
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Divergent hetero-[8+n] higher order cycloadditions of tropothione and enals catalyzed by N-heterocyclic carbenes†
Joanna Dybowskaa,
Artur Przydacza,
Weronika Olczyka,
Lesław Sieroń b,
Anna Skrzyńska*a and
Łukasz Albrecht*a
b,
Anna Skrzyńska*a and
Łukasz Albrecht*a
aInstitute of Organic Chemistry, Faculty of Chemistry, Lodz University of Technology, Żeromskiego 116, 90-924 Łódź, Poland. E-mail: anna.skrzynska@p.lodz.pl; lukasz.albrecht@p.lodz.pl; Web: https://www.a-teamlab.p.lodz.pl
bInstitute of General and Ecological Chemistry, Faculty of Chemistry, Lodz University of Technology, Żeromskiego 116, 90-924 Łódź, Poland
First published on 21st July 2025
Abstract
Divergent asymmetric NHC-catalyzed [8+n] higher-order cycloadditions using tropothione as an electron-poor 8π component were developed. The base-dependent selectivity of the synthetic approach allowed obtaining heterocyclic products bearing either γ- or δ-thiolactone rings with high enantioselectivity. The impact of base on NHC intermediate isomerization was explained by DFT studies. The diastereodivergency of the methodology was confirmed with both diastereomers being easy to isolate with very good results.
The introduction of orbital symmetry selection rules by Woodward and Hoffmann, which explain the course of cycloadditions, proved to be a seminal moment in organic chemistry.1 This important class of reactions is classified based on the number of electrons participating in the bond forming process. Transformations involving more than 6π-electrons overall are described as higher-order cycloadditions. Despite significant advancements in the understanding of “classical” cycloadditions, higher-order cycloadditions—especially when integrated with advanced principles of asymmetric organocatalysis—continue to be a dynamic and developing area of research providing valuable access to unique, chiral building blocks.2–4
Among various organocatalysts, N-heterocyclic carbenes stand out as highly effective tools in asymmetric synthesis, offering a versatile activation strategy that unlocks access to a broad range of non-classical reactivities.5 A variety of NHC-bound nucleophiles, obtained through the reaction of an NHC catalyst with an appropriate aldehyde, undergo reactions with carbon–heteroatom or carbon–carbon double bonds, leading to the formation of (hetero)cyclic systems.6 The effectiveness of NHC catalysis has also been demonstrated in higher-order cycloadditions, where it enables enantioselective reactions and facilitates the efficient construction of complex (hetero)polycyclic structures in a single step.7
Organocatalytic higher-order cycloadditions (HOCs) involving N-heterocyclic carbenes can be divided into two main categories. The first group relies on the generation of electron-rich NHC-bound hetero-higherenes,4 such as aza-fulvenes and aza-o-quinone methides derived from N-heteroaromatic aldehydes (Scheme 1, top). The second group includes HOCs in which NHC-derived nucleophilic intermediates act as higherenophiles4 in organocatalytic reactions with tropone and its electron-poor derivatives (Scheme 1, top). In this field, several examples of [8+n] cycloadditions have been reported; however, the asymmetric variants remain limited (Scheme 1, centre). The first study, conducted in a non-asymmetric setting, was reported by Nair's group. It utilized tropone as the 8π component in an [8+4]-annulation involving homoenolate intermediates derived from enals.8
 | ||
| Scheme 1 Organocatalytic higher-order cycloaddition reactions involving N-heterocyclic carbenes and the objectives of our studies. | ||
A similar approach, employing nucleophilic NHC-bound higherenophiles, was explored by Ye and co-workers, who developed an oxidative N-heterocyclic carbene-mediated [8+2]-cycloaddition between an enolate intermediate generated from simple alkyl aldehydes and tropone.9 Besides the above accomplishments, Pericas et al. successfully demonstrated that the chiral NHC-enolate intermediate reacted with tropone to deliver enantioenriched cycloadducts via a highly periselective [8+2] cycloaddition, representing a significant advancement in the development of stereoselective HOC reactions.10 Among the electron-poor troponoid systems, only azaheptafulvenes have been successfully used as hetero-8π-components in periselective [8+2] HOC.11
In our effort to further develop NHC-catalyzed HOCs we turned our attention to another tropone derivative – tropothione. This sulfur analogue is known for its reactivity as an electron-rich 8π-higherene.12 In reactions with LUMO-activated iminium ions, generated under aminocatalytic conditions, tropothione facilitates the efficient formation of hetero-[8+2] cycloaddition products (Scheme 1).12f Taking into account the low polarization of the tropothione system, we envisioned the possibility to inverse its most commonly observed reactivity.
Herein we report an unprecedent usage of tropothione as an electron-poor component in [8+n] higher-order cycloaddition involving NHC-bound higherenophiles. The developed approach enables the selective formation of [8+2] or [8+4] cycloaddition products via enolate or homoenolate intermediates, respectively, generated under NHC conditions (Scheme 1, bottom). Importantly, our work constitutes a significant contribution to NHC-catalyzed C–S bond formation, which remains underexplored compared to its more established applications in forming C–C and C–X bonds.13
Optimization studies were performed using tropothione 1 and trans-cinnamaldehyde 2a as model reactants. As the formation of three isomeric products cis-3a, trans-3a and 4a was observed (see Scheme 1, bottom), key reaction parameters enabling their selective formation were established (for details of screening studies, see the ESI†). For each of the three products the scope and limitations of developed synthetic methods were explored (see Schemes 2–4).
 | ||
| Scheme 2 [8+2]-Cycloaddition of tropothione 1 and α,β-unsaturated aldehyde 2 – scope leading to cis-3. | ||
 | ||
| Scheme 3 [8+2]-Cycloaddition of tropothione 1 and α,β-unsaturated aldehyde 2 – scope leading to trans-3. | ||
 | ||
| Scheme 4 [8+4]-Cycloaddition of tropothione 1 and α,β-unsaturated aldehyde 2 – scope leading to 4. | ||
Primarily the described methodology provided access to two diastereomeric products. It was cis-diastereoselective with the formation of cis-3 being accomplished after 2–18 hours (Scheme 2). Products cis-3 were obtained in good yields and with excellent enantioselectivity.
Increasing the cesium carbonate amount and prolonging the reaction time resulted in cis to trans epimerization of cis-3 and after 24–72 hours trans-3 was observed in the reaction mixture as the sole product (Scheme 3). This is in accordance with previous studies by Pericas et al.10 A control experiment using cis-3a in the presence of cesium carbonate was also performed (for details, see the ESI†).
All products trans-3a–q were afforded in good or very good yields, regardless of the position of substituents on the phenyl ring or the presence of a heteroaromatic furan ring. A slight decrease of enantioselectivity was observed for derivatives trans-3b–d and trans-3o containing strong electron-donating methoxy groups. The loss in enantiomeric excess during cis to trans epimerization was also noted. This suggests that the epimerization of the β-stereocenter also takes place but to a much smaller extent than the epimerization of the α-stereocenter. The reaction using aliphatic crotonaldehyde 2r was also performed and afforded product trans-3r with excellent diastereoselectivity, but moderate enantioselectivity and in 49% yield.
A method for the synthesis of [8+4]-cycloadduct 4 was also developed (with the selection of base, solvent and temperature being of key importance). A range of compounds 4a–h bearing a δ-thiolactone ring were obtained (Scheme 4). Unfortunately, the stereoselectivity of the process was not as satisfactory as that for the presented [8+2]-cycloaddition. The best results were observed for compound 4b containing a strong electron-donating methoxy group in the ortho position of the phenyl ring. For derivatives with electron-withdrawing substituents, the heteroaromatic furan ring or aliphatic methyl group, products 4e–h were obtained in a notably lower yield.
To demonstrate the applicability of the developed synthetic method, cycloadduct 3a was subjected to selected transformations (Scheme 5). Chemoselective reduction of double bonds on the cycloheptatriene ring was performed affording 6 in a very good 73% yield, with great diastereoselectivity (Scheme 5, top). Compound 3a was also employed as an electron-rich diene in the hetero-Diels–Alder reaction with triazole derivative 7 (Scheme 5, top). Reaction proceeded smoothly, giving product 8 in a good 66% yield and with good diastereoselectivity. Cycloadduct 4a was also subjected to the hetero-Diels–Alder reaction with 7. To our surprise, different product 9 was obtained (Scheme 5, middle). Its formation can be rationalized assuming that the cascade reaction involving two pericyclic reactions took place. Initial 6π-electrocyclic disrotatory ring closure led to cyclopropane and cyclohexadiene ring formation. Subsequent Diels–Alder cycloaddition provided 9 in a highly diastereoselective manner. The absolute configurations of transformation products 8 and 9 were assigned based on X-ray single crystal analysis.14 Due to the retention of the configuration on stereogenic centers in the thiolactone ring during the performed transformations of 3a and 4a, the stereochemistry of all compounds 3 and 4 was assigned by analogy. Experiments on a 1 mmol scale were also conducted, and product 3a was obtained with the same stereoselectivity as initially, but in slightly lower yields (Scheme 5, bottom).
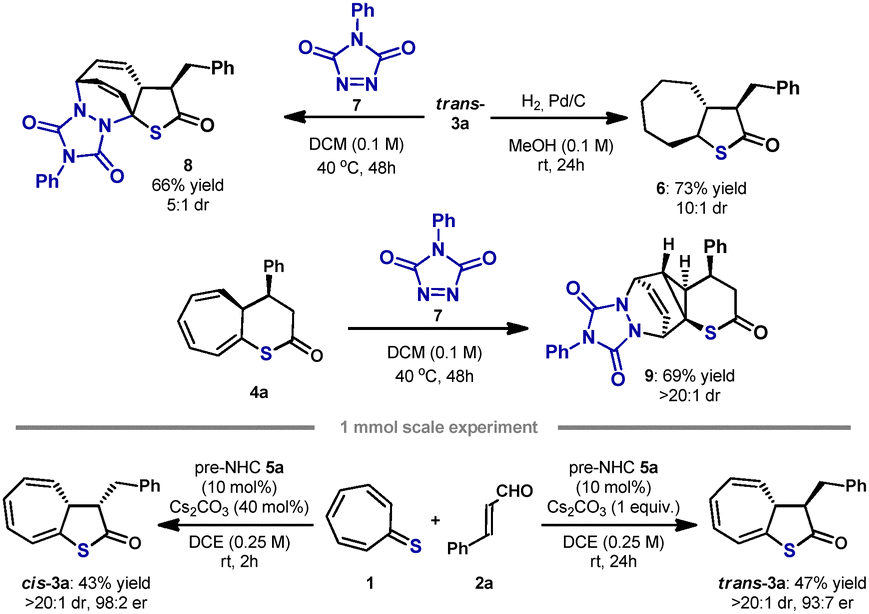 | ||
| Scheme 5 [8+2]-Cycloaddition of tropothione 1 and α,β-unsaturated aldehyde 2 – transformation of products 3a and 4a. | ||
To explain the base-dependent selectivity of the reported synthetic approach DFT studies were performed (Scheme 6). Since [8+4] and [8+2] HOCs proceed through different interchangeable intermediates (homoenolate INT1 and enolate INT2, respectively) we envisioned that the type of base may affect the proton transfer process that regulates our transformation of INT1 into INT2. Importantly, according to studies, INT2 is 19.0 kcal mol−1 more stable than its precursor INT1. Indeed, the presence of bicarbonate lowers the energy barrier for proton transfer to 16.6 kcal mol−1 as compared to 25.1 kcal mol−1 for unassisted intramolecular isomerization. The ability of bicarbonate relies on its bidentate character – an H-bond donor and acceptor – which results in a concerted mechanism of H-transfer. On the other hand, triethylamine used in the [8+4] protocol, as a bulky single H-bond donor, cannot assist in isomerization (i.e. the transition state for concerted proton transfer involving triethylamine could not be localized), and thus the process occurs in an intramolecular fashion. These findings are in line with experimental results: [8+2] HOC proceeds at room temperature which enables the intramolecular H-transfer to take place and leads to the thermodynamically more stable INT2. In contrast, the [8+4] HOC is conducted at 5 °C, as low temperature prevents the energetically costly intramolecular isomerization. A detailed mechanism is presented in Scheme 6.
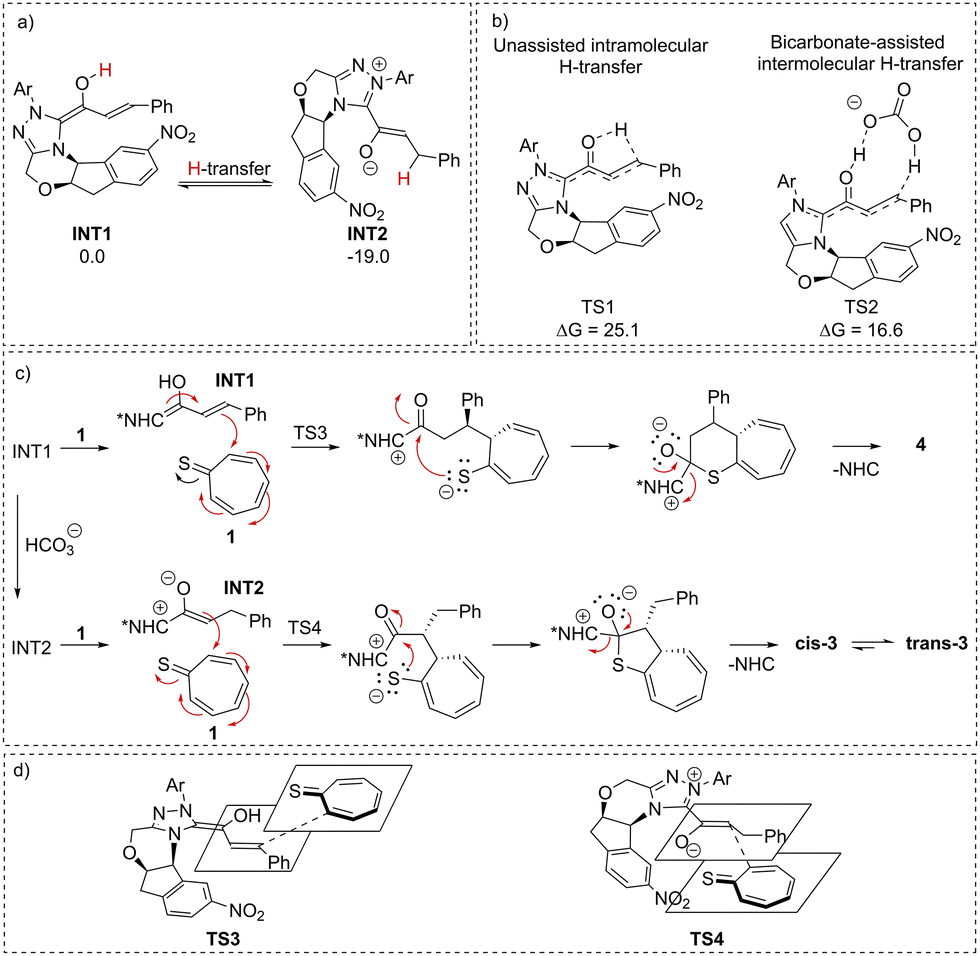 | ||
| Scheme 6 (a) Relative energies of two isomeric NHC-bound intermediates; (b) energy barriers for intramolecular (left) and carbonate-assisted (right) proton transfer for INT1 to INT2 isomerization; (c) proposed mechanism for investigated [8+4] (top) and [8+2] (bottom) higher-order cycloadditions; and (d) proposed models of transition states explaining the stereochemical outcome of the presented HOCs. | ||
In summary, we have developed divergent asymmetric [8+n] higher-order cycloadditions based on NHC catalysis. The reactivity inverts the classical reactivity of tropothione that serves as an electron-poor 8π component in cycloadditions described. By the selection of the base, the chemoselectivity of the process could be controlled, resulting in heterocyclic products bearing either γ- or δ-thiolactone rings with high enantioselectivity. This observation was explained following DFT studies. The diastereodivergency of the method was achievable by control of the reaction time with both diastereomers accessible with very good results. The applicability of the methodology was confirmed in interesting transformations leading to diverse polycyclic products.
This project was realized within the Opus programme (grant number: UMO-2021/41/B/ST4/03385) from the National Science Centre, Poland. This contribution has been completed while the first author (JD) was the Doctoral Candidate in the Interdisciplinary Doctoral School at the Lodz University of Technology, Poland.
Conflicts of interest
There are no conflicts to declare.Data availability
Data for this article are available at the Lodz University of Technology Research Data Repository at https://doi.org/10.34658/RDB.7FQRXV.Notes and references
- R. B. Woodward and R. Hoffmann, The Conservation of Orbital Symmetry, Wiley, 1970 Search PubMed
.
-
(a) R. B. Woodward and R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1969, 8, 781 CrossRef CAS
- For selected reviews, see:
(a) J. H. Rigby, Acc. Chem. Res., 1993, 26, 579–585 CrossRef CAS
- For nomenclature of higherene and higherenophiles, see: S. Frankowski, M. Romaniszyn, A. Skrzyńska and Ł. Albrecht, Chem. Eur. J., 2020, 26, 2120–2132 CrossRef CAS PubMed
- For selected reviews, see:
(a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed
- For selected reviews, see:
(a) T. Okano, Heterocycl. Commun., 2013, 19, 311–326 CrossRef CAS
-
(a) X. Yang, G. Luo, L. Zhou, B. Liu, X. Zhang, H. Gao, Z. Jin and Y. R. Chi, ACS Catal., 2019, 9, 10971–10976 CrossRef CAS
- V. Nair, M. Poonoth, S. Vellalath, E. Suresh and R. Thirumalai, J. Org. Chem., 2006, 71, 8964–8965 CrossRef CAS PubMed
- F. Xia, Z.-H. Gao, C.-L. Zhang and S. Ye, Adv. Synth. Catal., 2019, 361, 2291–2294 CrossRef CAS
- S. Wang, C. Rodríguez-Escrich, M. Fianchini, F. Maseras and M. A. Pericàs, Org. Lett., 2019, 21, 3187–3192 CrossRef CAS PubMed
- C. He, Z. Li, H. Zhou and J. Xu, Org. Lett., 2019, 21, 8022–8026 CrossRef CAS PubMed
- For selected examples, see:
(a) T. Machiguchi, T. Hasegawa, S. Itoh and H. Mizuno, J. Am. Chem. Soc., 1989, 111, 1920–1921 CrossRef CAS
- J. Lv, Z. Jin, H. Wang and Y. R. Chi, ACS Sustainable Chem. Eng., 2022, 10, 13901–13916 CrossRef CAS
- CCDC 2430652 (8) and 2430653 (9) contain the supplementary crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2430652 and 2430653. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc02536b |
| This journal is © The Royal Society of Chemistry 2025 |