin Cu0.3Zn0.7S achieves an impressive ethylene selectivity of 53.6% with a production rate of 8.45 μmol g−1 h−1 feeding with gaseous CO2 and H2O, without any additives or sacrificial agents.
The actual molar ratios of Cu and Zn were determined by ICP-OES, which closely matched the nominal values (Table S1, ESI†). The XRD patterns of CuS and ZnS are in agreement with JCPDS card No. 24-0060 and No. 03-1093, respectively (Fig. 1a). The multiple phases in CuxZn1−xS (x = 0.2, 0.3 and 0.4) came out, attributed to the introduction of Cu into the ZnS lattice. As the Cu content increased, the intensities of the multiphase signals were strengthened. The diffraction peaks exhibited a positive shift to higher 2θ value relative to pristine ZnS, while a negative shift to lower 2θ value was observed as compared to pristine CuS (Fig. S1, ESI†), demonstrating the successful formation of CuxZn1−xS solid solutions.13,15 Similar phenomena have also been reported for metal sulfides with twin crystal structures.13,15,16 The SEM and TEM images shown in Fig. S2 and S3 (ESI†) indicate that pristine ZnS and CuS exhibit typical nanoparticulate morphologies with average sizes of ∼5 nm and ∼52 nm, respectively. In contrast, the Cu0.3Zn0.7S solid solution displays an irregular nanostructure with a more homogeneous spatial distribution and pronounced interparticle voids (Fig. S4a, ESI†), which facilitates surface photochemical reactions by enhancing mass transport and light-matter interactions.17 The EDS mapping (Fig. S4b, ESI†) of Cu0.3Zn0.7S indicates a homogeneous distribution of Cu, Zn, and S elements, confirming the formation of the CuxZn1−xS solid solution.15 The TEM image of Cu0.3Zn0.7S reveals distinct striped features (alternating black and white bands) on the surface (Fig. 1b), which are not observed in pristine CuS or ZnS and can be attributed to the twin crystal structure.15,18 The HRTEM image (Fig. 1c) and the corresponding inverse Fourier transform (IFFT) pattern (Fig. 1d) reveal abundant, well-ordered zigzag structures with mirror symmetry, indicative of twin boundaries.15 The stacking faults are primarily originated from the dislocations of the (020) crystal planes of the cubic zinc-blende (ZB) structure, with an interplanar spacing of approximately 0.28 nm. Additionally, the lattice fringes with a lattice distance of 0.32 nm can be indexed to the (111) plane of sphalerite ZnS. Notably, a twin angle of 56.5° is observed between the (111) plane and the twin boundary, consistent with the characteristic geometry of the twin domains (Fig. 1d).12,13,18 Furthermore, the corresponding fast Fourier transform (FFT) pattern (Fig. 1e) displays two sets of symmetrically arranged diffraction spots, with additional reflections located at one-third positions between the main spots, verifying the presence of a well-ordered nano twin structure.12,13,19
 | ||
| Fig. 1 CuxZn1−xS structural characterizations. (a) XRD patterns of CuS, ZnS, and CuxZn1−xS. (b) TEM image, (c) HRTEM image, (d) inverse Fourier transform (IFFT) pattern of the selected area, and (e) FFT pattern of Cu0.3Zn0.7S. | ||
The XPS survey spectra calibrated with C 1s (284.8 eV) indicate that Cu0.3Zn0.7S contains Cu, Zn and S elements, as shown in Fig. S5 (ESI†). For the high resolution S 2p spectrum of Cu0.3Zn0.7S, two peaks located at 161.65 and 162.95 eV can be assigned to S 2p3/2 and S 2p1/2 of sulfide.20 Compared to ZnS, the peaks exhibit a noticeable shift to higher binding energies, suggesting the formation of Cu–S bonds in Cu0.3Zn0.7S. Meanwhile, the Zn 2p binding energies in Cu0.3Zn0.7S (1021.62 and 1044.66 eV) exhibit a slight negative shift compared to pristine ZnS (1021.76 and 1044.81 eV). In contrast, the Cu 2p peaks display an opposite trend, shifting positively from 931.79 and 951.65 eV for CuS to 932.19 and 952.06 eV for Cu0.3Zn0.7S. This contrasting shift behaviour suggests a redistribution of electron density upon the introduction of Cu into the ZnS lattice, wherein electrons tend to transfer from Cu to Zn.11,13 Consequently, the formation of a Cu0.3Zn0.7S solid solution alters the local electronic structure. It should be noted that the satellite peaks typically associated with Cu2+ are significantly suppressed in this type of material, which can be primarily attributed to the strong covalency of the Cu–S bond and the delocalized electronic structure.21,22
The CO2PR performance of the as-synthesized photocatalysts was investigated in a sealed gas–solid reaction system fed with gaseous CO2 and H2O, without any co-catalysts or sacrificial agents.23 As shown in Fig. 2a, pristine ZnS and CuS exhibit a CO production rate of 7.69 and 13.58 μmol g−1 h−1, respectively. CuS also produces trace amounts of CH4 (0.14 μmol g−1 h−1) and C2H4 (0.18 μmol g−1 h−1), owing to the unique electronic configuration of the Cu active sites that partially modulate the reaction pathway.4 The corresponding product and electron selectivity of C2H4 by CuS are 1.30% and 7.71%, respectively. Impressively, the overall photocatalytic performance of CuxZn1−xS is significantly enhanced, especially for C2H4. The optimized Cu0.3Zn0.7S achieves the CO, CH4 and C2H4 production rates of 37.60, 1.57 and 8.45 μmol g−1 h−1, respectively. The product and electron selectivity of C2H4 increase to 17.7% and 53.6% (Fig. 2b), respectively. It is worth emphasizing that the C2H4 selectivity steadily increases with the Cu content, highlighting the critical role of Cu in steering product selectivity toward a multi-carbon product. Specifically, the C2H4 production rate of Cu0.3Zn0.7S is 46 times higher than that of CuS and among the top in the reported data (Table S2, ESI†). The apparent quantum yield (AQY) for Cu0.3Zn0.7S reaches 1.16% at 365 nm and decreases drastically with increasing the wavelength, consistent with the optical absorption profile. This decline is attributed to the limited light-harvesting capability in the long-wavelength region of the solar spectrum.24 No products were detected in the dark or when any of the key components (photocatalyst, CO2, or H2O) were absent (Fig. S6, ESI†), confirming that the products originate solely from CO2PR. The 13CO2 isotope-labelling experiments (Fig. S7, ESI†) also confirm that C2H4, CH4 and CO originate from CO2 photoreduction. In addition, the oxidation half-reaction product of O2 can be verified (Fig. S8, ESI†).23,25 After three cycles (Fig. 2d), the performance of Cu0.3Zn0.7S generating C2H4 remains 12.36 μmol g−1, with an electron selectivity of 41.2%. No significant changes in the XRD and TEM results before and after the cycling stability test are observed (Fig. S9 and S10, ESI†). However, the Cu+/Cu2+ ratio increases, as characterized by XPS (Fig. S11, ESI†), which may contribute to the observed changes in photocatalytic performance.11,26
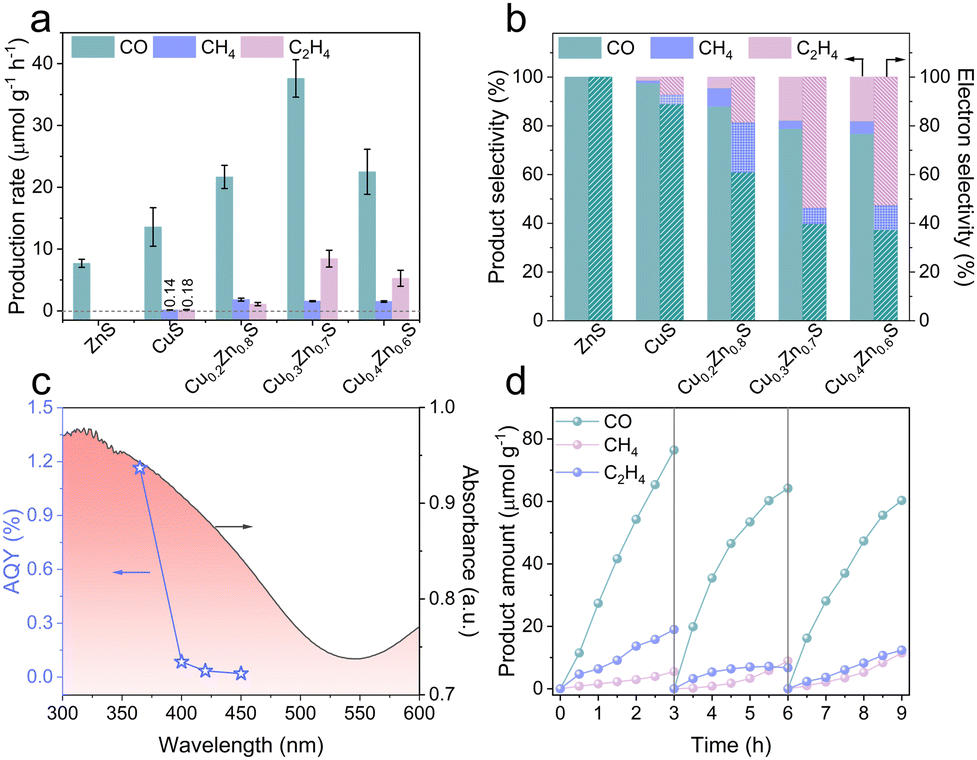 | ||
| Fig. 2 CO2PR performance: (a) production rates and (b) electron and product selectivity of the as-prepared photocatalysts. (c) DRS spectrum and AQY of Cu0.3Zn0.7S. (d) Cycling performance of Cu0.3Zn0.7S. | ||
As depicted in Fig. 3a and Fig. S12 (ESI†), ZnS exhibits strong absorption in the UV region with an absorption edge of around 365 nm, corresponding to a wide band gap of approximately 3.58 eV. Meanwhile, CuS shows broad absorption across the entire spectral range, indicative of a narrow band gap of about 1.73 eV. With increasing the Cu content, the light absorption of CuxZn1−xS gradually approaches that of CuS. The corresponding band gaps were calculated as 2.38, 2.02, and 1.88 eV for CuxZn1−xS (x = 0.2, 0.3, and 0.4), respectively. The valence band positions (Ev) of CuS, ZnS, and CuxZn1−xS were determined to be 0.87, 1.88, 1.01, 1.04 and 1.16 V (vs. NHE, pH = 7), respectively, from the UPS spectra (Fig. S13, ESI†). Consequently, the band structures of all synthesized samples are summarized in Fig. S14 (ESI†). Notably, the Cu/Zn ratio exerts a profound influence on the optical absorption, crystal structure, and electronic band configuration, which collectively determine the catalytic performance. Among them, the Cu0.3Zn0.7S with the optimal visible-light absorption and the highest catalytic activity, is selected as a representative for comparison with pristine ZnS and CuS to elucidate the underlying mechanism.
 | ||
| Fig. 3 (a) UV-vis DRS spectra of all samples. (b) Photocurrent response, (c) EIS spectra, (d) steady-state PL spectra, and (e) TRPL spectra of ZnS, CuS and Cu0.3Zn0.7S. (f) Schematic diagram of twin boundary dependent “back-to-back” potential in twin Cu0.3Zn0.7S. | ||
First of all, the charge-transfer dynamics is visited. A series of photoelectrochemical characterizations were conducted. As shown in Fig. 3b, all samples display an excellent photocurrent response under intermittent light irradiation. Cu0.3Zn0.7S exhibits the highest transient photocurrent density, indicative of its superior charge separation and transport. In addition, the EIS in terms of Nyquist plots (Fig. 3c) shows that Cu0.3Zn0.7S exhibits the smallest impedance, implying the lowest interfacial charge transfer resistance. Steady-state photoluminescence (PL) spectra (Fig. 3d) show significantly quenched emission for Cu0.3Zn0.7S as compared to pristine ZnS and CuS, suggesting effective suppression of charge recombination. Moreover, time-resolved photoluminescence (TRPL) analysis (Fig. 3e and Table S3, ESI†) reveals a prolonged average lifetime of photoinduced carriers in Cu0.3Zn0.7S (7.04 ns), compared to ZnS (2.48 ns) and CuS (4.17 ns), further confirming enhanced charge transfer. These results demonstrate that the Cu0.3Zn0.7S with twin homojunctions can effectively accelerate the generation and transfer of photo-excited electrons. The homojunctions formed by twin boundaries are arranged quasi-periodically and in parallel along specific crystallographic orientations, as evidenced by Fig. 1c. These periodic twin domains generate a series of spatially aligned, “back-to-back” electric fields within the Cu0.3Zn0.7S nanocrystals, as shown in Fig. 3f, which facilitate the directional migration of charge carriers. Specifically, photogenerated holes are preferentially trapped at twin boundaries, while electrons remain delocalized within the bulk domain.12,13,18 This spatial separation effectively suppresses charge recombination and significantly prolongs carrier lifetimes, thereby enhancing the participation of photogenerated carriers in surface redox reactions and boosting overall photocatalytic activity.
Prior to elucidating the reaction pathway of CO2PR by Cu0.3Zn0.7S, CO2-TPD analysis was conducted to probe the surface adsorption sites. As shown in Fig. 4a, CuS displays negligible CO2 desorption, suggesting a lack of effective adsorption sites. The desorption peak was located at 340 °C for ZnS, indicative of moderate CO2 binding strength.27 Remarkably, Cu0.3Zn0.7S shows a pronounced desorption peak at 323.1 °C, with substantially higher intensity than either CuS or ZnS, implying enhanced CO2 adsorption capacity. Furthermore, an additional desorption peak emerges at 426.2 °C for Cu0.3Zn0.7S, which can be attributed to the presence of strong chemisorption sites, likely associated with active centres induced by twin boundaries.28 Then, in situ DRIFTS measurements were carried out to experimentally probe the complicated process of CO2PR over twin Cu0.3Zn0.7S. As shown in Fig. 4b, several characteristic peaks, including HCO3− (1210 cm−1), m-CO32− (1288 cm−1), b-CO32− (1354 cm−1), CO2− (1675 cm−1) and *H2O (1630 cm−1), clearly indicate the co-adsorption and activation of CO2 and H2O on the surface of Cu0.3Zn0.7S.3,5 The *COOH, serving as a crucial intermediate in the conversion of CO2 to *CO and subsequent C–C coupling, is observed at 1579 cm−1.29 Besides, obvious linear-absorbed *CO peaks at 2016 and 2070 cm−1 and bridge-absorbed *CO peaks at 1833 and 1908 cm−1, can be observed, which directly represents the formation of *CO.30 The peaks located at 1084, 1170, 1487, and 1716 cm−1 are attributed to *CHO, a key intermediate for the formation of hydrocarbon products.3,5,31 In addition, the asymmetric vibration of *OCCO observed at 1530 cm−1 provides compelling evidence for the pathway of C2 product generation, suggesting that the C2 species originate from the coupling of *CO intermediates.5,7,31 Furthermore, the key C–C coupling intermediates, including *OCCOH (1570 cm−1),4 *OCCHOH (1305 cm−1),5 and *C2H4 (1418 and 1457 cm−1)5,31 were also detected, which provides direct evidence for a multiple electron-proton transfer process to generate C2H4. In parallel, the signals with respect to *CH3O (1028 and 1117 cm−1)32 and *CH2 (1375 cm−1)5 originating from *CHO intermediates, were also observed, suggesting their protonation pathways towards CH4 production. Similar spectral features were also observed for CuS (Fig. S15, ESI†), whereas no characteristic intermediates associated with hydrocarbon products were detected on ZnS (Fig. S16, ESI†). Notably, as shown in Fig. S17 (ESI†), only a linear adsorption *CO peak at 2077 cm−1 was observed for the ZnS sample, with no detection of bridge-bonded *CO species that are more readily activated, indicating the limited capability of ZnS for C–C coupling.30 In contrast, Cu0.3Zn0.7S exhibited significantly stronger *CO adsorption peaks with both linear and bridge adsorption compared to CuS. These results highlight the distinct advantage of the twin Cu0.3Zn0.7S in promoting CO adsorption and activation. CO-DRIFTS spectra (Fig. 4c) further reveal that for ZnS, only smooth gaseous CO features are detected at 2119 and 2171 cm−1,33 indicating that the ZnS surface exhibits negligible CO adsorption capability. In contrast, an adsorption peak at 2113 cm−1 is observed for CuS, which can be attributed to CO adsorption on Cu+ sites.33 Notably, in addition to the Cu+–CO adsorption for Cu0.3Zn0.7S, an additional adsorption peak appears at 2019 cm−1, which can be assigned to CO adsorption on Cu0 sites.34 This indicates that a mixed-valence state of Cu0/Cu+ exists in the twin Cu0.3Zn0.7S, which enhances the adsorption and activation of *CO intermediates to promote the C–C coupling. Moreover, CO-TPD analysis (Fig. S18, ESI†) shows that Cu0.3Zn0.7S exhibits the highest CO adsorption capacity and the lowest desorption temperature (∼320.4 °C), indicating a strong affinity to CO and excellent resistance to CO poisoning. These characteristics are conducive to enhancing *CO surface coverage and promoting the C–C coupling reactions. Based on the above results, the possible pathway of the conversion of CO2 to C2H4 can be deduced (Fig. 4d and Fig. S19, ESI†).
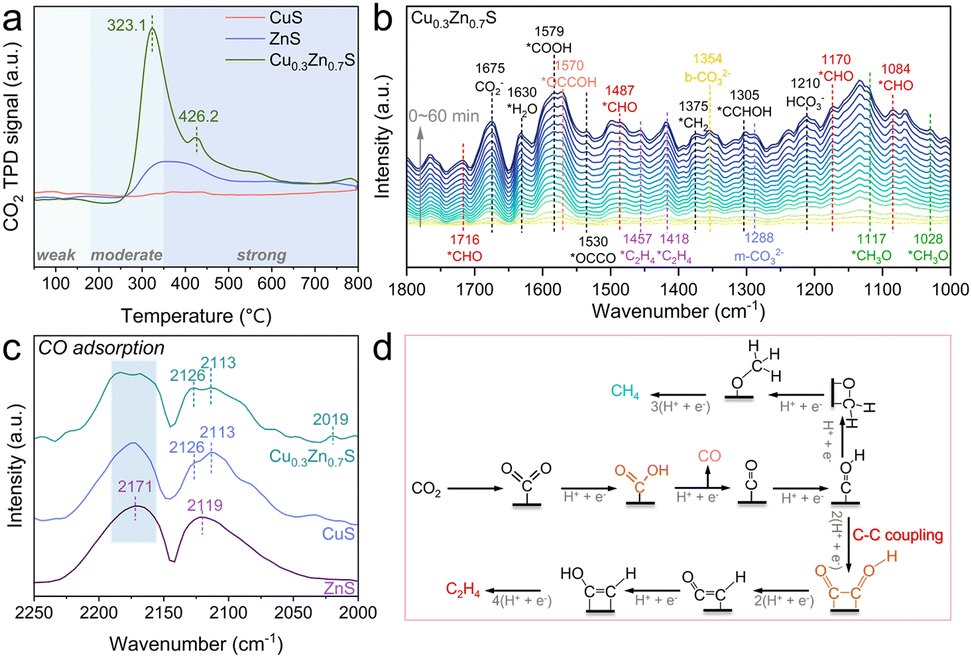 | ||
| Fig. 4 (a) CO2-TPD spectra of CuS, ZnS, and Cu0.3Zn0.7S. (b) In situ DRIFTS spectra for CO2PR on Cu0.3Zn0.7S. (c) In situ DRIFTS spectra for CO adsorbed on CuS, ZnS, and Cu0.3Zn0.7S. (d) Reaction pathway for the formation of CO, CH4 and C2H4 on Cu0.3Zn0.7S. | ||
In summary, this study reports the twin Cu0.3Zn0.7S solid solution for efficient CO2 photocatalytic reduction to C2H4. The formed surface “back-to-back” interfacial electric fields effectively promote the separation of photogenerated charge carriers and prolong the electron lifetime. Moreover, the presence of mixed-valence Cu species strengthens the adsorption of *CO intermediates, thereby facilitating the C–C coupling. This work provides new insights into rational design of twin-structured nanocrystal photocatalysts to boost the photocatalytic activity and selectivity of CO2 photoreduction to C2H4.
This work was financially supported by the National Natural Science Foundation of China (No. 51925601) and Innovative Research Group Project of the National Natural Science Foundation of China (No. 52021004).
Conflicts of interest
The authors declare no competing financial interest.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
- B. H. Zhao, et al., Nat. Sustainable, 2023, 6, 827–837 CrossRef
.
- E. Gomez, et al., Nat. Rev. Chem., 2019, 3, 638–649 CrossRef CAS
- Y. Xu, et al., Energy Environ. Sci., 2024, 17, 5060–5069 RSC
- W. Wang, et al., J. Am. Chem. Soc., 2021, 143, 2984–2993 CrossRef CAS PubMed
- Z. Xie, et al., Nat. Commun., 2024, 15, 2422 CrossRef CAS PubMed
- M. Xu, et al., Angew. Chem., Int. Ed., 2025, 64, e202506072 CrossRef CAS PubMed
- S. Gong, et al., Energy Environ. Sci., 2023, 16, 5956–5969 RSC
- W. Xie, et al., Adv. Mater., 2023, 35, 2208132 CrossRef CAS PubMed
- Y. Mao, et al., Adv. Sci., 2024, 11, 2401933 CrossRef CAS PubMed
- Y. Nie, et al., Appl. Catal., B, 2024, 345, 123704 CrossRef CAS
- J. Wang, et al., Adv. Funct. Mater., 2023, 33, 2213901 CrossRef CAS
- P. Guo, et al., ACS Energy Lett., 2023, 8, 494–501 CrossRef CAS
- K. Zhang, et al., Appl. Catal., B, 2024, 342, 123375 CrossRef CAS
- S. Sun, et al., Catal. Sci. Technol., 2020, 10, 4164–4178 RSC
- J. Chen, et al., Mater. Sci. Eng. R, 2024, 161, 100843 CrossRef
- Z. Lei, et al., J. Mater. Sci. Technol., 2025, 216, 81–92 CrossRef CAS
- C. Cheng, et al., Adv. Mater., 2021, 33, 2100317 CrossRef CAS PubMed
- M. Dan, et al., Adv. Mater., 2025, 37, 2415138 CrossRef CAS PubMed
- Y. Tang, et al., Energy Environ. Sci., 2024, 17, 7882–7894 RSC
- J. Li, et al., Chem. Commun., 2024, 60, 12393–12396 RSC
- S. Lie, et al., Mater. Horiz., 2025, 12, 2911–2921 RSC
- J. Zhang, et al., Angew. Chem., Int. Ed., 2025, 64, e202423861 CrossRef CAS PubMed
- B. Su, et al., Angew. Chem., Int. Ed., 2025, 64, e202505453 CrossRef PubMed
- R. Das, et al., J. Am. Chem. Soc., 2023, 145, 422–435 CrossRef CAS PubMed
- B. Su, et al., J. Am. Chem. Soc., 2023, 145, 27415–27423 CrossRef CAS PubMed
- X. Zhou, et al., J. Am. Chem. Soc., 2022, 144, 2079–2084 CrossRef CAS PubMed
- Y. Zhang, et al., ACS Mater. Lett., 2025, 7, 359–367 CrossRef CAS
- Y. Pu, et al., Appl. Catal., B, 2019, 254, 580–586 CrossRef CAS
- J. Zhu, et al., J. Am. Chem. Soc., 2021, 143, 18233–18241 CrossRef CAS PubMed
- C. Song, et al., Nat. Chem. Eng., 2024, 1, 638–649 CrossRef
- W. Song, et al., J. Am. Chem. Soc., 2024, 146, 29028–29039 CrossRef CAS PubMed
- M. Li, et al., J. Am. Chem. Soc., 2024, 146, 15538–15548 CrossRef CAS PubMed
- W. Lin, et al., ChemSusChem, 2021, 14, 3190–3197 CrossRef CAS PubMed
- I. Eswaramoorthi, et al., Appl. Catal., A, 2006, 313, 22–34 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc02418h |
| This journal is © The Royal Society of Chemistry 2025 |